Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis

 , ,
, , 
Abstract
:1. Introduction
2. Neuroinflammation and Oxidative Stress
3. Mitochondria and the Mechanism of Motor Neuron Death
4. Implications in the Therapy of ALS
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Preux, P.-M.; Marin, B. Epidemiology of amyotrophic lateral sclerosis: A review of literature. Rev. Neurol. 2016, 172, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Al Khleifat, A.; Al-Chalabi, A. What causes amyotrophic lateral sclerosis? F1000Research 2017, 6, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Morozova, N.; Weisskopf, M.G.; McCullough, M.L.; Munger, K.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Diet and amyotrophic lateral sclerosis. Epidemiology 2008, 19, 324–337. [Google Scholar] [CrossRef]
- Murdock, B.J.; Bender, D.E.; Segal, B.M.; Feldman, E.L. The dual roles of immunity in ALS: Injury overrides protection. Neurobiol. Dis. 2015, 77, 1–12. [Google Scholar] [CrossRef]
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Lacorte, E.; Ferrigno, L.; Leoncini, E.; Corbo, M.; Boccia, S.; Vanacore, N. Physical activity, and physical activity related to sports, leisure and occupational activity as risk factors for ALS: A systematic review. Neurosci. Biobehav. Rev. 2016, 66, 61–79. [Google Scholar] [CrossRef]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef]
- Gunnarsson, L.-G.; Bodin, L. Amyotrophic Lateral Sclerosis and Occupational Exposures: A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public Health 2018, 15, 2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2017, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T. Possible concurrence of TDP-43, tau and other proteins in amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Neuropathology 2018, 38, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanedo-Vazquez, D.; Bosque-Varela, P.; Sainz-Pelayo, A.; Riancho, J. Infectious agents and amyotrophic lateral sclerosis: Another piece of the puzzle of motor neuron degeneration. J. Neurol. 2019, 266, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Hamidou, B.; Marin, B.; Lautrette, G.; Nicol, M.; Camu, W.; Corcia, P.; Arnes-Bes, M.-C.; Tranchant, C.; Clavelou, P.; Hannequin, D.; et al. Exploring the diagnosis delay and ALS functional impairment at diagnosis as relevant criteria for clinical trial enrolment. Amyotroph Lateral Scler. Front. Degener 2017, 18, 519–527. [Google Scholar] [CrossRef]
- Gordon, P.H. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 2010, 7, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Radford, R.A.; Morsch, M.; Rayner, S.L.; Cole, N.J.; Pountney, D.L.; Chung, R.S. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci. 2015, 9, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef] [PubMed]
- Lasiene, J.; Yamanaka, K. Glial cells in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011, 2011, 718987. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xia, K.; Chen, L.; Fan, D. Increased Interleukin-6 Levels in the Astrocyte-Derived Exosomes of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 574. [Google Scholar] [CrossRef] [Green Version]
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; Volk, A.E.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A.; et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, M.C.; Fiala, M.; Dinglasan, L.A.V.; Liu, N.Q.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004, 5, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Beers, D.R.; Liao, B.; Henkel, J.S.; Appel, S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012, 48, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosi, N.; Cozzolino, M.; Carrì, M.T. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Redox (dys) Regulation. Antioxid. Redox Signal. 2018, 29, 15–36. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M.Y. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Shodai, A.; Morimura, T.; Ido, A.; Uchida, T.; Ayaki, T.; Takahashi, R.; Kitazawa, S.; Suzuki, S.; Shirouzu, M.; Kigawa, T.; et al. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2013, 288, 14886–14905. [Google Scholar] [CrossRef] [Green Version]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
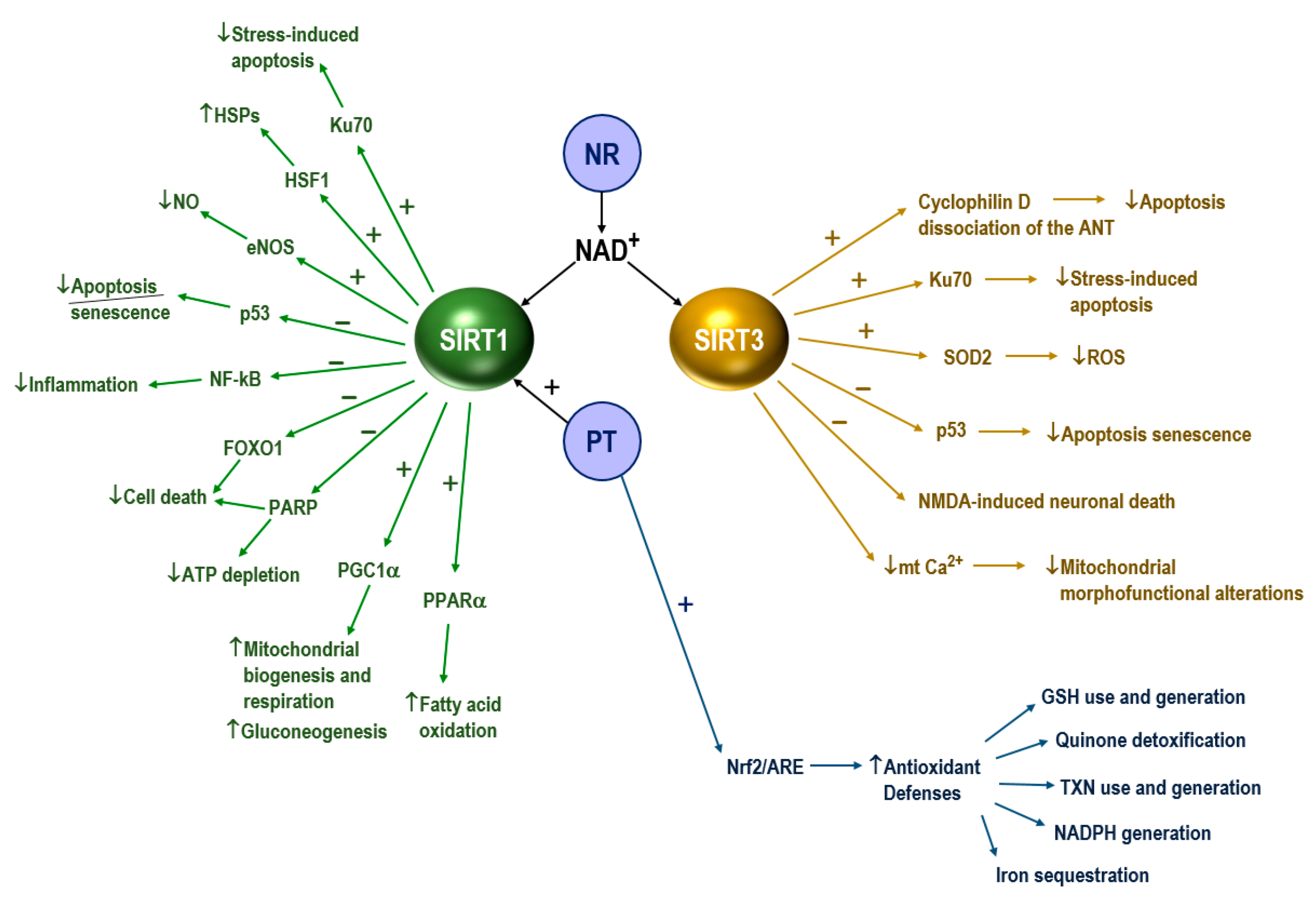
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef]
- Han, S.; Choi, J.-R.; Soon Shin, K.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Hunter, T.A.; Maxwell, N.; Pereira, K.; Whittaker, C.A.; Valdez, G.; Guarente, L.P. SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 2018, 17, e12839. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [PubMed] [Green Version]
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1α-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatori, I.; Valle, C.; Ferri, A.; Carrì, M.T. SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochem. Int. 2017, 109, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M.; Bilski, A.E.; Zhao, W. Sirtuins as therapeutic targets of ALS. Cell Res. 2013, 23, 1073–1074. [Google Scholar] [CrossRef]
- Harlan, B.A.; Pehar, M.; Sharma, D.R.; Beeson, G.; Beeson, C.C.; Vargas, M.R. Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1). J. Biol. Chem. 2016, 291, 10836–10846. [Google Scholar] [CrossRef] [Green Version]
- De la Rubia, J.E.; Drehmer, E.; Platero, J.L.; Benlloch, M.; Caplliure-Llopis, J.; Villaron-Casales, C.; de Bernardo, N.; AlarcÓn, J.; Fuente, C.; Carrera, S.; et al. Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: A randomized, double-blind, placebo-controlled human pilot study. Amyotroph Lateral Scler. Front. Degener. 2019, 20, 115–122. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Zhou, M.; Ottenberg, G.; Sferrazza, G.F.; Hubbs, C.; Fallahi, M.; Rumbaugh, G.; Brantley, A.F.; Lasmézas, C.I. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 2015, 138, 992–1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xu, T.-Y.; Guan, Y.-F.; Tian, W.-W.; Viollet, B.; Rui, Y.-C.; Zhai, Q.-W.; Su, D.-F.; Miao, C.-Y. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.B.; Jang, S.-Y.; Kang, H.T.; Wei, B.; Jeoun, U.-W.; Yoon, G.S.; Hwang, E.S. Modulation of Mitochondrial Membrane Potential and ROS Generation by Nicotinamide in a Manner Independent of SIRT1 and Mitophagy. Mol. Cells 2017, 40, 503–514. [Google Scholar]
- Estrela, J.M.; Ortega, A.; Mena, S.; Rodriguez, M.L.; Asensi, M. Pterostilbene: Biomedical applications. Crit Rev. Clin. Lab. Sci. 2013, 50, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar]
- Song, L.; Chen, L.; Zhang, X.; Li, J.; Le, W. Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1G93A mouse model of amyotrophic lateral sclerosis. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Wang, B.; Liu, H.; Yue, L.; Li, X.; Zhao, L.; Yang, X.; Wang, X.; Yang, Y.; Qu, Y. Neuroprotective effects of pterostilbene against oxidative stress injury: Involvement of nuclear factor erythroid 2-related factor 2 pathway. Brain Res. 2016, 1643, 70–79. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, L.; Yue, L.; Wang, B.; Li, X.; Guo, H.; Ma, Y.; Yao, C.; Gao, L.; Deng, J.; et al. Pterostilbene Attenuates Early Brain Injury Following Subarachnoid Hemorrhage via Inhibition of the NLRP3 Inflammasome and Nox2-Related Oxidative Stress. Mol. Neurobiol. 2017, 54, 5928–5940. [Google Scholar] [CrossRef]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Glutathione and apoptosis. Free Radic. Res. 2008, 42, 689–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytilineou, C.; Kokotos Leonardi, E.T.; Kramer, B.C.; Jamindar, T.; Olanow, C.W. Glial cells mediate toxicity in glutathione-depleted mesencephalic cultures. J. Neurochem. 1999, 73, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Xue, F.; Huang, J.-W.; Ding, P.-Y.; Zang, H.-G.; Kou, Z.-J.; Li, T.; Fan, J.; Peng, Z.-W.; Yan, W.-J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef]
- Mimoto, T.; Miyazaki, K.; Morimoto, N.; Kurata, T.; Satoh, K.; Ikeda, Y.; Abe, K. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012, 1446, 109–118. [Google Scholar] [CrossRef]
- Cheng, Y.; Di, S.; Fan, C.; Cai, L.; Gao, C.; Jiang, P.; Hu, W.; Ma, Z.; Jiang, S.; Dong, Y.; et al. SIRT1 activation by pterostilbene attenuates the skeletal muscle oxidative stress injury and mitochondrial dysfunction induced by ischemia reperfusion injury. Apoptosis 2016, 21, 905–916. [Google Scholar] [CrossRef]
- Jia, N.; Sun, Q.; Su, Q.; Chen, G. SIRT1-mediated deacetylation of PGC1α attributes to the protection of curcumin against glutamate excitotoxicity in cortical neurons. Biochem. Biophys. Res. Commun. 2016, 478, 1376–1381. [Google Scholar] [CrossRef]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.-S. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxid. Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Dai, S.-H.; Chen, T.; Wang, Y.-H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.-F.; Fei, Z.; Jiang, X.-F. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int. J. Mol. Sci. 2014, 15, 14591–14609. [Google Scholar] [CrossRef] [Green Version]
- Shulyakova, N.; Sidorova-Darmos, E.; Fong, J.; Zhang, G.; Mills, L.R.; Eubanks, J.H. Over-expression of the Sirt3 sirtuin Protects neuronally differentiated PC12 Cells from degeneration induced by oxidative stress and trophic withdrawal. Brain Res. 2014, 1587, 40–53. [Google Scholar] [CrossRef]
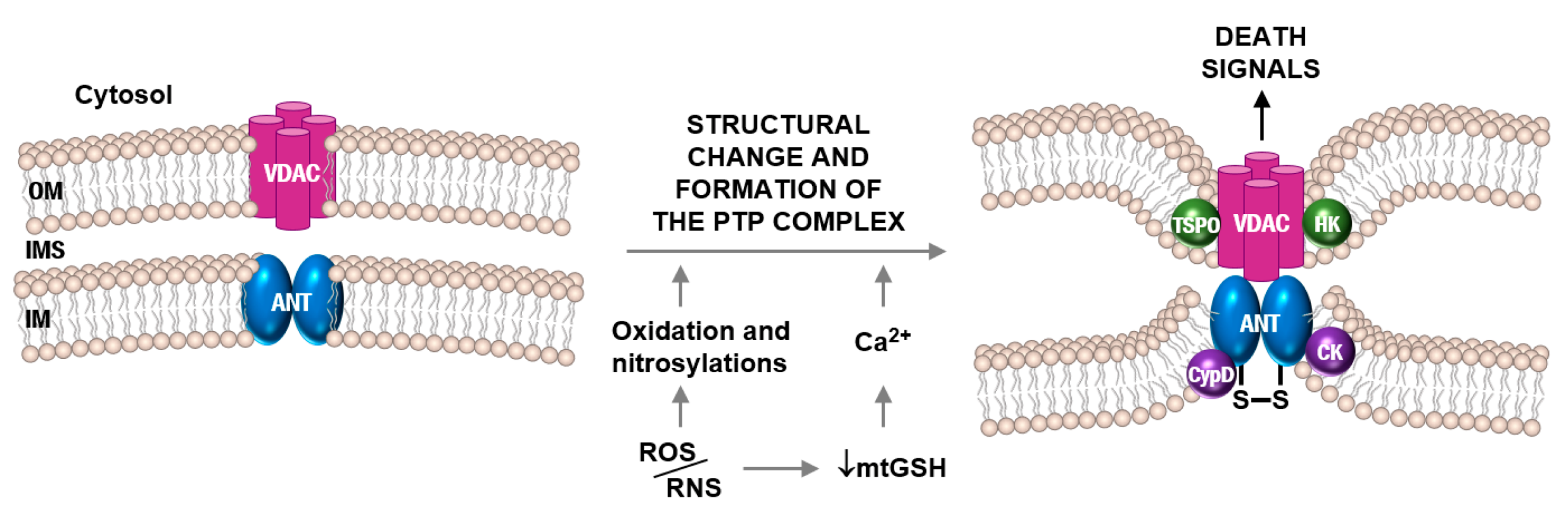
- Belzacq, A.-S.; Vieira, H.L.A.; Kroemer, G.; Brenner, C. The adenine nucleotide translocator in apoptosis. Biochimie 2002, 84, 167–176. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial pathobiology in ALS. J. Bioenerg. Biomembr. 2011, 43, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [Green Version]
- Vieira, H.L.; Belzacq, A.S.; Haouzi, D.; Bernassola, F.; Cohen, I.; Jacotot, E.; Ferri, K.F.; El Hamel, C.; Bartle, L.M.; Melino, G.; et al. The adenine nucleotide translocator: A target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 2001, 20, 4305–4316. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
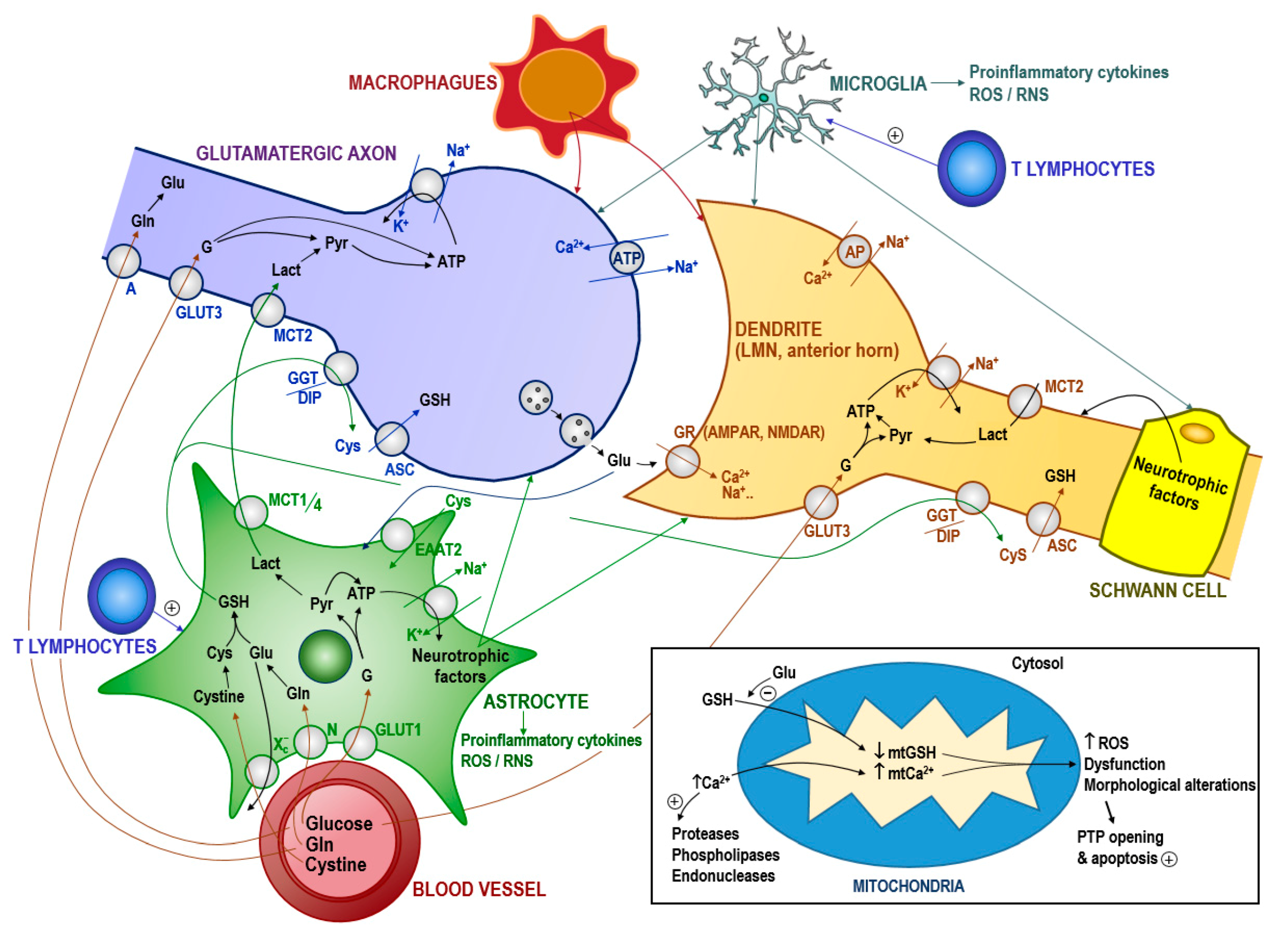
- Featherstone, D.E. Intercellular glutamate signaling in the nervous system and beyond. ACS Chem. Neurosci. 2010, 1, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Carretero, J.; Obrador, E.; Pellicer, J.A.; Pascual, A.; Estrela, J.M. Mitochondrial glutathione depletion by glutamine in growing tumor cells. Free Radic. Biol. Med. 2000, 29, 913–923. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.-Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [Green Version]
- Minich, T.; Riemer, J.; Schulz, J.B.; Wielinga, P.; Wijnholds, J.; Dringen, R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J. Neurochem. 2006, 97, 373–384. [Google Scholar] [CrossRef]
- Nicotera, P.; Bellomo, G.; Orrenius, S. Calcium-mediated mechanisms in chemically induced cell death. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 449–470. [Google Scholar] [CrossRef]
- Beaudet, M.-J.; Yang, Q.; Cadau, S.; Blais, M.; Bellenfant, S.; Gros-Louis, F.; Berthod, F. High yield extraction of pure spinal motor neurons, astrocytes and microglia from single embryo and adult mouse spinal cord. Sci. Rep. 2015, 5, 16763. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Navarro, J.; Mompo, J.; Asensi, M.; Pellicer, J.A.; Estrela, J.M. Glutathione and the rate of cellular proliferation determine tumour cell sensitivity to tumour necrosis factor in vivo. Biochem. J. 1997, 325 Pt 1, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Benlloch, M.; Mena, S.; Ferrer, P.; Obrador, E.; Asensi, M.; Pellicer, J.A.; Carretero, J.; Ortega, A.; Estrela, J.M. Bcl-2 and Mn-SOD antisense oligodeoxynucleotides and a glutamine-enriched diet facilitate elimination of highly resistant B16 melanoma cells by tumor necrosis factor-alpha and chemotherapy. J. Biol. Chem. 2006, 281, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Bergmeyer, H.U.; Bernt, E. UV-Assay with Pyruvate and NADH. In Methods of Enzymatic Analysis, 2nd ed.; Bergmeyer, H.U., Ed.; Academic Press: Cambridge, MA, USA, 1974; pp. 574–579. [Google Scholar]
- Obrador, E.; Valles, S.L.; Benlloch, M.; Sirerol, J.A.; Pellicer, J.A.; Alcácer, J.; Coronado, J.A.-F.; Estrela, J.M. Glucocorticoid receptor knockdown decreases the antioxidant protection of B16 melanoma cells: An endocrine system-related mechanism that compromises metastatic cell resistance to vascular endothelium-induced tumor cytotoxicity. PLoS ONE 2014, 9, e96466. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, CD001447. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-J.; Wang, K.-Y.; Wang, W.-C. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience 2004, 125, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharm. 2017, 18, 735–738. [Google Scholar] [CrossRef]
- Scott, A. Drug therapy: On the treatment trail for ALS. Nature 2017, 550, S120–S121. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, e437. [Google Scholar] [CrossRef] [Green Version]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.C.; Gaillard, P.J.; de Boer, M.; Appeldoorn, C.; Dorland, R.; Sibson, N.R.; Turner, M.R.; Anthony, D.C.; Stolp, H.B. CNS-targeted glucocorticoid reduces pathology in mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2014, 2, 66. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-P.; Lin, F.-C.; Lee, J.K.-W.; Lee, C.T.-C. Aspirin use associated with amyotrophic lateral sclerosis: A total population-based case-control study. J. Epidemiol. 2015, 25, 172–177. [Google Scholar] [CrossRef]
- Espejo-Porras, F.; Fernández-Ruiz, J.; de Lago, E. Analysis of endocannabinoid receptors and enzymes in the post-mortem motor cortex and spinal cord of amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler. Front. Degener 2018, 19, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could Conservative Iron Chelation Lead to Neuroprotection in Amyotrophic Lateral Sclerosis? Antioxid. Redox Signal. 2018, 29, 742–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, K.M.; Sheikh, I.S.; Smith, G.M. Targeting Neurotrophins to Specific Populations of Neurons: NGF, BDNF, and NT-3 and Their Relevance for Treatment of Spinal Cord Injury. Int. J. Mol. Sci. 2017, 18, 548. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Kurotani, T.; Komatsu, Y.; Kawanokuchi, J.; Kato, H.; Mitsuma, N.; Suzumura, A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004, 46, 404–411. [Google Scholar] [CrossRef]
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antivir. Res. 2013, 99, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, M.O.; Heuberger, J.A.A.C.; Sleutjes, B.T.H.M.; Ziagkos, D.; van den Berg, L.H.; Ferguson, T.A.; Franssen, H.; Groeneveld, G.J. Acute Effects of Riluzole and Retigabine on Axonal Excitability in Patients With Amyotrophic Lateral Sclerosis: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. Clin. Pharmacol. Ther. 2018, 104, 1136–1145. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Motor Neurons | ||||||
|---|---|---|---|---|---|---|
| WT | FUS R521C | |||||
| cyt | mt | Total | cyt | mt | Total | |
| H2O2 (nmol/106 cells·min) | 0.42 ± 0.17 | 1.06 ± 0.33 ** | ||||
| O2•− (ΔFL1, a.u.) | 2.11 ± 0.66 | 4.33 ± 1.20 * | ||||
| Glutamate (nmol/106 cells) | 28.5 ± 4.3 | 5.6 ± 1.4 | 41.7 ± 5.9 ** | 7.2 ± 1.8 | ||
| GSH (nmol/106 cells) | 21.2 ± 2.9 | 4.3 ± 1.2 | 15.0 ± 2.4 ** | 2.1 ± 0.5 ** | ||
| GSSG (nmol/106 cells) | 0.5 ± 0.2 | 0.2 ± 0.05 | 1.0 ± 0.3 * | 0.4 ± 0.1 ** | ||
| Ca2+ (nmol/mg protein) | 1087 ± 184 | 512 ± 96 | 1740 ± 260 ** | 826 ± 160 * | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090901
Obrador E, Salvador R, López-Blanch R, Jihad-Jebbar A, Vallés SL, Estrela JM. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants. 2020; 9(9):901. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090901
Chicago/Turabian StyleObrador, Elena, Rosario Salvador, Rafael López-Blanch, Ali Jihad-Jebbar, Soraya L. Vallés, and José M. Estrela. 2020. "Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis" Antioxidants 9, no. 9: 901. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090901