Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment

 , , and
, , and
Abstract
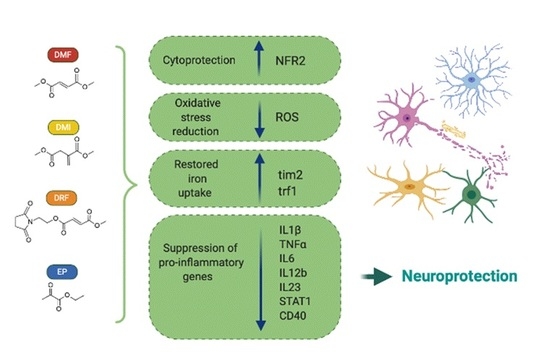
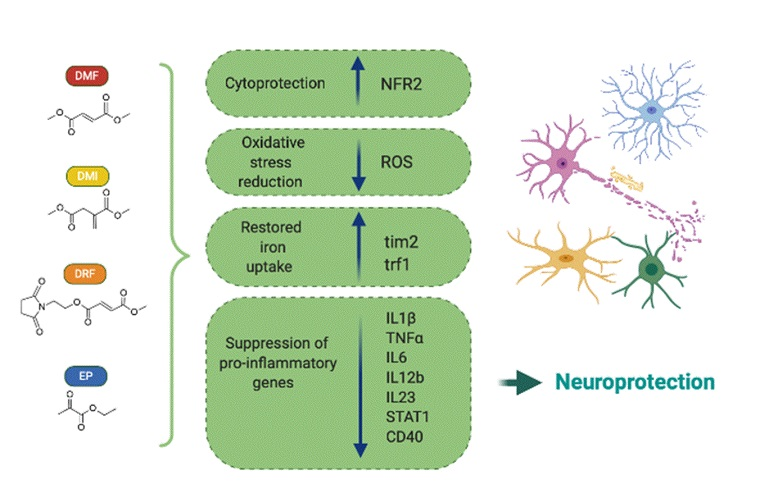
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
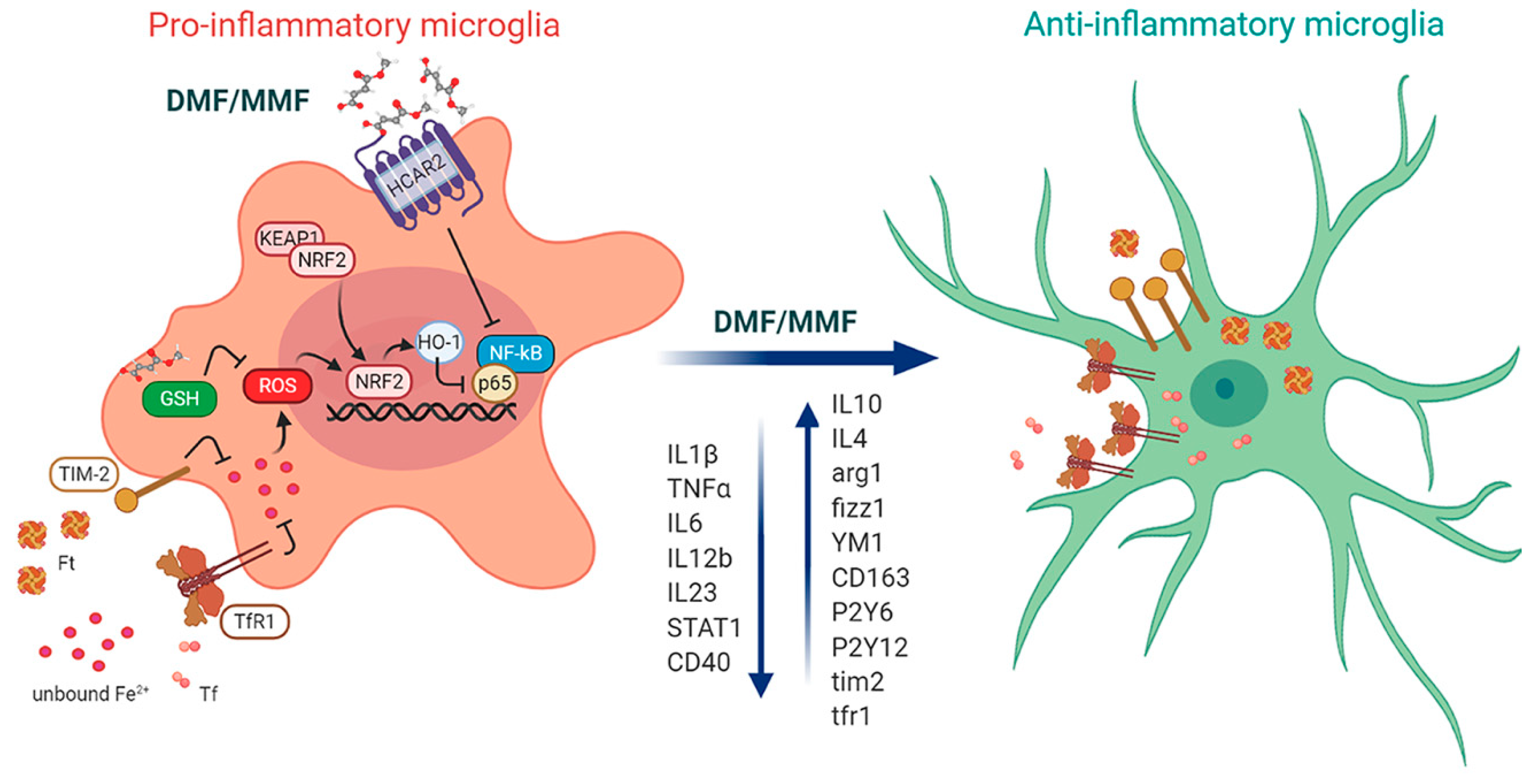
2. Biochemical Targets of DMF
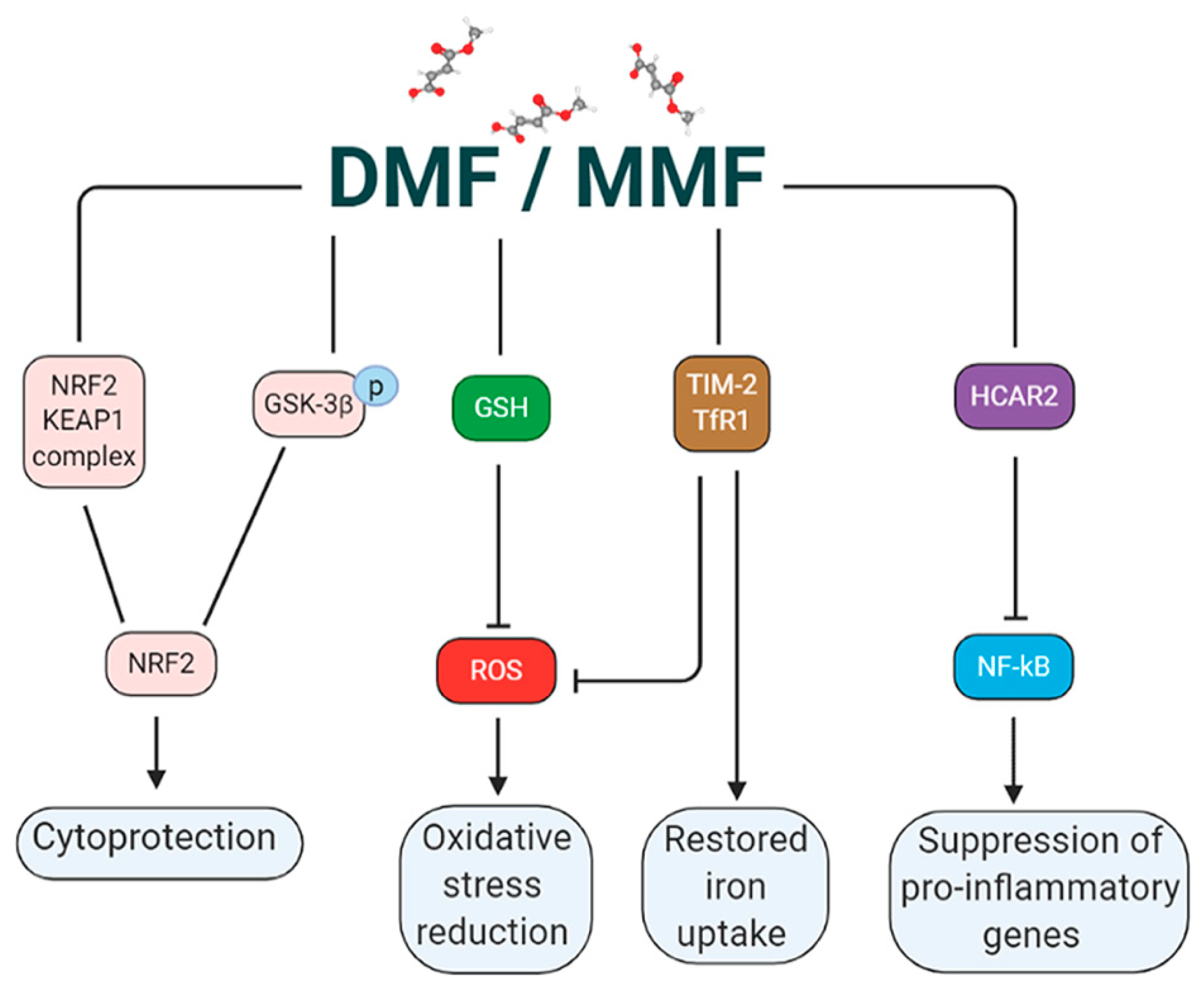
2.1. DMF Activates the NRF2 Pathway via Multiple Mechanisms
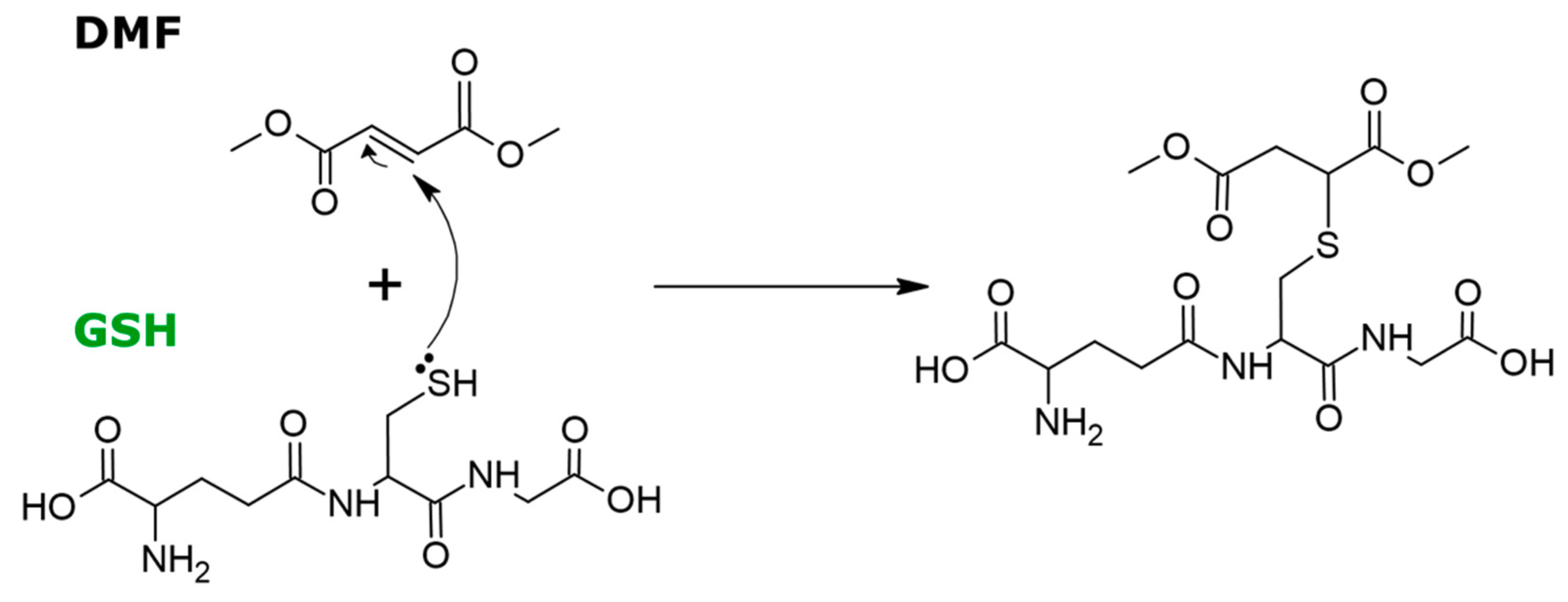
2.2. DMF Modulates GSH
2.3. DMF Activates HCAR2 in a NRF2 Independent Pathway
3. DMF Modulates Microglia Functions under Homeostatic and Pathological Conditions
4. DMF Targets in Neurodegenerative Disorders
5. DMF as Regulator of Iron Metabolism for the Treatment of Neurodegenerative Diseases
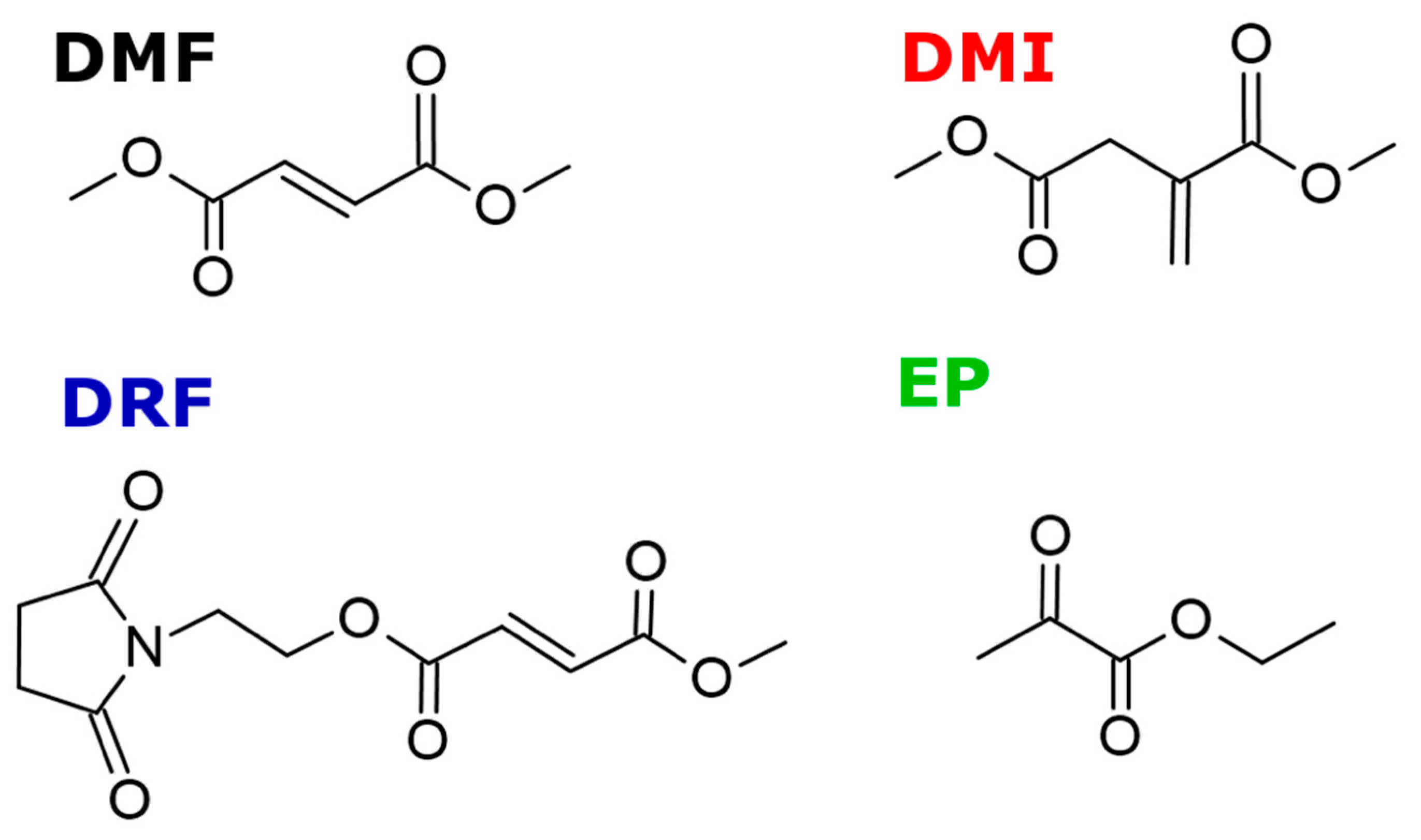
6. Potential DMF Alternatives: Emerging Anti-Inflammatory and Anti-Oxidant Compounds
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation List
References
- Mrowietz, U.; Barker, J.; Boehncke, W.H.; Iversen, L.; Kirby, B.; Naldi, L.; Reich, K.; Tanew, A.; van de Kerkhof, P.C.M.; Warren, R.B. Clinical use of dimethyl fumarate in moderate-to-severe plaque-type psoriasis: A European expert consensus. J. Eur. Acad. Dermatol. Venereol. 2018, 32 (Suppl. 3), 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheremata, W.; Brown, A.D.; Rammohan, K.W. Dimethyl fumarate for treating relapsing multiple sclerosis. Expert Opin. Drug Saf. 2015, 14, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Ciccarelli, O.; Barkhof, F.; Bodini, B.; De Stefano, N.; Golay, X.; Nicolay, K.; Pelletier, D.; Pouwels, P.J.; Smith, S.A.; Wheeler-Kingshott, C.A.; et al. Pathogenesis of multiple sclerosis: Insights from molecular and metabolic imaging. Lancet Neurol. 2014, 13, 807–822. [Google Scholar] [CrossRef]
- Yshii, L.; Gebauer, C.; Bernard-Valnet, R.; Liblau, R. Neurons and T cells: Understanding this interaction for inflammatory neurological diseases. Eur. J. Immunol. 2015, 45, 2712–2720. [Google Scholar] [CrossRef]
- Hoffmann, F.; Meinl, E. B cells in multiple sclerosis: Good or bad guys. An article for 28 May 2014-World MS Day 2014. Eur. J. Immunol. 2014, 44, 1247–1250. [Google Scholar] [CrossRef]
- Nishihara, H.; Soldati, S.; Mossu, A.; Rosito, M.; Rudolph, H.; Muller, W.A.; Latorre, D.; Sallusto, F.; Sospedra, M.; Martin, R.; et al. Human CD4. Fluids Barriers CNS 2020, 17, 3. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, F.J.; Khademi, M.; Aram, J.; Ammann, S.; Kockum, I.; Constantinescu, C.; Gran, B.; Piehl, F.; Olsson, T.; Codarri, L.; et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat. Commun. 2014, 5, 5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Ak, M.; Mrowietz, U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine—Preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem. 2007, 15, 333–342. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Dringen, R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem. Int. 2010, 57, 460–467. [Google Scholar] [CrossRef]
- Palte, M.J.; Wehr, A.; Tawa, M.; Perkin, K.; Leigh-Pemberton, R.; Hanna, J.; Miller, C.; Penner, N. Improving the Gastrointestinal Tolerability of Fumaric Acid Esters: Early Findings on Gastrointestinal Events with Diroximel Fumarate in Patients with Relapsing-Remitting Multiple Sclerosis from the Phase 3, Open-Label EVOLVE-MS-1 Study. Adv. Ther. 2019, 36, 3154–3165. [Google Scholar] [CrossRef] [Green Version]
- Miljković, D.; Blaževski, J.; Petković, F.; Djedović, N.; Momčilović, M.; Stanisavljević, S.; Jevtić, B.; Mostarica Stojković, M.; Spasojević, I. A comparative analysis of multiple sclerosis-relevant anti-inflammatory properties of ethyl pyruvate and dimethyl fumarate. J. Immunol. 2015, 194, 2493–2503. [Google Scholar] [CrossRef]
- Boyland, E.; Chasseaud, L.F. Enzyme-catalysed conjugations of glutathione with unsaturated compounds. Biochem. J. 1967, 104, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef]
- Piroli, G.G.; Manuel, A.M.; Patel, T.; Walla, M.D.; Shi, L.; Lanci, S.A.; Wang, J.; Galloway, A.; Ortinski, P.I.; Smith, D.S.; et al. Identification of Novel Protein Targets of Dimethyl Fumarate Modification in Neurons and Astrocytes Reveals Actions Independent of Nrf2 Stabilization. Mol. Cell. Proteom. 2019, 18, 504–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Gillard, G.O.; Collette, B.; Anderson, J.; Chao, J.; Scannevin, R.H.; Huss, D.J.; Fontenot, J.D. DMF, but not other fumarates, inhibits NF-κB activity in vitro in an Nrf2-independent manner. J. Neuroimmunol. 2015, 283, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Li, H.; Sheehy, A.; Cullen, P.; Allaire, N.; Scannevin, R.H. Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. J. Neuroimmunol. 2016, 299, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, 0120254. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, M.; Hoda, N. GSK3 Inhibitors in the Therapeutic Development of Diabetes, Cancer and Neurodegeneration: Past, Present and Future. Curr. Pharm. Des. 2017, 23, 4332–4350. [Google Scholar] [CrossRef]
- Wadhwa, P.; Jain, P.; Jadhav, H.R. Glycogen Synthase Kinase 3 (GSK3): Its Role and Inhibitors. Curr. Top. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Noack, H.; Possel, H.; Keilhoff, G.; Wolf, G. Glutathione levels in primary glial cultures: Monochlorobimane provides evidence of cell type-specific distribution. Glia 1999, 27, 152–161. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Gutterer, J.M.; Kussmaul, L.; Hamprecht, B.; Dringen, R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci. 2000, 22, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hollensworth, S.B.; Shen, C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Cooper, A.J.; Kristal, B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997, 378, 793–802. [Google Scholar]
- Landeck, L.; Asadullah, K.; Amasuno, A.; Pau-Charles, I.; Mrowietz, U. Dimethyl fumarate (DMF) vs. monoethyl fumarate (MEF) salts for the treatment of plaque psoriasis: A review of clinical data. Arch. Dermatol. Res. 2018, 310, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Chen, H.; Assmann, J.C.; Krenz, A.; Rahman, M.; Grimm, M.; Karsten, C.M.; Köhl, J.; Offermanns, S.; Wettschureck, N.; Schwaninger, M. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J. Clin. Investig. 2014, 124, 2188–2192. [Google Scholar] [CrossRef]
- Lin-Holderer, J.; Li, L.; Gruneberg, D.; Marti, H.H.; Kunze, R. Fumaric acid esters promote neuronal survival upon ischemic stress through activation of the Nrf2 but not HIF-1 signaling pathway. Neuropharmacology 2016, 105, 228–240. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration. Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rábano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195. [Google Scholar] [CrossRef] [Green Version]
- Lindenau, J.; Noack, H.; Asayama, K.; Wolf, G. Enhanced cellular glutathione peroxidase immunoreactivity in activated astrocytes and in microglia during excitotoxin induced neurodegeneration. Glia 1998, 24, 252–256. [Google Scholar] [CrossRef]
- Power, J.H.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chuikov, S.; Taitano, S.; Wu, Q.; Rastogi, A.; Tuck, S.J.; Corey, J.M.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl Fumarate Protects Neural Stem/Progenitor Cells and Neurons from Oxidative Damage through Nrf2-ERK1/2 MAPK Pathway. Int. J. Mol. Sci. 2015, 16, 13885–13907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiner, B.; Romanelli, E.; Liberski, P.; Ingold-Heppner, B.; Sobottka-Brillout, B.; Hartwig, T.; Chandrasekar, V.; Johannssen, H.; Zeilhofer, H.U.; Aguzzi, A.; et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015, 12, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Pars, K.; Gingele, M.; Kronenberg, J.; Prajeeth, C.K.; Skripuletz, T.; Pul, R.; Jacobs, R.; Gudi, V.; Stangel, M. Fumaric Acids Do Not Directly Influence Gene Expression of Neuroprotective Factors in Highly Purified Rodent Astrocytes. Brain Sci. 2019, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Kurian, M.A.; McNeill, A.; Lin, J.P.; Maher, E.R. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev. Med. Child. Neurol. 2011, 53, 394–404. [Google Scholar] [CrossRef]
- Schneider, S.A.; Hardy, J.; Bhatia, K.P. Syndromes of neurodegeneration with brain iron accumulation (NBIA): An update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov. Disord. 2012, 27, 42–53. [Google Scholar] [CrossRef]
- Todorich, B.; Zhang, X.; Connor, J.R. H-ferritin is the major source of iron for oligodendrocytes. Glia 2011, 59, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Sosa, J.C.; Gardeck, A.M.; Baez, A.S.; Lee, C.H.; Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagani, F.; Testi, C.; Grimaldi, A.; Corsi, G.; Cortese, B.; Basilico, B.; Baiocco, P.; De Panfilis, S.; Ragozzino, D.; Di Angelantonio, S. Dimethyl Fumarate Reduces Microglia Functional Response to Tissue Damage and Favors Brain Iron Homeostasis. Neuroscience 2019. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Carmichael, S.T. Glia in neurodegeneration: Drivers of disease or along for the ride. Neurobiol. Dis. 2020, 142, 104957. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Garretti, F.; Sulzer, D.; Sette, A. Roles for the adaptive immune system in Parkinson’s and Alzheimer’s diseases. Curr. Opin. Immunol. 2019, 59, 115–120. [Google Scholar] [CrossRef]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Grimaldi, A.; Pediconi, N.; Oieni, F.; Pizzarelli, R.; Rosito, M.; Giubettini, M.; Santini, T.; Limatola, C.; Ruocco, G.; Ragozzino, D.; et al. Neuroinflammatory Processes, A1 Astrocyte Activation and Protein Aggregation in the Retina of Alzheimer’s Disease Patients, Possible Biomarkers for Early Diagnosis. Front. Neurosci. 2019, 13, 925. [Google Scholar] [CrossRef] [Green Version]
- Rygiel, K. Novel strategies for Alzheimer’s disease treatment: An overview of anti-amyloid beta monoclonal antibodies. Indian J. Pharmacol. 2016, 48, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bona, D.; Scapagnini, G.; Candore, G.; Castiglia, L.; Colonna-Romano, G.; Duro, G.; Nuzzo, D.; Iemolo, F.; Lio, D.; Pellicanò, M.; et al. Immune-inflammatory responses and oxidative stress in Alzheimer’s disease: Therapeutic implications. Curr. Pharm. Des. 2010, 16, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, P.; Bouchachia, I.; Goebels, N.; Henke, N.; Hofstetter, H.H.; Issberner, A.; Kovacs, Z.; Lewerenz, J.; Lisak, D.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 2012, 9, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33. [Google Scholar] [CrossRef]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat. Rev. Neurol. 2015, 11, 25–40. [Google Scholar] [CrossRef]
- Zhao, N.; Yang, X.; Calvelli, H.R.; Cao, Y.; Francis, N.L.; Chmielowski, R.A.; Joseph, L.B.; Pang, Z.P.; Uhrich, K.E.; Baum, J.; et al. Antioxidant Nanoparticles for Concerted Inhibition of α-Synuclein Fibrillization, and Attenuation of Microglial Intracellular Aggregation and Activation. Front. Bioeng. Biotechnol. 2020, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. Erratum: T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 549, 292. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.; Shi, H.; Zhang, C.; Ren, M.; Han, M.; Wei, X.; Zhang, X.; Lou, H. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 2015, 286, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P.V. Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease. Neural Regen. Res. 2018, 13, 1493–1506. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Özdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef]
- Group, W.; Group, E.M.-A.S. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K.; et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017, 2, 89530. [Google Scholar] [CrossRef]
- Vucic, S.; Ryder, J.; Mekhael, L.; Rd, H.; Mathers, S.; Needham, M.; Dw, S.; Mc, K.; TEALS Study Group. Phase 2 randomized placebo controlled double blind study to assess the efficacy and safety of tecfidera in patients with amyotrophic lateral sclerosis (TEALS Study): Study protocol clinical trial (SPIRIT Compliant). Medicine 2020, 99, 18904. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Chen, X.; Li, S.; Shang, H. Abnormal Serum Iron-Status Indicator Changes in Amyotrophic Lateral Sclerosis (ALS) Patients: A Meta-Analysis. Front. Neurol. 2020, 11, 380. [Google Scholar] [CrossRef]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural Transm. 2011, 118, 301–314. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, F.; Hametner, S.; Yao, B.; van Gelderen, P.; Merkle, H.; Cantor, F.K.; Lassmann, H.; Duyn, J.H. Tracking iron in multiple sclerosis: A combined imaging and histopathological study at 7 Tesla. Brain 2011, 134, 3602–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartzokis, G.; Tishler, T.A. MRI evaluation of basal ganglia ferritin iron and neurotoxicity in Alzheimer’s and Huntingon’s disease. Mol. Cell. Biol. 2000, 46, 821–833. [Google Scholar]
- Mehta, V.; Pei, W.; Yang, G.; Li, S.; Swamy, E.; Boster, A.; Schmalbrock, P.; Pitt, D. Iron is a sensitive biomarker for inflammation in multiple sclerosis lesions. PLoS ONE 2013, 8, 57573. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.A.; Connor, J.R. Ferritin, transferrin, and iron in selected regions of the adult and aged rat brain. J. Comp. Neurol. 1993, 338, 97–113. [Google Scholar] [CrossRef]
- Connor, J.R.; Menzies, S.L. Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: The role of iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Ortiz, E.; Pasquini, J.M.; Thompson, K.; Felt, B.; Butkus, G.; Beard, J.; Connor, J.R. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J. Neurosci. Res. 2004, 77, 681–689. [Google Scholar] [CrossRef]
- Zhang, X.; Surguladze, N.; Slagle-Webb, B.; Cozzi, A.; Connor, J.R. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia 2006, 54, 795–804. [Google Scholar] [CrossRef]
- Hallgren, B.; Sourander, P. The effect of age on the non-haemin iron in the human brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Craelius, W.; Migdal, M.W.; Luessenhop, C.P.; Sugar, A.; Mihalakis, I. Iron deposits surrounding multiple sclerosis plaques. Arch. Pathol. Lab. Med. 1982, 106, 397–399. [Google Scholar] [PubMed]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Götz, M.E.; Double, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Adlard, P.A.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Finkelstein, D.I. Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: Implications for Parkinson’s disease. Mol. Neurodegener. 2014, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef] [Green Version]
- Connor, J.R. Myelin breakdown in Alzheimer’s disease: A commentary. Neurobiol. Aging 2004, 25, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Chen, K.M.; Ling, H.W.; Sun, F.; Li, X.; Wan, T.; Chai, W.M.; Zhang, H.; Zhan, Y.; Guan, Y.J. Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J. Magn. Reson. Imaging 2009, 29, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Langkammer, C.; Ropele, S.; Pirpamer, L.; Fazekas, F.; Schmidt, R. MRI for iron mapping in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.Z.; Zhong, W.D.; Wang, W.; Zhan, C.J.; Wang, C.Y.; Qi, J.P.; Wang, J.Z.; Lei, T. Quantitative MR phase-corrected imaging to investigate increased brain iron deposition of patients with Alzheimer disease. Radiology 2009, 253, 497–504. [Google Scholar] [CrossRef]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Takanashi, M.; Mochizuki, H.; Yokomizo, K.; Hattori, N.; Mori, H.; Yamamura, Y.; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Parkinsonism Relat. Disord. 2001, 7, 311–314. [Google Scholar] [CrossRef]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Arawaka, S.; Saito, Y.; Murayama, S.; Mori, H. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 1998, 51, 887–889. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef] [Green Version]
- Ostrerova-Golts, N.; Petrucelli, L.; Hardy, J.; Lee, J.M.; Farer, M.; Wolozin, B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 2000, 20, 6048–6054. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Hsu, L.J.; Xia, Y.; Takeda, A.; Sisk, A.; Sundsmo, M.; Masliah, E. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 1999, 10, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeineh, M.M.; Chen, Y.; Kitzler, H.H.; Hammond, R.; Vogel, H.; Rutt, B.K. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [Green Version]
- Bou-Abdallah, F. The iron redox and hydrolysis chemistry of the ferritins. Biochim. Biophys. Acta 2010, 1800, 719–731. [Google Scholar] [CrossRef]
- Harrison, P.M.; Banyard, S.H.; Hoare, R.J.; Russell, S.M.; Treffry, A. The structure and function of ferritin. Ciba Found. Symp. 1976, 19–40. [Google Scholar] [CrossRef]
- Honarmand Ebrahimi, K.; Bill, E.; Hagedoorn, P.L.; Hagen, W.R. The catalytic center of ferritin regulates iron storage via Fe(II)-Fe(III) displacement. Nat. Chem. Biol. 2012, 8, 941–948. [Google Scholar] [CrossRef]
- Montemiglio, L.C.; Testi, C.; Ceci, P.; Falvo, E.; Pitea, M.; Savino, C.; Arcovito, A.; Peruzzi, G.; Baiocco, P.; Mancia, F.; et al. Cryo-EM structure of the human ferritin-transferrin receptor 1 complex. Nat. Commun. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Testi, C.; Boffi, A.; Montemiglio, L.C. Structural analysis of the transferrin receptor multifaceted ligand(s) interface. Biophys. Chem. 2019, 254, 106242. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.X.; Duce, J.A. The iron regulatory capability of the major protein participants in prevalent neurodegenerative disorders. Front. Pharmacol. 2014, 5, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef]
- Todorich, B.; Zhang, X.; Slagle-Webb, B.; Seaman, W.E.; Connor, J.R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 2008, 107, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Goldstein, E.Z.; Sahinkaya, F.R.; Wei, P.; Popovich, P.G.; McTigue, D.M. Ferritin stimulates oligodendrocyte genesis in the adult spinal cord and can be transferred from macrophages to NG2 cells in vivo. J. Neurosci. 2012, 32, 5374–5384. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- McNamara, N.B.; Miron, V.E. Microglia in developing white matter and perinatal brain injury. Neurosci. Lett. 2020, 714, 134539. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [Green Version]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. Macrophages: Central regulators of iron balance. Metallomics 2014, 6, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I. Drug development based on the metals hypothesis of Alzheimer’s disease. J. Alzheimers Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Guo, C.; Wang, T.; Zheng, W.; Shan, Z.Y.; Teng, W.P.; Wang, Z.Y. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, Y.X.; Wang, T.; Zhong, M.L.; Yang, Z.H.; Hao, L.J.; Chai, R.; Zhang, S. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Sangchot, P.; Sharma, S.; Chetsawang, B.; Porter, J.; Govitrapong, P.; Ebadi, M. Deferoxamine attenuates iron-induced oxidative stress and prevents mitochondrial aggregation and alpha-synuclein translocation in SK-N-SH cells in culture. Dev. Neurosci. 2002, 24, 143–153. [Google Scholar] [CrossRef]
- Kronenberg, J.; Pars, K.; Brieskorn, M.; Prajeeth, C.K.; Heckers, S.; Schwenkenbecher, P.; Skripuletz, T.; Pul, R.; Pavlou, A.; Stangel, M. Fumaric Acids Directly Influence Gene Expression of Neuroprotective Factors in Rodent Microglia. Int. J. Mol. Sci. 2019, 20, 325. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Taraboletti, A.; Shriver, L.P. Dimethyl fumarate modulates antioxidant and lipid metabolism in oligodendrocytes. Redox Biol. 2015, 5, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Litjens, N.H.; van Strijen, E.; van Gulpen, C.; Mattie, H.; van Dissel, J.T.; Thio, H.B.; Nibbering, P.H. In vitro pharmacokinetics of anti-psoriatic fumaric acid esters. BMC Pharmacol. 2004, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Kuo, P.C.; Weng, W.T.; Scofield, B.A.; Paraiso, H.C.; Brown, D.A.; Wang, P.Y.; Yu, I.C.; Yen, J.H. Dimethyl itaconate, an itaconate derivative, exhibits immunomodulatory effects on neuroinflammation in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Miljković, D.; Spasojević, I. Multiple sclerosis: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2013, 19, 2286–2334. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Englert, J.A.; Yang, R.; Delude, R.L.; Fink, M.P. Ethyl pyruvate inhibits nuclear factor-kappaB-dependent signaling by directly targeting p65. J. Pharmacol. Exp. Ther. 2005, 312, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.K.; Fink, M.P. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochem. Pharmacol. 2010, 80, 151–159. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, S.W.; Jin, Y.; Kim, I.D.; Lee, J.K. Ethyl pyruvate-mediated Nrf2 activation and hemeoxygenase 1 induction in astrocytes confer protective effects via autocrine and paracrine mechanisms. Neurochem. Int. 2012, 61, 89–99. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, H.K.; Shin, J.H.; Lee, J.K. Up-down regulation of HO-1 and iNOS gene expressions by ethyl pyruvate via recruiting p300 to Nrf2 and depriving It from p65. Free Radic. Biol. Med. 2013, 65, 468–476. [Google Scholar] [CrossRef]
- Fink, M.P. Ethyl pyruvate: A novel anti-inflammatory agent. J. Intern. Med. 2007, 261, 349–362. [Google Scholar] [CrossRef]
- Kalariya, N.M.; Reddy, A.B.; Ansari, N.H.; VanKuijk, F.J.; Ramana, K.V. Preventive effects of ethyl pyruvate on endotoxin-induced uveitis in rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5144–5152. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, H.; Song, J.; Dong, H.; Yao, L.; Liang, Z.; Lv, Y.; Zou, F.; Cai, S. Ethyl pyruvate decreases airway neutrophil infiltration partly through a high mobility group box 1-dependent mechanism in a chemical-induced murine asthma model. Int. Immunopharmacol. 2014, 21, 163–170. [Google Scholar] [CrossRef]
- Kim, J.B.; Yu, Y.M.; Kim, S.W.; Lee, J.K. Anti-inflammatory mechanism is involved in ethyl pyruvate-mediated efficacious neuroprotection in the postischemic brain. Brain Res. 2005, 1060, 188–192. [Google Scholar] [CrossRef]
- Su, X.; Wang, H.; Zhu, L.; Zhao, J.; Pan, H.; Ji, X. Ethyl pyruvate ameliorates intracerebral hemorrhage-induced brain injury through anti-cell death and anti-inflammatory mechanisms. Neuroscience 2013, 245, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, H.L.; Pu, H.J.; Shi, Y.J.; Zhang, J.; Zhang, W.T.; Wang, G.H.; Hu, X.M.; Leak, R.K.; Chen, J.; et al. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. CNS Neurosci. Ther. 2015, 21, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Khasnavis, S.; Jana, A.; Roy, A.; Mazumder, M.; Bhushan, B.; Wood, T.; Ghosh, S.; Watson, R.; Pahan, K. Suppression of nuclear factor-κB activation and inflammation in microglia by physically modified saline. J. Biol. Chem. 2012, 287, 29529–29542. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosito, M.; Testi, C.; Parisi, G.; Cortese, B.; Baiocco, P.; Di Angelantonio, S. Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment. Antioxidants 2020, 9, 700. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9080700
Rosito M, Testi C, Parisi G, Cortese B, Baiocco P, Di Angelantonio S. Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment. Antioxidants. 2020; 9(8):700. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9080700
Chicago/Turabian StyleRosito, Maria, Claudia Testi, Giacomo Parisi, Barbara Cortese, Paola Baiocco, and Silvia Di Angelantonio. 2020. "Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment" Antioxidants 9, no. 8: 700. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9080700