Preventing the Interaction between Coronaviruses Spike Protein and Angiotensin I Converting Enzyme 2: An In Silico Mechanistic Case Study on Emodin as a Potential Model Compound


Abstract
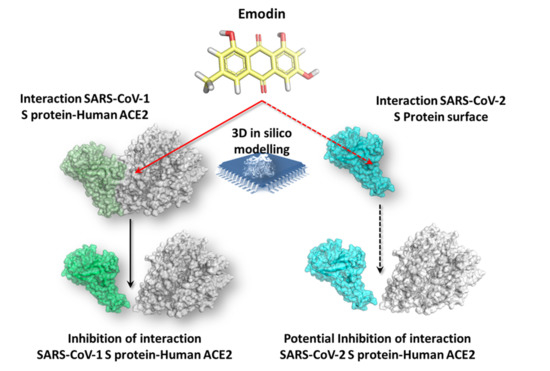
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Design of S Protein Models
2.2. Computational Alanine Scanning
2.3. Docking Simulation
2.4. Pharmacophoric Analysis
2.5. Molecular Dynamics
2.6. Statistical Analysis
3. Results and Discussion
3.1. Analysis of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-1) S Protein-Angiotensin I Converting Enzyme 2 (ACE2) Complex
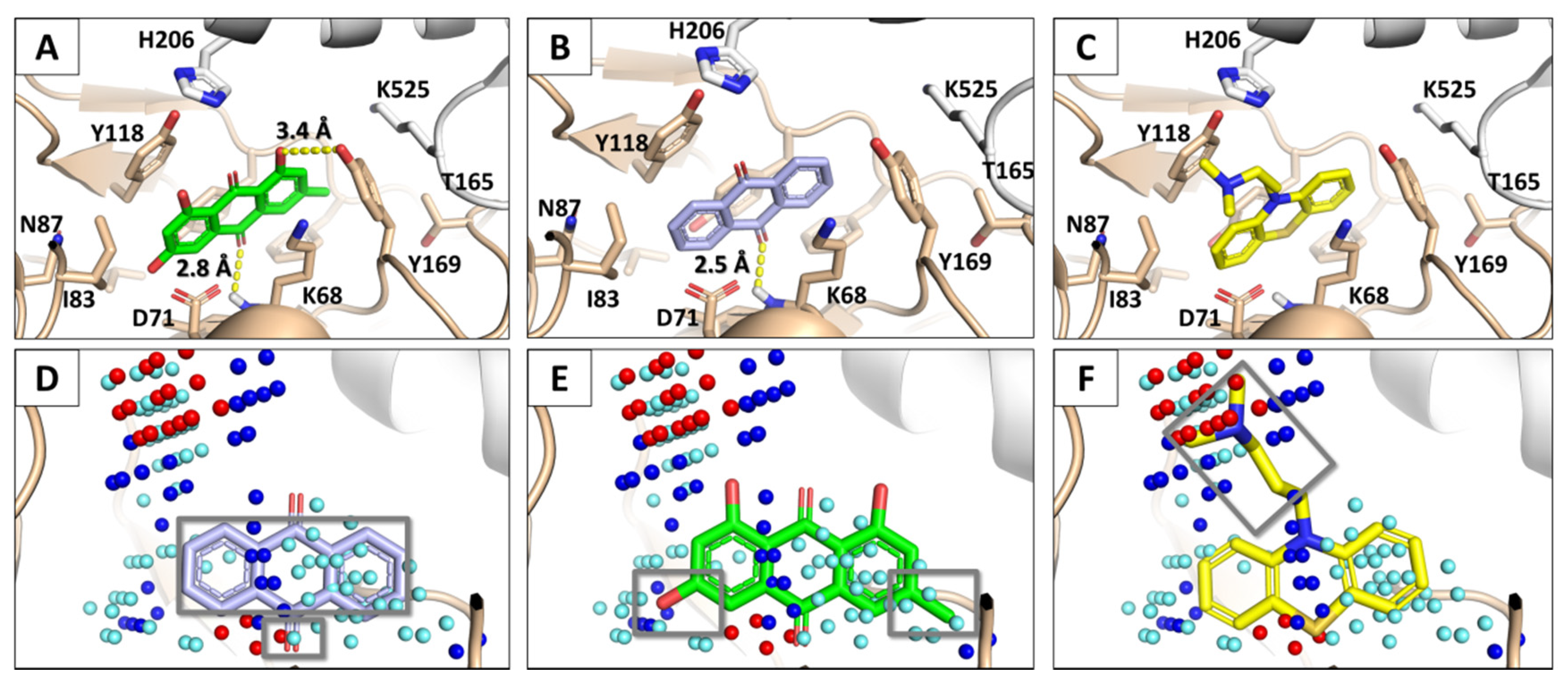
3.2. Docking Simulations of SARS-CoV-1 S Protein–ACE2 Complex
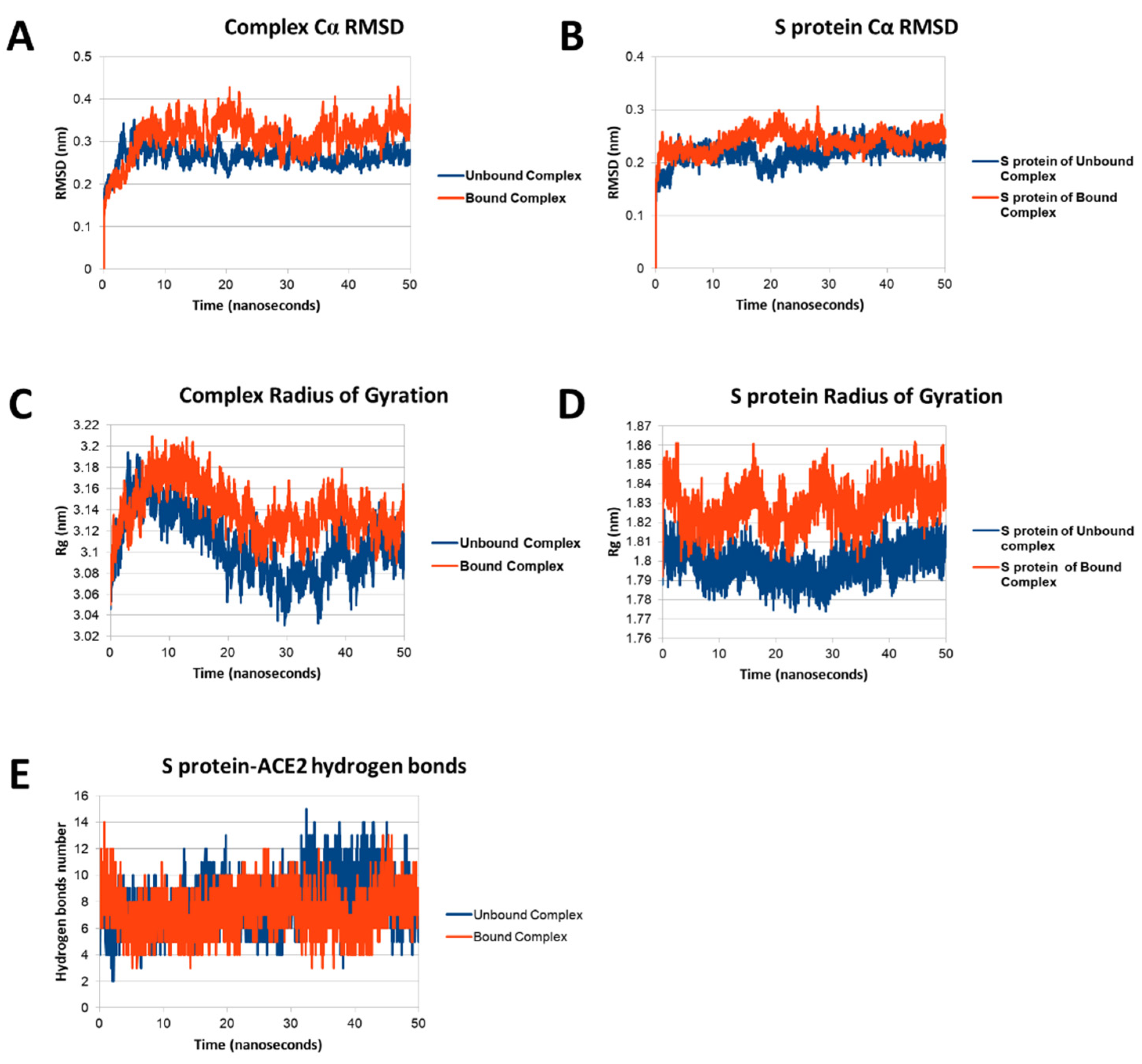
3.3. Molecular Dynamics of SARS-CoV-1 S Protein
3.4. Molecular Dynamics of SARS-CoV-2 S Protein
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin I converting enzyme 2 |
| MD | Molecular dynamic |
| PDB | Protein Data Bank |
| RBD | Receptor Binding Domain |
| RMSD | Root mean square deviation |
| SARS | Severe acute respiratory syndrome |
References
- Cui, Y.T.; Lu, P.R.; Song, G.; Liu, Q.; Zhu, D.; Liu, X.B. Involvement of PI3K/Akt, ERK and p38 signaling pathways in emodin-mediated extrinsic and intrinsic human hepatoblastoma cell apoptosis. Food Chem. Toxicol. 2016, 92, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gomez, J.V.; Gimeno-Adelantadob, J.V.; Jimenez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC-MS/MS and UPLC-QTOF-MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Beed, F.; Boni, S.; Abass, A.; Mukunzi, A.; Krska, R. Quantitation of multiple mycotoxins and cyanogenic glucosides in cassava samples from Tanzania and Rwanda by an LC-MS/MS-based multi-toxin method. Food Addit. Contam. A 2015, 32, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Ezekiel, C.N.; Sulyok, M.; Ogara, I.M.; Abia, W.A.; Warth, B.; Sarkanj, B.; Turner, P.C.; Krska, R. Mycotoxins in uncooked and plate-ready household food from rural northern Nigeria. Food Chem. Toxicol. 2019, 128, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Fu, J.; Yin, X.B.; Cao, S.L.; Li, X.C.; Lin, L.F.; Huyiligeqi; Ni, J. Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Dewanjee, S.; Chakraborty, P.; Mukherjee, B.; De Feo, V. Plant-Based Antidiabetic Nanoformulations: The Emerging Paradigm for Effective Therapy. Int. J. Mol. Sci. 2020, 21, 2217. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.Y.; Wu, S.L.; Chen, J.C.; Li, C.C.; Hsiang, C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef]
- Wu, Y.S.; Lin, W.H.; Hsu, J.T.A.; Hsieh, H.P. Antiviral Drug Discovery Against SARS-CoV. Curr. Med. Chem. 2006, 13, 2003–2020. [Google Scholar] [CrossRef]
- Elshabrawy, H.A. SARS-CoV-2: An Update on Potential Antivirals in Light of SARS-CoV Antiviral Drug Discoveries. Vaccines 2020, 8, 335. [Google Scholar] [CrossRef]
- Li, S.Y.; Chen, C.; Zhang, H.Q.; Guo, H.Y.; Wang, H.; Wang, L.; Zhang, X.; Hua, S.N.; Yu, J.; Xiao, P.G.; et al. Identification of natural compounds with antiviral activities against SARS-associated coronavirus. Antivir. Res. 2005, 67, 18–23. [Google Scholar] [CrossRef]
- Schwarz, S.; Wang, K.; Yu, W.J.; Sun, B.; Schwarz, W. Emodin inhibits current through SARS-associated coronavirus 3a protein. Antivir. Res. 2011, 90, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, S.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. Broad-Spectrum Coronavirus Fusion Inhibitors to Combat COVID-19 and Other Emerging Coronavirus Diseases. Int. J. Mol. Sci. 2020, 21, 384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.D.; Hou, Y.; Shen, J.Y.; Huang, Y.; Martin, W.; Cheng, F.X. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talarico, C.; Gervasoni, S.; Manelfi, C.; Pedretti, A.; Vistoli, G.; Beccari, A.R. Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel. Int. J. Mol. Sci. 2020, 21, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.H.; Chen, L.X.; Mei, H.; Zhang, D.; Shi, T.T.; Kuang, Z.Y.; Heng, Y.; Xu, L.; Pan, X.C. In Silico Prediction of the Dissociation Rate Constants of Small Chemical Ligands by 3D-Grid-Based VolSurf Method. Int. J. Mol. Sci. 2020, 21, 2456. [Google Scholar] [CrossRef] [Green Version]
- Poot Velez, A.H.; Fontove, F.; Del Rio, G. Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes. Int. J. Mol. Sci. 2020, 21, 4787. [Google Scholar] [CrossRef]
- Dellafiora, L.; Filipello, V.; Dall’Asta, C.; Finazzi, G.; Galaverna, G.; Losio, M.N. A Structural Study on the Listeria Monocytogenes Internalin A-Human E-cadherin Interaction: A Molecular Tool to Investigate the Effects of Missense Mutations. Toxins 2020, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Dellafiora, L.; Oswald, I.P.; Dorne, J.L.; Galaverna, G.; Battilani, P.; Dall’Asta, C. An in silico structural approach to characterize human and rainbow trout estrogenicity of mycotoxins: Proof of concept study using zearalenone and alternariol. Food Chem. 2020, 312, 10. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Song, W.F.; Gui, M.; Wang, X.Q.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellafiora, L.; Mena, P.; Del Rio, D.; Cozzini, P. Modeling the effect of phase II conjugations on topoisomerase I poisoning: Pilot study with luteolin and quercetin. J. Agric. Food Chem. 2014, 62, 5881–5886. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.J.; Gindulyte, A.; He, J.; He, S.Q.; Li, Q.L.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acid Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational alanine scanning of protein-protein interface. Sci. STKE 2004, 219, pI2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortemme, T.; Baker, D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [Green Version]
- Rollinger, J.M.; Schuster, D.; Baier, E.; Ellmerer, E.P.; Langer, T.; Stuppner, H. Taspine: Bioactivity-guided isolation and molecular ligand-target insight of a potent acetylcholinesterase inhibitor from Magnolia x soulangiana. J. Nat. Prod. 2006, 69, 1341–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Potential interaction of natural dietary bioactive compounds with COX-2. J. Mol. Graph. Model. 2011, 30, 157–166. [Google Scholar] [CrossRef]
- Gaudreault, F.; Morency, L.P.; Najmanovich, R.J. NRGsuite: A PyMOL plugin to perform docking simulations in real time using FlexAID. Bioinformatics 2015, 31, 3856–3858. [Google Scholar] [CrossRef] [Green Version]
- Chartier, M.; Najmanovich, R. Detection of Binding Site Molecular Interaction Field Similarities. J. Chem. Inf. Model. 2015, 55, 1600–1615. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D.J. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Blau, C.; Grubmuller, H. g_contacts: Fast contact search in bio-molecular ensemble data. Comput. Phys. Commun. 2013, 184, 2856–2859. [Google Scholar] [CrossRef] [Green Version]
- Krumm, B.E.; Lee, S.; Bhattacharya, S.; Botos, I.; White, C.F.; Du, H.J.; Vaidehi, N.; Grisshammer, R. Structure and dynamics of a constitutively active neurotensin receptor. Sci. Rep. 2016, 6, 38564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.C.; Kulig, W.; Poojari, C.; Rog, T.; Strodel, B. Physiologically-relevant levels of sphingomyelin, but not GM1, induces a β-sheet-rich structure in the amyloid-β(1-42) monomer. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.C.; Li, X.Q.; Song, T.; Zhao, Y.; Feng, Y.G. An Anthraquinone Scaffold for Putative, Two-Face Bim BH3 alpha-Helix Mimic. J. Med. Chem. 2012, 55, 10735–10741. [Google Scholar] [CrossRef]
- Vincendeau, M.; Hadian, K.; Messias, A.C.; Brenke, J.K.; Halander, J.; Griesbach, R.; Greczmiel, U.; Bertossi, A.; Stehle, R.; Nagel, D.; et al. Inhibition of Canonical NF-kappa B Signaling by a Small Molecule Targeting NEMO-Ubiquitin Interaction. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Yan, Y.Y.; Zheng, L.S.; Zhang, X.; Chen, L.K.; Singh, S.; Wang, F.; Zhang, J.Y.; Liang, Y.J.; Dai, C.L.; Gu, L.Q.; et al. Blockade of Her2/neu Binding to Hsp90 by Emodin Azide Methyl Anthraquinone Derivative Induces Proteasomal Degradation of Her2/neu. Mol. Pharm. 2011, 8, 1687–1697. [Google Scholar] [CrossRef]
- Ibarra, A.A.; Bartlett, G.J.; Hegedus, Z.; Dutt, S.; Hobor, F.; Horner, K.A.; Hetherington, K.; Spence, K.; Nelson, A.; Edwards, T.A.; et al. Predicting and Experimentally Validating Hot-Spot Residues at Protein-Protein Interfaces. ACS Chem. Biol. 2019, 14, 2252–2263. [Google Scholar] [CrossRef]
- Nerukh, D.; Okimoto, N.; Suenaga, A.; Taiji, M. Ligand Diffusion on Protein Surface Observed in Molecular Dynamics Simulation. J. Phys. Chem. Lett. 2012, 3, 3476–3479. [Google Scholar] [CrossRef]
- Del Favero, G.; Mayer, R.M.; Dellafiora, L.; Janker, L.; Niederstaetter, L.; Dall’Asta, C.; Gerner, C.; Marko, D. Structural Similarity with Cholesterol Reveals Crucial Insights into Mechanisms Sustaining the Immunomodulatory Activity of the Mycotoxin Alternariol. Cells 2020, 9, E847. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.L.; Guo, X.N.; Cong, Y.L.; Feng, G.Q.; Li, Y.C.; Zhang, J.Z.H. Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water. Front. Chem. 2019, 7, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.; Smith, J.C.; Sanson, A. Protein unfolding transitions in an intrinsically unstable annexin domain: Molecular dynamics simulation and comparison with nuclear magnetic resonance data. Biophys. J. 2002, 83, 681–698. [Google Scholar] [CrossRef] [Green Version]
- Rocco, A.G.; Mollica, L.; Ricchiuto, P.; Baptista, A.M.; Gianazza, E.; Eberini, I. Characterization of the protein unfolding processes induced by urea and temperature. Biophys. J. 2008, 94, 2241–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, M.N.; Braga, A.C.S.; Campos, G.R.F.; Souza, M.M.; de Matos, R.P.A.; Lopes, T.Z.; Candido, N.M.; Lima, M.L.D.; Machado, F.C.; de Andrade, S.T.Q.; et al. Natural Products Isolated from Oriental Medicinal Herbs Inactivate Zika Virus. Viruses 2019, 11, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellafiora, L.; Dorne, J.L.C.M.; Galaverna, G.; Dall’Asta, C. Preventing the Interaction between Coronaviruses Spike Protein and Angiotensin I Converting Enzyme 2: An In Silico Mechanistic Case Study on Emodin as a Potential Model Compound. Appl. Sci. 2020, 10, 6358. https://0-doi-org.brum.beds.ac.uk/10.3390/app10186358
Dellafiora L, Dorne JLCM, Galaverna G, Dall’Asta C. Preventing the Interaction between Coronaviruses Spike Protein and Angiotensin I Converting Enzyme 2: An In Silico Mechanistic Case Study on Emodin as a Potential Model Compound. Applied Sciences. 2020; 10(18):6358. https://0-doi-org.brum.beds.ac.uk/10.3390/app10186358
Chicago/Turabian StyleDellafiora, Luca, Jean Lou C M Dorne, Gianni Galaverna, and Chiara Dall’Asta. 2020. "Preventing the Interaction between Coronaviruses Spike Protein and Angiotensin I Converting Enzyme 2: An In Silico Mechanistic Case Study on Emodin as a Potential Model Compound" Applied Sciences 10, no. 18: 6358. https://0-doi-org.brum.beds.ac.uk/10.3390/app10186358