Oxidative Stress and Neuroinflammation as a Pivot in Drug Abuse. A Focus on the Therapeutic Potential of Antioxidant and Anti-Inflammatory Agents and Biomolecules

 , and
, and
Abstract
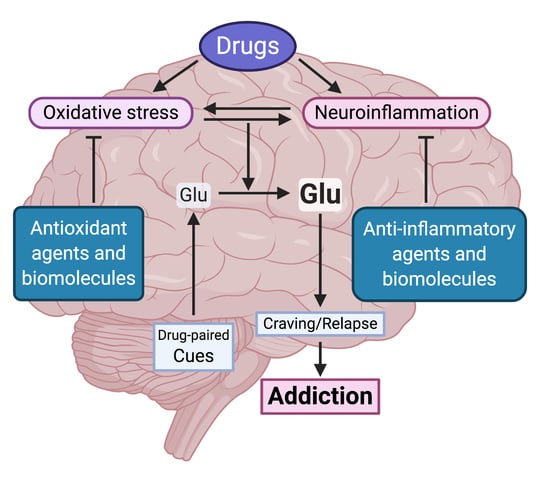
:
1. Introduction
1.1. Drug Abuse and Current Treatments
1.2. Drugs of Abuse Activate the Brain Reward System
2. The Oxidative Stress and Inflammation Role in Drug Consumption
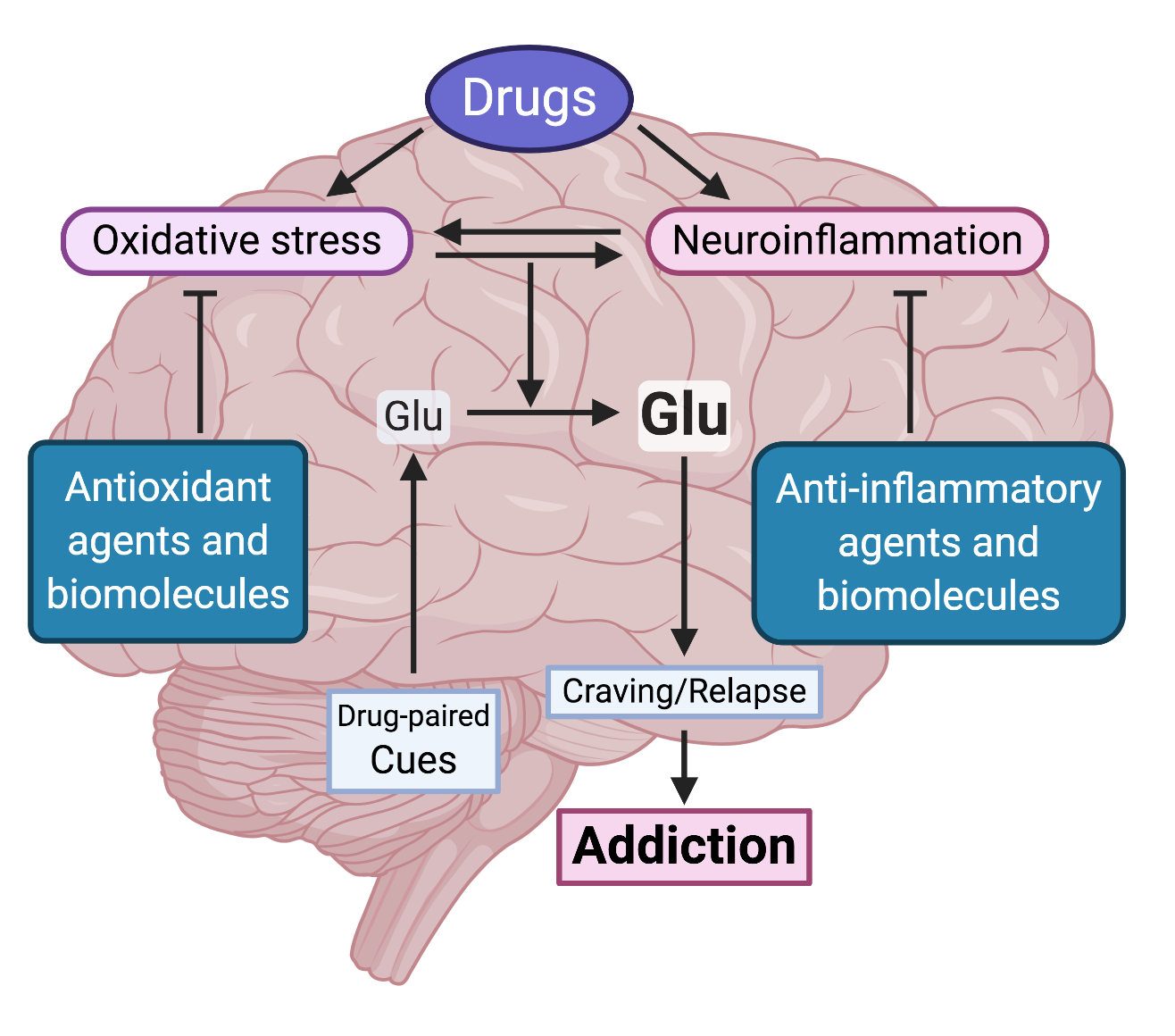
2.1. The Brain Oxidative Stress and Neuroinflammation Vicious Cycle
2.2. Drug Consumption Promotes Brain Oxidative Stress and Neuroinflammation
2.2.1. Dopamine Oxidation
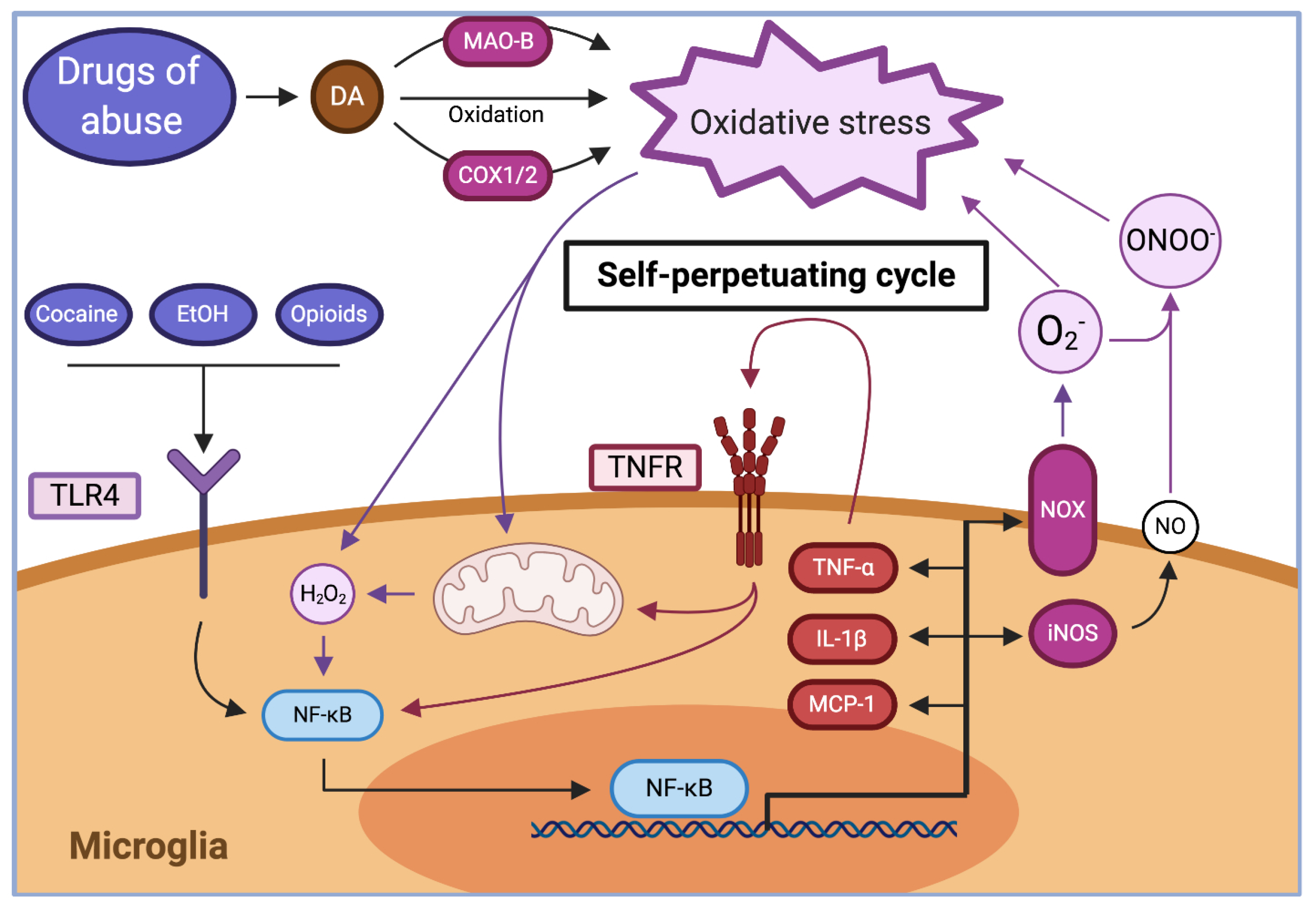
2.2.2. Inhibition of System Xc− Activity
2.2.3. Drug-Induced Mitochondrial Dysfunction
2.2.4. Peripheral Inflammation Contributes to Neuroinflammation
2.2.5. Activation of Toll-like Receptors
2.3. Effect of Oxidative Stress and Inflammation on Glutamate Signaling
2.3.1. Glutamate Transport Activity Is Impaired by Brain Oxidative Stress and Inflammation Components
2.3.2. Drugs of Abuse Modify the Extracellular Levels of Glutamate
2.3.3. The Recovery of GLT-1 and System Xc− Activities Inhibits Drug Seeking and Reinstatement
3. Evidence for the Therapeutic Potential of Antioxidant and Anti-inflammatory Agents for the Treatment of Chronic Drug Abuse
3.1. N-Acetylcysteine (NAC)
3.2. Ibudilast
3.3. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
4. Biomolecules as Potential Treatments of Drug Abuse
4.1. Mesenchymal Stem Cells and Their Products
4.1.1. MSCs Intra-Cranial Administration Reduces Ethanol Intake and Relapse in Rats
4.1.2. MSCs-Derived Secretome: A Safer Product Recapitulates the Effect of Living MSCs Administration
4.1.3. MSCs-Derived Secretome Administration Reduces Drug Consumption and Relapse in Animal Models
4.2. MicroRNA
4.2.1. MicroRNA Expressed After Pro-inflammatory or Anti-inflammatory Signals in the Brain
4.2.2. Pro- and Anti-Inflammatory miRNAs in Neurodegenerative Diseases. The case of miR-155, miR-146, and miR-124
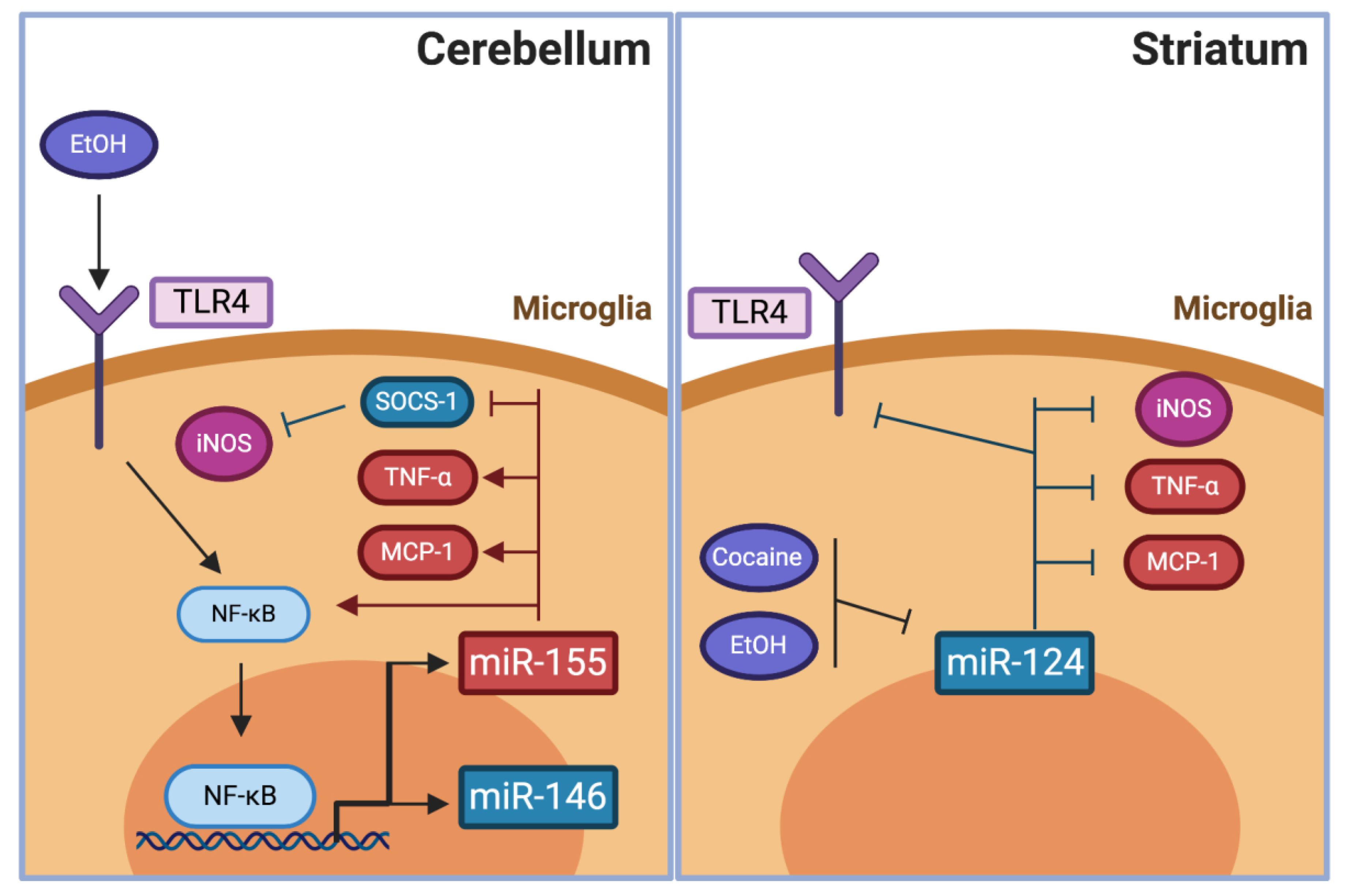
4.2.3. The potential of Pro- and Anti-Inflammatory miRNA in the Modulation of Drug Abuse
MiR-155
MiR-146
MiR-124
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- WHO. WHO Report on the Global Tobacco Epidemic 2019: Offer Help to Quit Tobacco Use; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- UNODC. World Drug Report 2019; UNODC: Vienna, Austria, 2019. [Google Scholar]
- Bhalla, I.P.; Stefanovics, E.A.; Rosenheck, R.A. Clinical epidemiology of single versus multiple substance use disorders: Polysubstance use disorder. Med. Care 2017, 55, S24–S32. [Google Scholar] [CrossRef] [PubMed]
- Van Skike, C.; Maggio, S.; Reynolds, A.; Casey, E.; Bardo, M.; Dwoskin, L.; Prendergast, M.; Nixon, K. Critical needs in drug discovery for cessation of alcohol and nicotine polysubstance abuse. Progress Neuro Psychopharmacol. Biol. Psychiatr. 2016, 65, 269–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIDA. Trends & Statistics. Available online: https://www.drugabuse.gov/related-topics/trends-statistics (accessed on 8 June 2020).
- Peacock, A.; Leung, J.; Larney, S.; Colledge, S.; Hickman, M.; Rehm, J.; Giovino, G.A.; West, R.; Hall, W.; Griffiths, P. Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction 2018, 113, 1905–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soyka, M.; Müller, C.A. Pharmacotherapy of alcoholism–an update on approved and off-label medications. Exp. Opin. Pharmacother. 2017, 18, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Coronado, N.; Walker, A.J.; Berk, M.; Dodd, S. Current and emerging pharmacotherapies for cessation of tobacco smoking. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 235–258. [Google Scholar] [CrossRef] [PubMed]
- FDA. Information about Medication-Assisted Treatment (Mat). Available online: https://www.fda.gov/drugs/information-drug-class/information-about-medication-assisted-treatment-mat (accessed on 8 June 2020).
- Siefried, K.J.; Acheson, L.S.; Lintzeris, N.; Ezard, N. Pharmacological treatment of methamphetamine/amphetamine dependence: A systematic review. CNS Drugs 2020, 34, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, V.; Bowling, K.C. Topiramate in the treatment of cocaine use disorder. Bull. Am. Soc. Hosp. Pharm. 2018, 75, e13–e22. [Google Scholar] [CrossRef]
- Jonas, D.E.; Amick, H.R.; Feltner, C.; Bobashev, G.; Thomas, K.; Wines, R.; Kim, M.M.; Shanahan, E.; Gass, C.E.; Rowe, C.J.; et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: A systematic review and meta-analysis. JAMA 2014, 311, 1889–1900. [Google Scholar] [CrossRef]
- Jordan, C.J.; Xi, Z.-X. Discovery and development of varenicline for smoking cessation. Exp. Opin. Drug Discov. 2018, 13, 671–683. [Google Scholar] [CrossRef]
- Ebbert, J.O.; Croghan, I.T.; Sood, A.; Schroeder, D.R.; Hays, J.T.; Hurt, R.D. Varenicline and bupropion sustained-release combination therapy for smoking cessation. Nicotine Tob. Res. 2009, 11, 234–239. [Google Scholar] [CrossRef]
- Dahan, A. Opioid-induced respiratory effects: New data on buprenorphine. Palliat. Med. 2006, 20 (Suppl. 1), s3–s8. [Google Scholar]
- Koehl, J.L.; Zimmerman, D.E.; Bridgeman, P.J. Medications for management of opioid use disorder. Am. J. Health Syst. Pharm. 2019, 76, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.O.; Kunins, H.V.; Roose, R.J.; Elam, R.T.; Sohler, N.L. Barriers to obtaining waivers to prescribe buprenorphine for opioid addiction treatment among hiv physicians. J. Gen. Intern. Med. 2007, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, F.; Marie, N. Management of opioid addiction with opioid substitution treatments: Beyond methadone and buprenorphine. Front. Psychiatr. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Koob, G.F.; McLellan, A.T. Neurobiologic advances from the brain disease model of addiction. N. Eng. J. Med. 2016, 374, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikemoto, S. Dopamine reward circuitry: Two projection systems from the ventral midbrain to the nucleus accumbens–olfactory tubercle complex. Brain Res. Rev. 2007, 56, 27–78. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.W.; North, R.A. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992, 12, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Sora, I.; Hall, F.S.; Andrews, A.M.; Itokawa, M.; Li, X.-F.; Wei, H.-B.; Wichems, C.; Lesch, K.-P.; Murphy, D.L.; Uhl, G.R. Molecular mechanisms of cocaine reward: Combined dopamine and serotonin transporter knockouts eliminate cocaine place preference. Proc. Natl. Acad. Sci. USA 2001, 98, 5300–5305. [Google Scholar] [CrossRef] [Green Version]
- Budygin, E.A.; John, C.E.; Mateo, Y.; Jones, S.R. Lack of cocaine effect on dopamine clearance in the core and shell of the nucleus accumbens of dopamine transporter knock-out mice. J. Neurosci. 2002, 22, RC222-RC222. [Google Scholar] [CrossRef]
- Jones, S.R.; Gainetdinov, R.R.; Wightman, R.M.; Caron, M.G. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 1998, 18, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Israel, Y.; Quintanilla, M.E.; Karahanian, E.; Rivera-Meza, M.; Herrera-Marschitz, M. The “first hit” toward alcohol reinforcement: Role of ethanol metabolites. Alcohol Clin. Exp. Res. 2015, 39, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Berrios-Carcamo, P.; Quintanilla, M.E.; Herrera-Marschitz, M.; Vasiliou, V.; Zapata-Torres, G.; Rivera-Meza, M. Racemic salsolinol and its enantiomers act as agonists of the mu-opioid receptor by activating the gi protein-adenylate cyclase pathway. Front. Behav. Neurosci. 2016, 10, 253. [Google Scholar] [PubMed] [Green Version]
- Berríos-Cárcamo, P.; Rivera-Meza, M.; Herrera-Marschitz, M.; Zapata-Torres, G. Molecular modeling of salsolinol, a full gi protein agonist of the μ-opioid receptor, within the receptor binding site. Chem. Biol. Drug Design 2019, 94, 1467–1477. [Google Scholar]
- Xie, G.; Hipolito, L.; Zuo, W.; Polache, A.; Granero, L.; Krnjevic, K.; Ye, J.H. Salsolinol stimulates dopamine neurons in slices of posterior ventral tegmental area indirectly by activating mu-opioid receptors. J. Pharmacol. Exp. Ther. 2012, 341, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, B.; Siemes, S.; Wallmichrath, I. Short communication inhibition of gabaergic neurotransmission in the ventral tegmental area by cannabinoids. Eur. J. Neurosci. 2002, 15, 2057–2061. [Google Scholar] [CrossRef]
- Pons, S.; Fattore, L.; Cossu, G.; Tolu, S.; Porcu, E.; McIntosh, J.; Changeux, J.; Maskos, U.; Fratta, W. Crucial role of α4 and α6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J. Neurosci. 2008, 28, 12318–12327. [Google Scholar] [CrossRef] [Green Version]
- Pontieri, F.E.; Tanda, G.; Orzi, F.; Di Chiara, G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 1996, 382, 255–257. [Google Scholar] [CrossRef]
- Koob, G.F. Neurobiology of addiction. Focus 2011, 9, 55–65. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron 2011, 69, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Di Chiara, G.; Bassareo, V. Reward system and addiction: What dopamine does and doesn’t do. Curr. Opin. Pharmacol. 2007, 7, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.; Roberts-Wolfe, D.; Kalivas, P.W. The nucleus accumbens: Mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornish, J.L.; Kalivas, P.W. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J. Neurosci. 2000, 20, RC89-RC89. [Google Scholar] [CrossRef] [Green Version]
- Bechard, A.R.; Knackstedt, L.A. Glutamatergic neuroplasticity in addiction. In Neural Mechanisms of Addiction; Elsevier: Amsterdam, The Netherlands, 2019; pp. 61–74. [Google Scholar]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the mechanisms of cns immune privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Kraft, A.D.; Harry, G.J. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of tnf. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by apc/c–cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.-L.; Wang, H.-H.; Wu, W.-B.; Chu, P.-J.; Yang, C.-M. Transforming growth factor-β1 induces matrix metalloproteinase-9 and cell migration in astrocytes: Roles of ros-dependent erk-and jnk-nf-κb pathways. J. Neuroinflamm. 2010, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Min, J.-S.; Kim, B.; Chae, U.-B.; Yun, J.W.; Choi, M.-S.; Kong, I.-K.; Chang, K.-T.; Lee, D.-S. Mitochondrial ros govern the lps-induced pro-inflammatory response in microglia cells by regulating mapk and nf-κb pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. Tnf and ros crosstalk in inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic lps causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Cahill, C.M.; Taylor, A.M. Neuroinflammation-a co-occurring phenomenon linking chronic pain and opioid dependence. Curr. Opin. Behav. Sci. 2017, 13, 171–177. [Google Scholar] [CrossRef]
- Leclercq, S.; De Timary, P.; Delzenne, N.M.; Stärkel, P. The link between inflammation, bugs, the intestine and the brain in alcohol dependence. Transl. Psychiatr. 2017, 7, e1048. [Google Scholar] [CrossRef] [PubMed]
- Hofford, R.S.; Russo, S.J.; Kiraly, D.D. Neuroimmune mechanisms of psychostimulant and opioid use disorders. Eur. J. Neurosci. 2019, 50, 2562–2573. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Link, J.; Dennis, L.E.; McCready, H.; Huckans, M.; Hoffman, W.F.; Loftis, J.M. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol. Biochem. Behav. 2019, 179, 34–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, X.; Bai, Y.; Zhu, X.; Wu, X.; Chao, J.; Duan, M.; Buch, S.; Chen, L.; Yao, H. Involvement of sigma-1 receptor in astrocyte activation induced by methamphetamine via up-regulation of its own expression. J. Neuroinflamm. 2015, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.; Zhang, Y.; Du, L.; Zhou, R.; Wu, X.; Shen, K.; Yao, H. Molecular mechanisms underlying the involvement of the sigma-1 receptor in methamphetamine-mediated microglial polarization. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.-L.; Buch, S. Cocaine-mediated downregulation of mir-124 activates microglia by targeting klf4 and tlr4 signaling. Molecular Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Baliño, P.; Aragón, C.M.; Guerri, C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of tlr4 and tlr2. Neuropharmacology 2015, 89, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal role of tlr4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Morales, P.; Ezquer, F.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Commonality of ethanol and nicotine reinforcement and relapse in wistar-derived uchb rats: Inhibition by n-acetylcysteine. Alcohol. Clin. Exp. Res. 2018, 42, 1988–1999. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Santapau, D.; Berríos-Cárcamo, P.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Intranasal mesenchymal stem cell secretome administration markedly inhibits alcohol and nicotine self-administration and blocks relapse-intake: Mechanism and translational options. Stem Cell Res. Ther. 2019, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Loram, L.C.; Ramos, K.; De Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamberletti, E.; Gabaglio, M.; Prini, P.; Rubino, T.; Parolaro, D. Cortical neuroinflammation contributes to long-term cognitive dysfunctions following adolescent delta-9-tetrahydrocannabinol treatment in female rats. Eur. Neuropsychopharmacol. 2015, 25, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Cutando, L.; Busquets-Garcia, A.; Puighermanal, E.; Gomis-González, M.; Delgado-García, J.M.; Gruart, A.; Maldonado, R.; Ozaita, A. Microglial activation underlies cerebellar deficits produced by repeated cannabis exposure. J. Clin. Investig. 2013, 123, 2816–2831. [Google Scholar] [CrossRef] [PubMed]
- Munoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Parkinson’s Dis. 2012, 2012. [Google Scholar] [CrossRef]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, oxidative stress and protein–quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew. Chem. Int. Ed. 2019, 58, 6512–6527. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Approaches to prevent dopamine quinone-induced neurotoxicity. Neurochem. Res. 2009, 34, 698–706. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Hastings, T.G. Enzymatic oxidation of dopamine: The role of prostaglandin h synthase. J. Neurochem. 1995, 64, 919–924. [Google Scholar] [CrossRef]
- Ramkissoon, A.; Wells, P.G. Human prostaglandin h synthase (hphs)-1-and hphs-2-dependent bioactivation, oxidative macromolecular damage, and cytotoxicity of dopamine, its precursor, and its metabolites. Free Radic. Biol. Med. 2011, 50, 295–304. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. CCS 2013, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Skrabalova, J.; Drastichova, Z.; Novotny, J. Morphine as a potential oxidative stress-causing agent. Mini Rev. Org. Chem. 2013, 10, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, D.M.; Francescutti-Verbeem, D.M.; Thomas, D.M. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: Relationship to methamphetamine-induced nerve ending damage. Ann. N. Y. Acad. Sci. 2006, 1074, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.A.; Xi, Z.-X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosc. 2002, 22, 9134–9141. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking outside the cleft to understand synaptic activity: Contribution of the cystine-glutamate antiporter (system Xc−) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.A.; McFarland, K.; Lake, R.W.; Shen, H.; Tang, X.-C.; Toda, S.; Kalivas, P.W. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 2003, 6, 743–749. [Google Scholar] [CrossRef]
- Madayag, A.; Lobner, D.; Kau, K.S.; Mantsch, J.R.; Abdulhameed, O.; Hearing, M.; Grier, M.D.; Baker, D.A. Repeated n-acetylcysteine administration alters plasticity-dependent effects of cocaine. J. Neurosci. 2007, 27, 13968–13976. [Google Scholar] [CrossRef] [Green Version]
- Amaral, V.C.S.; Morais-Silva, G.; Laverde, C.F.; Marin, M.T. Susceptibility to extinction and reinstatement of ethanol-induced conditioned place preference is related to differences in astrocyte cystine-glutamate antiporter content. Neurosci. Res. 2020, in press. [Google Scholar] [CrossRef]
- Knackstedt, L.A.; LaRowe, S.; Mardikian, P.; Malcolm, R.; Upadhyaya, H.; Hedden, S.; Markou, A.; Kalivas, P.W. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol. Psychiatr. 2009, 65, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Ghasemitarei, M.; Yusupov, M.; Razzokov, J.; Shokri, B.; Bogaerts, A. Effect of oxidative stress on cystine transportation by xc− antiporter. Arch. Biochem. Biophys. 2019, 674, 108114. [Google Scholar] [CrossRef]
- Sadakierska-Chudy, A.; Frankowska, M.; Filip, M. Mitoepigenetics and drug addiction. Pharmacol. Ther. 2014, 144, 226–233. [Google Scholar] [CrossRef]
- Mansouri, A.; Demeilliers, C.; Amsellem, S.; Pessayre, D.; Fromenty, B. Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart, and skeletal muscles: Protective effects of antioxidants. J. Pharmacol. Exp. Ther. 2001, 298, 737–743. [Google Scholar] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Silva, L.; Silva, A.M.; Moreno, A.J.; Oliveira, C.R.; Santos, M.S. Mitochondrial complex i dysfunction induced by cocaine and cocaine plus morphine in brain and liver mitochondria. Toxicol. Lett. 2013, 219, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex i inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [Green Version]
- Thangaraj, A.; Periyasamy, P.; Guo, M.-L.; Chivero, E.T.; Callen, S.; Buch, S. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy 2020, 16, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Chivero, E.T.; Ahmad, R.; Thangaraj, A.; Periyasamy, P.; Kumar, B.; Kroeger, E.; Feng, D.; Guo, M.-L.; Roy, S.; Dhawan, P. Cocaine induces inflammatory gut milieu by compromising the mucosal barrier integrity and altering the gut microbiota colonization. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Narvaez, J.C.; Magalhães, P.V.; Fries, G.R.; Colpo, G.D.; Czepielewski, L.S.; Vianna, P.; Chies, J.A.B.; Rosa, A.R.; Von Diemen, L.; Vieta, E. Peripheral toxicity in crack cocaine use disorders. Neurosci. Lett. 2013, 544, 80–84. [Google Scholar] [CrossRef]
- Moreira, F.P.; Medeiros, J.R.C.; Lhullier, A.C.; De Mattos Souza, L.D.; Jansen, K.; Portela, L.V.; Lara, D.R.; Da Silva, R.A.; Wiener, C.D.; Oses, J.P. Cocaine abuse and effects in the serum levels of cytokines il-6 and il-10. Drug Alcohol Depend. 2016, 158, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Meier, P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl. Res. 2007, 149, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, X.; Ma, Q.; Xu, Q.; Duan, W.; Lei, J.; Zhang, L.; Wu, Z. Chronic alcohol exposure exacerbates inflammation and triggers pancreatic acinar-to-ductal metaplasia through pi3k/akt/ikk. Int. J. Mol. Med. 2015, 35, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav. Immun. 2015, 44, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.; Benavidez, J.M.; Geil, C.; Perra, S.; Morikawa, H.; Harris, R. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav. Immun. 2011, 25, S92–S105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, S.; Cani, P.D.; Neyrinck, A.M.; Stärkel, P.; Jamar, F.; Mikolajczak, M.; Delzenne, N.M.; De Timary, P. Role of intestinal permeability and inflammation in the Biol.ical and behavioral control of alcohol-dependent subjects. Brain Behav. Immun. 2012, 26, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; De Saeger, C.; Delzenne, N.; De Timary, P.; Stärkel, P. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. Biol. Psychiatr. 2014, 76, 725–733. [Google Scholar] [CrossRef]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Cho, Y.; Ambade, A.; Szabo, G. Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J. Neuroinflamm. 2018, 15, 298. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Innate gut microbiota is required for the acquisition of ethanol intake and relapse binge-drinking by wistar-derived high drinker rats. 43rd annual poster abstracts of the research society on alcoholism jointly with the international society for biomedical research on alcoholism, June 2020. Alcohol. Clin. Exp. Res. 2020, 44 (Suppl. 1), 6A–199A. [Google Scholar]
- Northcutt, A.; Hutchinson, M.; Wang, X.; Baratta, M.; Hiranita, T.; Cochran, T.; Pomrenze, M.; Galer, E.; Kopajtic, T.; Li, C. Dat isn’t all that: Cocaine reward and reinforcement require toll-like receptor 4 signaling. Mol. Psychiatr. 2015, 20, 1525–1537. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, M.R.; Zhang, Y.; Shridhar, M.; Evans, J.H.; Buchanan, M.M.; Zhao, T.X.; Slivka, P.F.; Coats, B.D.; Rezvani, N.; Wieseler, J.; et al. Evidence that opioids may have toll-like receptor 4 and md-2 effects. Brain Behav. Immun. 2010, 24, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Eidson, L.N.; Inoue, K.; Young, L.J.; Tansey, M.G.; Murphy, A.Z. Toll-like receptor 4 mediates morphine-induced neuroinflammation and tolerance via soluble tumor necrosis factor signaling. Neuropsychopharmacology 2017, 42, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Sun, X.; Jiang, L.; Hu, L.; Han, Y.; Qian, C.; Song, C.; Qian, Y.; Liu, W. Metformin reduces morphine tolerance by inhibiting microglial-mediated neuroinflammation. J. Neuroinflamm. 2016, 13, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. Hmgb1 signals through toll-like receptor (tlr) 4 and tlr2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Crews, F.T. Release of neuronal hmgb1 by ethanol through decreased hdac activity activates brain neuroimmune signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [Green Version]
- Crews, F.T.; Qin, L.; Sheedy, D.; Vetreno, R.P.; Zou, J. High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatr. 2013, 73, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol induces tlr 4/tlr 2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Northcutt, A.; Hiranita, T.; Wang, X.; Lewis, S.; Thomas, J.; Van Steeg, K.; Kopajtic, T.; Loram, L.; Sfregola, C. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J. Neurosci. 2012, 32, 11187–11200. [Google Scholar] [CrossRef] [Green Version]
- Yue, K.; Tanda, G.; Katz, J.L.; Zanettini, C. A further assessment of a role for toll-like receptor 4 in the reinforcing and reinstating effects of opioids. Behav. Pharmacol. 2020, 31, 186–195. [Google Scholar] [CrossRef]
- Brown, K.T.; Levis, S.C.; O’Neill, C.E.; Northcutt, A.L.; Fabisiak, T.J.; Watkins, L.R.; Bachtell, R.K. Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking. Brain Behav. Immun. 2018, 67, 130–138. [Google Scholar] [CrossRef]
- Janova, H.; Böttcher, C.; Holtman, I.R.; Regen, T.; Van Rossum, D.; Götz, A.; Ernst, A.S.; Fritsche, C.; Gertig, U.; Saiepour, N. Cd 14 is a key organizer of microglial responses to cns infection and injury. Glia 2016, 64, 635–649. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Chernis, J.; Da Costa, A.; Mayfield, J.; Harris, R.A. Ethanol consumption in mice lacking cd14, tlr2, tlr4, or myd88. Alcohol. Clin. Exp. Res. 2017, 41, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Blednov, Y.A.; Ponomarev, I.; Geil, C.; Bergeson, S.; Koob, G.F.; Harris, R.A. Neuroimmune regulation of alcohol consumption: Behavioral validation of genes obtained from genomic studies. Addict. Biol. 2012, 17, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef]
- Herrera-Marschitz, M.; You, Z.B.; Goiny, M.; Meana, J.; Silveira, R.; Godukhin, O.; Chen, Y.; Espinoza, S.; Pettersson, E.; Loidl, C. On the origin of extracellular glutamate levels monitored in the basal ganglia of the rat by in vivo microdialysis. J. Neurochem. 1996, 66, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Melendez, R.I.; Vuthiganon, J.; Kalivas, P.W. Regulation of extracellular glutamate in the prefrontal cortex: Focus on the cystine glutamate exchanger and group i metabotropic glutamate receptors. J. Pharmacol. Exp. Ther. 2005, 314, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter glt-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Robinson, M. Review article the family of sodium-dependent glutamate transporters: A focus on the glt-1/eaat2 subtype. Neurochem. Int. 1998, 33, 479–491. [Google Scholar] [CrossRef]
- Trotti, D.; Rossi, D.; Gjesdal, O.; Levy, L.M.; Racagni, G.; Danbolt, N.C.; Volterra, A. Peroxynitrite inhibits glutamate transporter subtypes. J. Biol. Chem. 1996, 271, 5976–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volterra, A.; Trotti, D.; Tromba, C.; Floridi, S.; Racagni, G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 1994, 14, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Trotti, D.; Rizzini, B.L.; Rossi, D.; Haugeto, O.; Racagni, G.; Danbolt, N.C.; Volterra, A. Neuronal and glial glutamate transporters possess an sh-based redox regulatory mechanism. Eur. J. Neurosci. 1997, 9, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-hydroxy-nonenal—A bioactive lipid peroxidation product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralles, V.J.; Martínez-López, I.; Zaragozá, R.; Borrás, E.; García, C.; Pallardó, F.V.; Viña, J.R. Na+ dependent glutamate transporters (eaat1, eaat2, and eaat3) in primary astrocyte cultures: Effect of oxidative stress. Brain Res. 2001, 922, 21–29. [Google Scholar] [CrossRef]
- Lu, M.; Hu, L.-F.; Hu, G.; Bian, J.-S. Hydrogen sulfide protects astrocytes against h2o2-induced neural injury via enhancing glutamate uptake. Free Radic. Biol. Med. 2008, 45, 1705–1713. [Google Scholar] [CrossRef]
- Chen, Y.; Ying, W.; Simma, V.; Chen, Y.; Copin, J.C.; Chan, P.H.; Swanson, R.A. Overexpression of cu, zn superoxide dismutase attenuates oxidative inhibition of astrocyte glutamate uptake. J. Neurochem. 2000, 75, 939–945. [Google Scholar] [CrossRef]
- Sorg, O.; Horn, T.F.; Yu, N.; Gruol, D.L.; Bloom, F.E. Inhibition of astrocyte glutamate uptake by reactive oxygen species: Role of antioxidant enzymes. Mol. Med. 1997, 3, 431–440. [Google Scholar] [CrossRef]
- Muscoli, C.; Dagostino, C.; Ilari, S.; Lauro, F.; Gliozzi, M.; Bardhi, E.; Palma, E.; Mollace, V.; Salvemini, D. Posttranslational nitration of tyrosine residues modulates glutamate transmission and contributes to n-methyl-d-aspartate-mediated thermal hyperalgesia. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Xie, W.; Le, W.; Beers, D.R.; He, Y.; Henkel, J.S.; Simpson, E.P.; Yen, A.A.; Xiao, Q.; Appel, S.H. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004, 63, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.Y.; Crews, F.T. Tnfα potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by nfκb inhibition. Brain Res. 2005, 1034, 11–24. [Google Scholar] [CrossRef]
- Szymocha, R.; Akaoka, H.; Dutuit, M.; Malcus, C.; Didier-Bazes, M.; Belin, M.-F.; Giraudon, P. Human t-cell lymphotropic virus type 1-infected t lymphocytes impair catabolism and uptake of glutamate by astrocytes via tax-1 and tumor necrosis factor alpha. J. Virol. 2000, 74, 6433–6441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.-C.; Sontheimer, H. Cytokine modulation of glial glutamate uptake: A possible involvement of nitric oxide. Neuroreport 1996, 7, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Fine, S.M.; Angel, R.A.; Perry, S.W.; Epstein, L.G.; Rothstein, J.D.; Dewhurst, S.; Gelbard, H.A. Tumor necrosis factor α inhibits glutamate uptake by primary human astrocytes implications for pathogenesis of hiv-1 dementia. J. Biol. Chem. 1996, 271, 15303–15306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Ehrlich, L.; Peterson, P.K. Interleukin-1 and tumor necrosis factor-α synergistically mediate neurotoxicity: Involvement of nitric oxide and of n-methyl-d-aspartate receptors. Brain Behav. Immun. 1995, 9, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of eaat2 by nf-κb: A role for n-myc in tnfα-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Pekarskaya, O.; Bencheikh, M.; Chao, W.; Gelbard, H.A.; Ghorpade, A.; Rothstein, J.D.; Volsky, D.J. Reduced expression of glutamate transporter eaat2 and impaired glutamate transport in human primary astrocytes exposed to hiv-1 or gp120. Virology 2003, 312, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Moussawi, K.; Pacchioni, A.; Moran, M.; Olive, M.F.; Gass, J.T.; Lavin, A.; Kalivas, P.W. N-acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci. 2009, 12, 182–189. [Google Scholar] [CrossRef] [Green Version]
- LaRowe, S.D.; Myrick, H.; Hedden, S.; Mardikian, P.; Saladin, M.; McRae, A.; Brady, K.; Kalivas, P.W.; Malcolm, R. Is cocaine desire reduced by n-acetylcysteine? Am. J. Psychiatr. 2007, 164, 1115–1117. [Google Scholar] [CrossRef]
- Moussawi, K.; Kalivas, P.W. Group ii metabotropic glutamate receptors (mglu2/3) in drug addiction. Eur. J. Pharmacol. 2010, 639, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.M.; McFarland, K.; Melendez, R.I.; Kalivas, P.W.; Seamans, J.K. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J. Neurosci. 2005, 25, 6389–6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Ezquer, M.; Olivares, B.; Santapau, D.; Herrera-Marschitz, M.; Israel, Y. N-acetylcysteine and acetylsalicylic acid inhibit alcohol consumption by different mechanisms: Combined protection. Front. Behav. Neurosci. 2020, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Parsegian, A.; See, R.E. Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology 2014, 39, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lominac, K.D.; Sacramento, A.D.; Szumlinski, K.K.; Kippin, T.E. Distinct neurochemical adaptations within the nucleus accumbens produced by a history of self-administered vs non-contingently administered intravenous methamphetamine. Neuropsychopharmacology 2012, 37, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Griffin III, W.C.; Haun, H.L.; Hazelbaker, C.L.; Ramachandra, V.S.; Becker, H.C. Increased extracellular glutamate in the nucleus accumbens promotes excessive ethanol drinking in ethanol dependent mice. Neuropsychopharmacology 2014, 39, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.C.; Ramachandra, V.S.; Knackstedt, L.A.; Becker, H.C. Repeated cycles of chronic intermittent ethanol exposure increases basal glutamate in the nucleus accumbens of mice without affecting glutamate transport. Front. Pharmacol. 2015, 6, 27. [Google Scholar] [CrossRef]
- Ding, Z.M.; Rodd, Z.A.; Engleman, E.A.; Bailey, J.A.; Lahiri, D.K.; McBride, W.J. Alcohol drinking and deprivation alter basal extracellular glutamate concentrations and clearance in the mesolimbic system of alcohol-preferring (p) rats. Addict. Biol. 2013, 18, 297–306. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Hoberg, M.D.; Vidensky, S.; Chung, D.S. Β-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef]
- Lewerenz, J.; Albrecht, P.; Tien, M.L.T.; Henke, N.; Karumbayaram, S.; Kornblum, H.I.; Wiedau-Pazos, M.; Schubert, D.; Maher, P.; Methner, A. Induction of nrf2 and xct are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem. 2009, 111, 332–343. [Google Scholar] [CrossRef]
- Knackstedt, L.A.; Melendez, R.I.; Kalivas, P.W. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatr. 2010, 67, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.J.; Arndt, D.L.; Cain, M.E. Dynamic interactions of ceftriaxone and environmental variables suppress amphetamine seeking. Brain Res. 2019, 1712, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Weiland, A.; Garcia, S.; Knackstedt, L.A. Ceftriaxone and cefazolin attenuate the cue-primed reinstatement of alcohol-seeking. Front. Pharmacol. 2015, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sari, Y.; Sakai, M.; Weedman, J.M.; Rebec, G.V.; Bell, R.L. Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol Alcohol. 2011, 46, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.C.; Yamamoto, B.K.; Hristov, A.M.; Sari, Y. Ceftriaxone attenuates ethanol drinking and restores extracellular glutamate concentration through normalization of glt-1 in nucleus accumbens of male alcohol-preferring rats. Neuropharmacology 2015, 97, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshehri, F.S.; Hakami, A.Y.; Althobaiti, Y.S.; Sari, Y. Effects of ceftriaxone on hydrocodone seeking behavior and glial glutamate transporters in p rats. Behav. Brain Res. 2018, 347, 368–376. [Google Scholar] [CrossRef]
- Alajaji, M.; Bowers, M.; Knackstedt, L.; Damaj, M. Effects of the beta-lactam antibiotic ceftriaxone on nicotine withdrawal and nicotine-induced reinstatement of preference in mice. Psychopharmacology 2013, 228, 419–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Schneider, R.; Bandiera, S.; Souza, D.G.; Bellaver, B.; Caletti, G.; Quincozes-Santos, A.; Elisabetsky, E.; Gomez, R. N-acetylcysteine prevents alcohol related neuroinflammation in rats. Neurochem. Res. 2017, 42, 2135–2141. [Google Scholar] [CrossRef]
- Swanepoel, T.; Möller, M.; Harvey, B.H. N-acetyl cysteine reverses bio-behavioural changes induced by prenatal inflammation, adolescent methamphetamine exposure and combined challenges. Psychopharmacology 2018, 235, 351–368. [Google Scholar] [CrossRef]
- Israel, Y.; Quintanilla, M.E.; Ezquer, F.; Morales, P.; Santapau, D.; Berríos-Cárcamo, P.; Ezquer, M.; Olivares, B.; Herrera-Marschitz, M. Aspirin and n-acetylcysteine co-administration markedly inhibit chronic ethanol intake and block relapse binge drinking: Role of neuroinflammation-oxidative stress self-perpetuation. Addiction Biol. 2019, e12853. [Google Scholar] [CrossRef]
- Lebourgeois, S.; González-Marín, M.C.; Jeanblanc, J.; Naassila, M.; Vilpoux, C. Effect of n-acetylcysteine on motivation, seeking and relapse to ethanol self-administration. Addict. Biol. 2018, 23, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Reichel, C.M.; Moussawi, K.; Do, P.H.; Kalivas, P.W.; See, R.E. Chronic n-acetylcysteine during abstinence or extinction after cocaine self-administration produces enduring reductions in drug seeking. J. Pharmacol. Exp. Ther. 2011, 337, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.; Neuhofer, D.; Chioma, V.C.; Garcia-Keller, C.; Schwartz, D.J.; Allen, N.; Scofield, M.D.; Ortiz-Ithier, T.; Kalivas, P.W. A model of δ9-tetrahydrocannabinol self-administration and reinstatement that alters synaptic plasticity in nucleus accumbens. Biol. Psychiatr. 2018, 84, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Kalivas, P.W. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue-and heroin-induced drug-seeking. Biol. Psychiatr. 2008, 63, 338–340. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.M. Acetaminophen and the us acute liver failure study group: Lowering the risks of hepatic failure. Hepatology 2004, 40, 6–9. [Google Scholar] [CrossRef]
- Duailibi, M.S.; Cordeiro, Q.; Brietzke, E.; Ribeiro, M.; LaRowe, S.; Berk, M.; Trevizol, A.P. N-acetylcysteine in the treatment of craving in substance use disorders: Systematic review and meta-analysis. Am. J. Addict. 2017, 26, 660–666. [Google Scholar] [CrossRef]
- Holdiness, M.R. Clinical pharmacokinetics of n-acetylcysteine. Clin. Pharmacokinet. 1991, 20, 123–134. [Google Scholar] [CrossRef]
- Evren, C.; Alniak, I. N-acetylcysteine in the treatment of substance use disorders. Neurol. Sci. 2020, 33, 1–7. [Google Scholar] [CrossRef]
- Tomko, R.L.; Jones, J.L.; Gilmore, A.K.; Brady, K.T.; Back, S.E.; Gray, K.M. N-acetylcysteine: A potential treatment for substance use disorders. Curr. Psychiatr. 2018, 17, 30. [Google Scholar]
- Kawasaki, A.; Hoshino, K.; Osaki, R.; Mizushima, Y.; Yano, S. Effect of ibudilast: A novel antiasthmatic agent, on airway hypersensitivity in bronchial asthma. J. Asthma 1992, 29, 245–252. [Google Scholar] [CrossRef]
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A review of its pharmacology, efficacy and safety in respiratory and neurological disease. Exp. Opin. Pharmacother. 2009, 10, 2897–2904. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.C.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; MacKenzie, F.L.; Nagasawa, M.; Stevens, P.A.; MacKenzie, S.J. The inhibitory profile of ibudilast against the human phosphodiesterase enzyme family. Eur. J. Pharmacol. 2006, 538, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, S.; Zhang, L.; Salem, M.; Greig, G.M.; Chan, C.C.; Natsumeda, Y.; Noguchi, K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sci. 2006, 78, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Suzumura, A.; Ito, A.; Yoshikawa, M.; Sawada, M. Ibudilast suppresses tnfα production by glial cells functioning mainly as type iii phosphodiesterase inhibitor in the cns. Brain Res. 1999, 837, 203–212. [Google Scholar] [CrossRef]
- Mizuno, T.; Kurotani, T.; Komatsu, Y.; Kawanokuchi, J.; Kato, H.; Mitsuma, N.; Suzumura, A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004, 46, 404–411. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Lewis, S.S.; Coats, B.D.; Skyba, D.A.; Crysdale, N.Y.; Berkelhammer, D.L.; Brzeski, A.; Northcutt, A.; Vietz, C.M.; Judd, C.M.; et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic av411 (ibudilast). Brain Behav. Immun. 2009, 23, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.L.; Lopez, M.F.; Cui, C.; Egli, M.; Johnson, K.W.; Franklin, K.M.; Becker, H.C. Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict. Biol. 2015, 20, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Beardsley, P.M.; Shelton, K.L.; Hendrick, E.; Johnson, K.W. The glial cell modulator and phosphodiesterase inhibitor, av411 (ibudilast), attenuates prime-and stress-induced methamphetamine relapse. Eur. J. Pharmacol. 2010, 637, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Rolan, P.; Gibbons, J.A.; He, L.; Chang, E.; Jones, D.; Gross, M.I.; Davidson, J.B.; Sanftner, L.M.; Johnson, K.W. Ibudilast in healthy volunteers: Safety, tolerability and pharmacokinetics with single and multiple doses. Brit. J. Clin. Pharmacol. 2008, 66, 792–801. [Google Scholar] [CrossRef]
- Ray, L.A.; Bujarski, S.; Shoptaw, S.; Roche, D.J.; Heinzerling, K.; Miotto, K. Development of the neuroimmune modulator ibudilast for the treatment of alcoholism: A randomized, placebo-controlled, human laboratory trial. Neuropsychopharmacology 2017, 42, 1776–1788. [Google Scholar] [CrossRef] [Green Version]
- Worley, M.J.; Heinzerling, K.G.; Roche, D.J.; Shoptaw, S. Ibudilast attenuates subjective effects of methamphetamine in a placebo-controlled inpatient study. Drug and alcohol dependence 2016, 162, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.; Baptista, S.; Martins, T.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Malva, J.O.; Silva, A.P. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: Preventive effect of indomethacin. Eur. J. Neurosci. 2010, 31, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Dal-Pont, G.C.; Tonin, P.T.; Varela, R.B.; Ferreira, C.L.; Gava, F.F.; Andersen, M.L.; Soares, J.C.; Quevedo, J. Coadministration of lithium and celecoxib attenuates the behavioral alterations and inflammatory processes induced by amphetamine in an animal model of mania. Pharmacol. Biochem. Behav. 2019, 183, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, O.; Barker, R.A.; Brustle, O.; Isacson, O.; Svendsen, C.N. Clinical translation of stem cells in neurodegenerative disorders. Cell Stem Cell 2012, 10, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.-C.; Shyu, W.-C.; Lin, S.-Z. Mesenchymal stem cells. Cell Transplant. 2011, 20, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.S.; Park, B.S.; Kim, H.K.; Park, J.S.; Kim, K.J.; Choi, J.S.; Chung, S.J.; Kim, D.D.; Sung, J.H. Evidence supporting antioxidant action of adipose-derived stem cells: Protection of human dermal fibroblasts from oxidative stress. J. Dermatol. Sci. 2008, 49, 133–142. [Google Scholar] [CrossRef]
- Hegyi, B.; Kornyei, Z.; Ferenczi, S.; Fekete, R.; Kudlik, G.; Kovacs, K.J.; Madarasz, E.; Uher, F. Regulation of mouse microglia activation and effector functions by bone marrow-derived mesenchymal stem cells. Stem Cells Develop. 2014, 23, 2600–2612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.-R.; Li, P.; Chen, F.-F.; Jiang, X.-D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Chen, A.; Fa, Z.; Ding, Z.; Xiao, L.; Wu, G.; Wang, Q.; Zhang, R. Bone marrow mesenchymal stem cells upregulate pi3k/akt pathway and down-regulate nf-κb pathway by secreting glial cell-derived neurotrophic factors to regulate microglial polarization and alleviate deafferentation pain in rats. Neurobiol. Dis. 2020, 143, 104945. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hong, J.M.; Moon, G.J.; Lee, P.H.; Ahn, Y.H.; Bang, O.Y.; Collaborators, S. A long-term follow-up study of intravenous autologous mesenchymal stem cell transplantation in patients with ischemic stroke. Stem Cells 2010, 28, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.M.; Barry, F.P.; Murphy, J.M.; Mahon, B.P. Mesenchymal stem cells avoid allogeneic rejection. J. Inflamm. 2005, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Lennon, D.P.; Eaton, V.; Maier, K.; Caplan, A.I.; Miller, S.D.; Miller, R.H. Human bone marrow-derived mesenchymal stem cells induce th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia 2009, 57, 1192–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Fernández, M.; Rodríguez-Frutos, B.; Ramos-Cejudo, J.; Otero-Ortega, L.; Fuentes, B.; Vallejo-Cremades, M.T.; Sanz-Cuesta, B.E.; Díez-Tejedor, E. Comparison between xenogeneic and allogeneic adipose mesenchymal stem cells in the treatment of acute cerebral infarct: Proof of concept in rats. J. Transl. Med. 2015, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Lanza, C.; Morando, S.; Voci, A.; Canesi, L.; Principato, M.C.; Serpero, L.D.; Mancardi, G.; Uccelli, A.; Vergani, L. Neuroprotective mesenchymal stem cells are endowed with a potent antioxidant effect in vivo. J. Neurochem. 2009, 110, 1674–1684. [Google Scholar] [CrossRef]
- Ohtaki, H.; Ylostalo, J.H.; Foraker, J.E.; Robinson, A.P.; Reger, R.L.; Shioda, S.; Prockop, D.J. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 14638–14643. [Google Scholar] [CrossRef] [Green Version]
- Laroni, A.; De Rosbo, N.K.; Uccelli, A. Mesenchymal stem cells for the treatment of neurological diseases: Immunoregulation beyond neuroprotection. Immunol. Lett. 2015, 168, 183–190. [Google Scholar] [CrossRef]
- Israel, Y.; Ezquer, F.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Herrera-Marschitz, M. Intracerebral stem cell administration inhibits relapse-like alcohol drinking in rats. Alcohol Alcohol. 2017, 52, 1–4. [Google Scholar] [CrossRef]
- Ezquer, F.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Lespay-Rebolledo, C.; Herrera-Marschitz, M.; Israel, Y. Activated mesenchymal stem cell administration inhibits chronic alcohol drinking and suppresses relapse-like drinking in high-alcohol drinker rats. Addict. Biol. 2019, 24, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.; Morales, P.; Quintanilla, M.E.; Santapau, D.; Lespay-Rebolledo, C.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Intravenous administration of anti-inflammatory mesenchymal stem cell spheroids reduces chronic alcohol intake and abolishes binge-drinking. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Woodbury, D.; Schwarz, E.J.; Prockop, D.J.; Black, I.B. Adult rat and human bone marrow stromal cells differentiate into neurons. J. Neurosci. Res. 2000, 61, 364–370. [Google Scholar] [CrossRef]
- Meyerrose, T.; Olson, S.; Pontow, S.; Kalomoiris, S.; Jung, Y.; Annett, G.; Bauer, G.; Nolta, J.A. Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv. Drug Deliv. Rev. 2010, 62, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.I.; Correa, D. The msc: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, F.G.; Carvalho, M.M.; Sousa, N.; Salgado, A.J. Mesenchymal stem cells secretome: A new paradigm for central nervous system regeneration? Cell. Mol. Life Sci. CMLS 2013, 70, 3871–3882. [Google Scholar] [CrossRef]
- Cantinieaux, D.; Quertainmont, R.; Blacher, S.; Rossi, L.; Wanet, T.; Noel, A.; Brook, G.; Schoenen, J.; Franzen, R. Conditioned medium from bone marrow-derived mesenchymal stem cells improves recovery after spinal cord injury in rats: An original strategy to avoid cell transplantation. PLoS ONE 2013, 8, e69515. [Google Scholar] [CrossRef]
- Saparov, A.; Ogay, V.; Nurgozhin, T.; Jumabay, M.; Chen, W.C. Preconditioning of human mesenchymal stem cells to enhance their regulation of the immune response. Stem Cells Int. 2016, 2016, 3924858. [Google Scholar] [CrossRef] [Green Version]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl. Med. 2020. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise review: Msc-derived exosomes for cell-free therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.; Quintanilla, M.E.; Morales, P.; Santapau, D.; Ezquer, M.; Kogan, M.J.; Salas-Huenuleo, E.; Herrera-Marschitz, M.; Israel, Y. Intranasal delivery of mesenchymal stem cell-derived exosomes reduces oxidative stress and markedly inhibits ethanol consumption and post-deprivation relapse drinking. Addict. Biol. 2019, 24, 994–1007. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. Microrna functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microrna biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian micrornas predominantly act to decrease target mrna levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by micrornas: Contributions of translational repression and mrna decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some micrornas downregulate large numbers of target mrnas. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microrna replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottoo, F.H.; Javed, N.; Rahman, J.; Abu-Izneid, T.; Khan, F.A. Targeted delivery of mirna based therapeuticals in the clinical management of glioblastoma multiforme. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative stress and micrornas in vascular diseases. Int. J. Mol. Sci. 2013, 14, 17319–17346. [Google Scholar] [CrossRef]
- Liu, G.; Abraham, E. Micrornas in immune response and macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Saba, R.; Gushue, S.; Huzarewich, R.L.; Manguiat, K.; Medina, S.; Robertson, C.; Booth, S.A. Microrna 146a (mir-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state. PLoS ONE 2012, 7, e30832. [Google Scholar] [CrossRef]
- Kong, H.; Yin, F.; He, F.; Omran, A.; Li, L.; Wu, T.; Wang, Y.; Peng, J. The effect of mir-132, mir-146a, and mir-155 on mrp8/tlr4-induced astrocyte-related inflammation. J. Mol. Neurosci. 2015, 57, 28–37. [Google Scholar] [CrossRef]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated expression profiles of mrna and mirna in polarized primary murine microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O. Targeting mi r-155 restores abnormal microglia and attenuates disease in sod 1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Guedes, J.R.; Pereira de Almeida, L.; Pedroso de Lima, M.C. Mir-155 modulates microglia-mediated immune response by down-regulating socs-1 and promoting cytokine and nitric oxide production. Immunology 2012, 135, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Tarassishin, L.; Loudig, O.; Bauman, A.; Shafit-Zagardo, B.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory mir-155 and mir-155. Glia 2011, 59, 1911–1922. [Google Scholar] [CrossRef] [Green Version]
- Korotkov, A.; Broekaart, D.W.; Van Scheppingen, J.; Anink, J.J.; Baayen, J.C.; Idema, S.; Gorter, J.A.; Aronica, E.; Van Vliet, E.A. Increased expression of matrix metalloproteinase 3 can be attenuated by inhibition of microrna-155 in cultured human astrocytes. J. Neuroinflamm. 2018, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.L.; Guedes, J.R.; De Lima, M.C.P. Role of micrornas in the regulation of innate immune cells under neuroinflammatory conditions. Curr. Opin. Pharmacol. 2016, 26, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific micrornas from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. Microrna-124 promotes microglia quiescence and suppresses eae by deactivating macrophages via the c/ebp-α–pu. 1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Friedman, A. Transcriptional control of granulocyte and monocyte development. Oncogene 2007, 26, 6816–6828. [Google Scholar] [CrossRef] [Green Version]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between micrornas and oxidative stress in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [Green Version]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. Microrna dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Henry, R.J.; Doran, S.J.; Barrett, J.P.; Meadows, V.E.; Sabirzhanov, B.; Stoica, B.A.; Loane, D.J.; Faden, A.I. Inhibition of mir-155 limits neuroinflammation and improves functional recovery after experimental traumatic brain injury in mice. Neurotherapeutics 2019, 16, 216–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetto, I.; Cagnon, V.; Lizarte, F.; Tirapelli, L.; Tirapelli, D.; Arantes, R.; Chuffa, L.; Martinez, F.; Martinez, M. Ethanol and caffeine consumption modulates the expression of mirnas in the cerebellum and plasma of uchb rats. Life Sci. 2019, 229, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Lippai, D.; Bala, S.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Chronic alcohol-induced microrna-155 contributes to neuroinflammation in a tlr4-dependent manner in mice. PLoS ONE 2013, 8, e70945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microrna-155 in macrophages contributes to increased tumor necrosis factor α (tnfα) production via increased mrna half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef] [Green Version]
- Lewohl, J.M.; Nunez, Y.O.; Dodd, P.R.; Tiwari, G.R.; Harris, R.A.; Mayfield, R.D. Up-regulation of micrornas in brain of human alcoholics. Alcohol. Clin. Exp. Res. 2011, 35, 1928–1937. [Google Scholar] [CrossRef] [Green Version]
- Novo-Veleiro, I.; González-Sarmiento, R.; Cieza-Borrella, C.; Pastor, I.; Laso, F.-J.; Marcos, M. A genetic variant in the microrna-146a gene is associated with susceptibility to alcohol use disorders. Eur. Psychiatr. 2014, 29, 288–292. [Google Scholar] [CrossRef]
- Guo, M.-L.; Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Buch, S. Cocaine-mediated downregulation of microglial mir-124 expression involves promoter DNA methylation. Epigenetics 2016, 11, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, V.; Dreyer, J.-L. Micrornas mir-124, let-7d and mir-181a regulate cocaine-induced plasticity. Mol. Cell. Neurosci. 2009, 42, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Bahi, A.; Dreyer, J.L. Striatal modulation of bdnf expression using micro rna 124a-expressing lentiviral vectors impairs ethanol-induced conditioned-place preference and voluntary alcohol consumption. Eur. J. Neurosci. 2013, 38, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, V.; Dreyer, J.-L. Regulation of mir-124, let-7d, and mir-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 2011, 36, 1149–1164. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism |
|---|---|
| Amphetamines | Substrates of the dopamine transporter. Promote the efflux of dopamine from cytosolic vesicles into the synaptic cleft [26]. |
| Cocaine | Blockage of the dopamine transporter, increasing the levels of dopamine in the synaptic cleft [24,25]. |
| Opioids | Activate µ-opioid receptors on GABAergic interneurons in the ventral tegmental area inhibiting them, which in turn disinhibits dopaminergic neurons that release dopamine in the nucleus accumbens [23]. |
| Ethanol | Acts as a pro-drug through its first metabolite acetaldehyde, which reacts with dopamine to form the tetrahydroisoquinoline adduct salsolinol [27], likely an agonist of the µ-opioid receptor [28,29]. Salsolinol activates opioid receptors on GABAergic interneurons in the ventral tegmental area inhibiting them, which in turn disinhibits dopaminergic neurons that release dopamine in the nucleus accumbens [30]. |
| Cannabinoids | Agonists of CB1 and CB2 receptors. Activate CB1 receptors on GABAergic interneurons in the ventral tegmental area, which in turn disinhibits dopaminergic neurons that release dopamine in the nucleus accumbens [31]. |
| Nicotine | Activate α4β2 or α6β2 nicotinic acetylcholine receptors in mesolimbic dopaminergic neurons [32], which promotes the release of dopamine in the nucleus accumbens [33]. |
| Mechanism | Drugs Involved | References |
|---|---|---|
| Oxidation of dopamine | Every drug that increases dopamine levels | [70,71,78] |
| Inhibition of system Xc− | Cocaine, ethanol, and nicotine | [81,82,83,84,85] |
| Drug-induced mitochondrial dysfunction | Ethanol, amphetamines, cocaine, morphine | [87,88,92] |
| Peripheral inflammation contributes to neuroinflammation | Ethanol, cocaine | [57,93,96] |
| Activation of Toll-like receptors | Cocaine, opioids, ethanol | [105,107,111] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berríos-Cárcamo, P.; Quezada, M.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y.; Ezquer, F. Oxidative Stress and Neuroinflammation as a Pivot in Drug Abuse. A Focus on the Therapeutic Potential of Antioxidant and Anti-Inflammatory Agents and Biomolecules. Antioxidants 2020, 9, 830. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090830
Berríos-Cárcamo P, Quezada M, Quintanilla ME, Morales P, Ezquer M, Herrera-Marschitz M, Israel Y, Ezquer F. Oxidative Stress and Neuroinflammation as a Pivot in Drug Abuse. A Focus on the Therapeutic Potential of Antioxidant and Anti-Inflammatory Agents and Biomolecules. Antioxidants. 2020; 9(9):830. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090830
Chicago/Turabian StyleBerríos-Cárcamo, Pablo, Mauricio Quezada, María Elena Quintanilla, Paola Morales, Marcelo Ezquer, Mario Herrera-Marschitz, Yedy Israel, and Fernando Ezquer. 2020. "Oxidative Stress and Neuroinflammation as a Pivot in Drug Abuse. A Focus on the Therapeutic Potential of Antioxidant and Anti-Inflammatory Agents and Biomolecules" Antioxidants 9, no. 9: 830. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090830