
Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials


1
Department of Bioengineering, Volgenau School of Engineering, George Mason University, Fairfax, VA 22030, USA
2
Institute for Advanced Biomedical Research, George Mason University, Manassas, VA 20110, USA
*
Author to whom correspondence should be addressed.
Appl. Sci. 2021, 11(4), 1885; https://0-doi-org.brum.beds.ac.uk/10.3390/app11041885
Submission received: 19 January 2021
/
Revised: 12 February 2021
/
Accepted: 15 February 2021
/
Published: 21 February 2021
(This article belongs to the Special Issue Mechanical Design in DNA Nanotechnology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:DNA hydrogels are self-assembled biomaterials that rely on Watson–Crick base pairing to form large-scale programmable three-dimensional networks of nanostructured DNA components. The unique mechanical and biochemical properties of DNA, along with its biocompatibility, make it a suitable material for the assembly of hydrogels with controllable mechanical properties and composition that could be used in several biomedical applications, including the design of novel multifunctional biomaterials. Numerous studies that have recently emerged, demonstrate the assembly of functional DNA hydrogels that are responsive to stimuli such as pH, light, temperature, biomolecules, and programmable strand-displacement reaction cascades. Recent studies have investigated the role of different factors such as linker flexibility, functionality, and chemical crosslinking on the macroscale mechanical properties of DNA hydrogels. In this review, we present the existing data and methods regarding the mechanical design of pure DNA hydrogels and hybrid DNA hydrogels, and their use as hydrogels for cell culture. The aim of this review is to facilitate further study and development of DNA hydrogels towards utilizing their full potential as multifeatured and highly programmable biomaterials with controlled mechanical properties.
1. Introduction
Deoxyribonucleic acid (DNA) nanotechnology uses DNA molecules as a building block to assemble various types of nanoscale architectures [1,2,3]. Considerable progress has been made in the DNA nanotechnology field in only a few decades, and now DNA is routinely used to synthetize complex one-dimensional (1D), two-dimensional (2D), and three-dimensional (3D) nanostructures [4,5,6,7,8], meshes [9,10], devices [11,12,13], and hybrid biomaterials [14,15,16,17], among others. These DNA architectures can be assembled using different techniques such as tile assembly [18,19], DNA origami [4], and DNA bricks [20]. DNA-based materials are developing rapidly because of the unique structural, mechanical, and biochemical features offered by DNA building blocks that are not found in any other polymeric materials. For instance, DNA-based assembly relies on the Watson–Crick base pairing mechanism to hybridize two single strands of DNA (ssDNA) with complementary sequences and form double stranded DNA (dsDNA) molecules, thus making DNA-based materials highly programmable, which allows for high specificity of assembly at the nanoscale using simple sequence design. The structural predictability provides the user with modeling capability and an exquisite control over the final assembly as well as great design flexibility [4,7,19,21]. In addition, the mechanical properties of DNA-based nanoarchitectures can be easily programmed by changing the type of DNA motifs used, the hybridization state, and their nanostructure design [22,23,24,25,26]. DNA materials are inherently biocompatible, self-assemble in solution, exhibit high structural stability, and their ease of modification makes them a great material to scaffold various organic and inorganic molecules with nanoscale precision to functionalize the nanoarchitectures [27,28,29,30]. While DNA building blocks have been used extensively to assemble several nanoscale objects, DNA as a material is now gaining more traction as a potential candidate for larger scale polymeric biomaterials assembly, particularly to build nanostructured functional soft hydrogels with sizes ranging from the nanoscale to the millimeter scale. Similar to the discrete DNA nanostructures, the structural, mechanical, and biochemical properties of these DNA hydrogels are highly controllable [31,32,33]. In the past decade, several studies have demonstrated the potential of these DNA hydrogels to be used in several biomedical applications [34] including drug delivery [35,36,37], cancer therapy [38,39,40], antigen delivery and immunomodulation [41,42], sensing [43,44,45], and extracellular matrix (ECM) mimics for tissue engineering [34,46]. In addition, using the unique biochemical properties of DNA, pure and hybrid DNA hydrogels can be designed to be responsive to specific stimuli, such as pH, temperature, biomolecules, and strand displacement [38,47,48]. For instance, introducing pH-sensitive crosslinker motifs such as the i-motif or the G-quadruplex inside the DNA hydrogel design has been used to control the stiffness of DNA-based hydrogels [31,49]. Careful sequence design of the linker or the hydrogel motifs allows for precisely controlling the melting temperature and the mechanical properties. Several methods are also available to induce DNA hydrogel crosslinking in order to control the structural stability and the mechanical properties of the hydrogels [38,47,50]. Thus, their mechanical, structural, and biochemical properties can be modulated in real time, which represents an immense advantage over other types of polymeric hydrogels that are static by nature.
This review focuses on the different methods used to assemble DNA hydrogels and the various designs used to control their mechanical properties. Particularly, it focuses on the crosslinking methods used and the relationship between the structure and the mechanical properties of the DNA-based hydrogels. Furthermore, the use of synthetic and biopolymers with DNA crosslinkers for modular mechanical properties is addressed. It also describes the various existing methods used to measure the mechanical properties of such soft hydrogels, the potential for using these hydrogels in biomedical research, and the next steps required for a broadened use of these DNA-based materials.
2. Structural DNA Hydrogel Design
As already demonstrated with techniques such as DNA tile assembly and DNA origami technology, DNA can be used as a building block to precisely design complex 1D, 2D, and 3D structures with controlled structural and mechanical features [4,13,23]. Similar strategies can be applied to assemble DNA nanogels and hydrogels with great design flexibility [32]. In this section, we classify the various types of DNA hydrogel designs as either entanglement-based constructs or based on the assembly of DNA nanostructures and present the mechanical properties for different design variations.
2.1. Entangled and Crosslinked
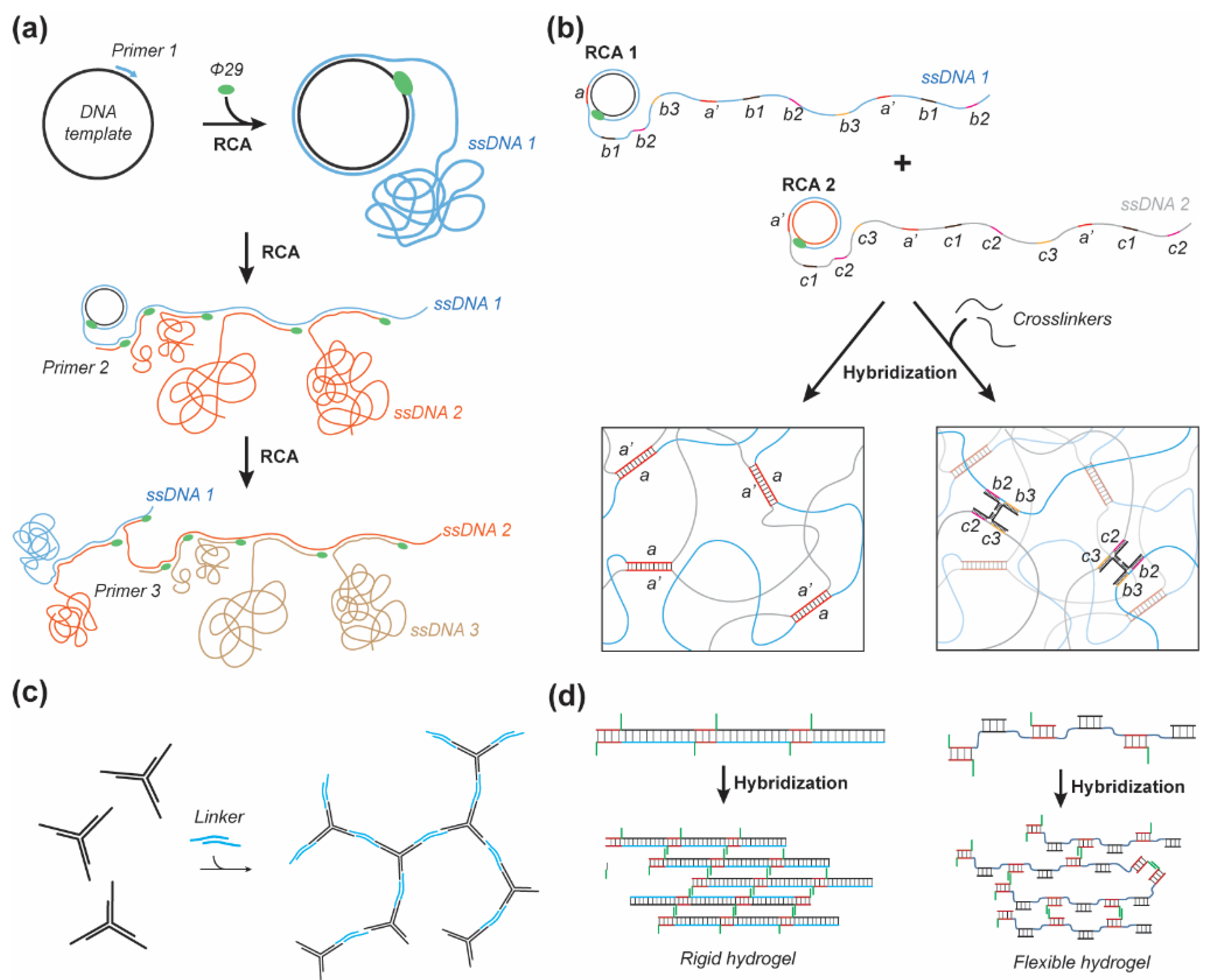
Entangled structures are commonly composed of rolling chain amplification (RCA) produced DNA concatemers [48,51,52] or DNA from salmon testes, which may be covalently crosslinked by ethylene glycol diglycidyl ether (EDGE) and oxidized alginate (OA) [53,54,55]. The source of DNA material is an important factor as synthetically synthesized DNA can cost a few hundred to a few thousand dollars for a few milligrams required for milliliter scale DNA hydrogel synthesis [56]; typically requiring 2–4 milligrams (mg) of DNA per 100 µL, or 2–4 percent weight/volume (%w/v) of hydrogel. However, RCA is able to produce a few hundred micrograms of DNA per milliliter of overnight reaction [57,58,59], though it is currently limited to short tandem repeats (~100 nucleotides [nt]) without control over the repeat number. Multiprimed chain amplification (MCA) is the subsequent priming of the concatemeric strand produced by RCA (Figure 1a); this approach allows for a significant amount of DNA to be produced with a multitude of complementary domains enabling partial hybridization between multiple strands ranging from a few hundred to thousands of nucleotides in length. Lee et al. demonstrated the ability of MCA to produce enough material to assemble a DNA hydrogel by entanglement which formed spheroidal clusters or “nests” measuring close to 1 micron in diameter (Figure 1a) [51]. This DNA hydrogel yielded a storage modulus (G’) ranging from 5–7 Pascals (Pa) at frequencies between 0.1–100 Hertz (Hz). Geng et al. found similar mechanical properties when using the same technique to produce a DNA hydrogel seeded with silver nanoclusters. The all-DNA MCA-based gel yielded a storage modulus ranging from 5–7 Pa over the course of 3 min and when heated up to 75 °C [52]. When silver nanoclusters were seeded onto cytosine-rich domains the storage modulus increased to G’ = 12–20 Pa and exhibited similar temperature independent rheological properties between 40 °C and 75 °C. Merindol et al. introduced a different approach to RCA-based hydrogels by synthesizing two independent concatemeric ssDNA scaffolds, thus voiding the MCA-step (Figure 1b). A set of complimentary short ssDNA strands were used to crosslink the independent scaffolds and control the strength of the linker by altering the sequence and length of the hybridizing regions. A fluorescence resonance energy transfer (FRET) dye pair was incorporated within the crosslinker to monitor the dissociation of the linkage and visualize strain in the material. This approach yielded temperature-dependent properties with diminishing storage moduli from 400, 160, and 6 Pa which melted between 60–80 °C for DNA concentrations of 1.4, 0.7, and 0.36% w/v, respectively [48]. Interestingly the 0.7% w/v DNA hydrogel showed an increase in its storage modulus by an order of magnitude, from ~10 to ~100 Pa by simply heating the hydrogel to 80 °C and cooling it to 10 °C over approximately 15 min. A second heating and cooling ramp showed no further increase in the mechanical properties. Melting the DNA hydrogel and cooling is thought to allow for reorganization of the internal structure by releasing entrapped complementary binding regions during heating and increasing the number of crosslinks upon cooling. An alternative approach to entangled DNA hydrogels is to use biologically sourced DNA, commonly acquired from salmon testes. Lee et al. were able to form a hydrogel by wet spinning DNA in a room temperature ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF4), which showed a fiber swelling of 600% from a diameter of ~100 μm in deionized water and diminished to 150% upon addition of 150 mM NaCl. The salt-induced deswelling is due to the neutralization of the negatively charged DNA backbone, thus diminishing the electrostatic repulsion between DNA strands [60]. Topuz and Okay studied the effect of EGDE as a crosslinker for DNA hydrogels and demonstrated increases in the storage modulus from around 300 to 3000 Pa using a 9.8% w/v DNA formulation [53]. Their hydrogel also showed an increase in stiffness as a result of network reorganization at 50 °C for 2 h. The fraction of increase was shown to be higher with higher EGDE crosslinker concentration ranging from 10–49% EGDE/DNA. Topuz and Okay also studied the effect of concentration on the heating–cooling induced stiffening of a pure DNA hydrogel. They observed stiffening in each sample of 3, 6, and 9.3% w/v. However, for concentrations below 9.3% w/v the gel was melted at 90 °C, whereas the 9.3% w/v the gel showed transient stiffening over the course of 10 min at 90 °C from a few hundred Pa to nearly 100,000 Pa after cooling, demonstrating the potential for DNA hydrogels of higher concentration to enter the kPa range. Similarly, the 6% w/v gel melted initially but returned to its gel state with an increasing storage modulus after 6 min at 90 °C and increased to a few thousand Pa after cooling, from its initial storage modulus of a few hundred Pa. Karacan et al. demonstrated a similar response to heating–cooling for a 5% w/v DNA solution whose storage modulus was increased from 20–200 Pa to 10 kPa, measured at frequencies of 0.1–100 Hz [54].
2.2. Nanostructured
Nanostructured DNA hydrogels are most commonly constructed from multivalent DNA nanostars (DNAns) (Figure 1c) [50,61,62,63]. Alternatively, short multidomain self-assembling strands have been used to construct complementary linear sections of dsDNA crosslinked by complementary overhangs (Figure 1d) [64,65]. Jiang et al. designed a single short DNA strand with 3–6 self-complementary binding domains of 12–20 nt in length to form a DNA hydrogel. The rheological characterization of 2% w/v DNA hydrogels formed from strands with 3, 4, 5, and 6 domains of 12 nt in length showed a storage modulus of ~3000, 1000, 7, and 7 Pa, respectively. Interestingly, when the 3 domain strand was tested with different length domains, 12, 16, 20 nt, a storage modulus of ~3000, 10, and 80 Pa was observed respectively [64]. This approach yielded counterintuitive results where an increase in valency and hybridization interaction yielded lower storage moduli, which demonstrates the need to consider the availability of binding sites in nanostructured designs. Pan et al. conducted an alternative study of linear self-assembling DNA hydrogels which further supports this need for flexibility in the internal structure to enhance macroscopic stiffness [65]. Specifically, 2–3 short DNA strands were designed to hybridize sequentially with one strand designed with a short 10 nt sticky overhang that could hybridize to another long tiled dsDNA strand with a self-complementary overhang. The length of the tiled dsDNA was designed to be rigid, with full complementary dsDNA regions between crosslinks, or flexible, with 30 nt thymidine repeats between crosslinks, with interchangeable parts (Figure 1d). These rigid and flexible designs were mixed in fractions between 100, 50, 25, and 0% of rigid linkers at approximately 4% w/v DNA content. The respective mixtures were found to have the corresponding storage moduli of 450, 600, 1800 and 1800 Pa at frequencies between 0.1–10 Hz and gelling temperatures of 40.1, 43.6, 49.4, and 48.7 °C [65]. This example as well demonstrated counterintuitive results where a greater fraction of flexible linkers increased the storage moduli, which was due to an increased number of hybridized crosslinks with the flexible linkers. However, when flexible linkers were introduced to DNAns based gels they caused a decrease in the storage modulus [66]. Stoev et al. studied the formation a trivalent DNAns hydrogel with rigid sticky ends and sticky ends with 6 nt poly-T region between the sticky end and the dsDNA arm of the structure [66]. Their design allowed for enough flexibility in the linkers that they could self-hybridize with linkers from another arm on the same structure, thus also limiting the degree of connectivity between different structures. The flexible system only showed soft gel-like rheological properties near the melting temperature (Tm) of the linker (52 °C) with a storage modulus < 1 Pa greater than the loss modulus at frequencies below 103 Hz, whereas the nonflexible linkers showed a high-frequency plateau modulus of 45 Pa at 104 Hz at 44 °C and melted at 52 °C [66].
With regards to DNAns based hydrogel designs, valency presents an additional degree of freedom for mechanical design. Conrad et al. studied the role of DNAns valency (f) ranging from 3 to 6 with 20 base-pair (bp) arm lengths and a 7 nt sticky end separated by a nonbinding adenine base to allow for a slightly greater degree of flexibility. This study of valency was inspired by Maxwell’s rigidity criterion, which predicts that a 3D network must have six beams emanating from each junction in order to be rigid. With ~500 uM of DNAns for 3, 4, 5, and 6 arms, corresponding to ~2.3, 3.0, 3.8, and 4.6% w/v, a plateau storage modulus of approximately 215, 1240, 2140, and 5810 Pa was observed at a frequency of 100 Hz [63]. Moreover, when valency was compared at equivalent volume fractions of DNA an increased storage moduli correlated with the increase in valency. The change in plateau storage modulus as a function of molar DNAns concentration was fitted to a power law for each valency and the corresponding exponents were 1.8, 1.3, 1.5, and 1.0 when, f = 3, 4, 5, 6, respectively. This demonstrates a greater dependence on concentration for a valence of 3 as compared to 6. Interestingly, valencies of 3 and 4 showed strain stiffening properties by almost 2-fold near 100% strain while 5 and 6 fractured at approximately 10% strain. Xing et al. explored a slightly different approach for the design of a DNA hydrogel based on trivalent DNAns by incorporating two DNAns with sticky ends which complement the other DNAns but not itself [62]. The arms of these DNAns were 15 bp and sticky ends were either 9 or 12 nt, with or without a 4 nt noncomplementary thymidine repeat to study the role of flexibility in the linkers. At a DNA content of 2% w/v with the 12 nt sticky ends, the gel exhibited a storage modulus of ~70 and 500 Pa with and without the 4 poly-T flexible joint, respectively, while the 9 nt sticky ends similarly yielded ~70 and 250 Pa, showing that the flexibility significantly reduces the plateau modulus but also diminishes the contribution from the higher affinity sticky ends.
3. Responsive DNA Linkers
3.1. Nucleic Acid Structural Motifs
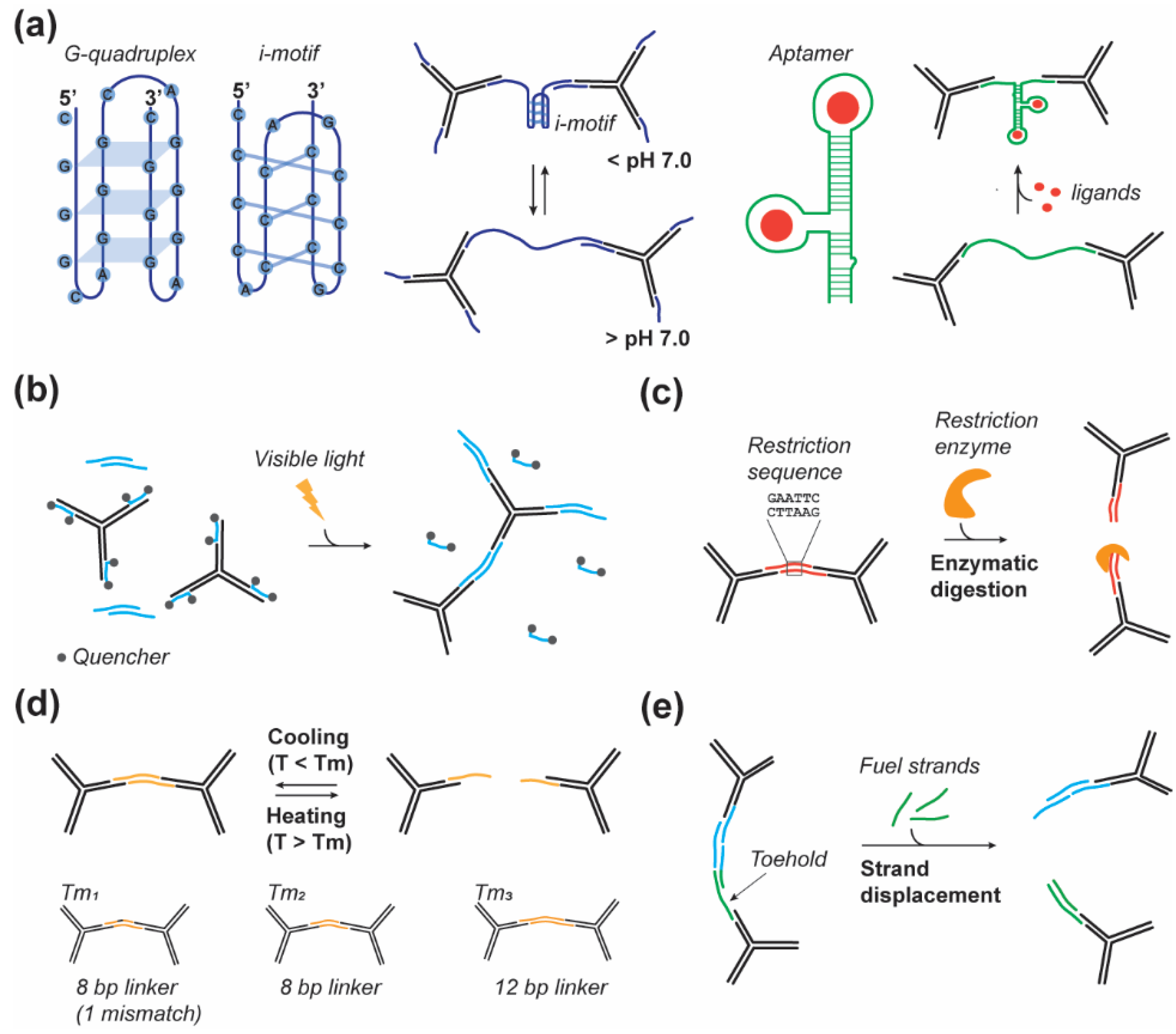
Nucleic acid structural motifs have been used as dynamic linkers to modulate the mechanical properties of pure DNA hydrogels or other polymeric hydrogels upon specific stimuli. These structural motifs are, in general, short sequences of DNA (or RNA) that can adopt various structural conformations, which can be canonical and follow the Watson–Crick base pairing rule, or noncanonical, not following the Watson–Crick base pairing [67,68]. These motifs, which play a significant role in biology, are sensitive to various environmental parameters such as temperature, pH, and salt concentration [69,70,71,72,73]. Their capacity to convert from an open (extended) state to a folded state upon specific conditions has allowed researchers to induce change in the hydrogels’ mechanical and structural properties (Figure 2a). The two most common noncanonical motifs used are the G-quadruplexes and the i-motifs. The G-quadruplex motifs are four-stranded helical structures that can form in G-rich nucleic acid sequences via Hoogsteen hydrogen-bonding between a multi-G-track (GGG) separated by another nucleotide [74]. The i-motifs, also called C-quadruplexes, are found in C-rich regions and are composed of four short strands maintained together by a noncanonical C:C base pair [72]. Both the G and C-quadruplexes are pH sensitive and exhibit high structural stability at acidic pH and are unfolded at physiological or basic pH (Figure 2a). They naturally became motifs of choice to control the structural and the mechanical properties of DNA-based hydrogels. For example, Cheng et al., in 2009 [49] and Zhou et al., in 2016 [31] used trivalent DNAns and linkers containing, respectively, one or two i-motifs to control the mechanical properties of DNA hydrogels exhibiting rapid actuation (~100 ms) [75] and a sharp stiffness increase at pH lower than pH 6.0. Zhou et al. demonstrated the reversibility of the mechanical properties with a G’ ranging from 250 Pa at pH 8.0 and 1000 Pa at pH 5.0 [31]. In another study, Hu et al. used another Hoogsten-type motifs called triplex DNA structures [76] to assemble pH-sensitive hydrogels that can be used to cyclically control the gel shape [15].
Aptamers represent another large category of structural nucleic acid motifs that can undergo distinct structural changes upon binding to a specific biomolecule target (Figure 2a) [77]. These aptamers are often used as capture strands but they can also be included in the linker design to render the mechanical properties of DNA-based hydrogels sensitive to specific biomolecules. For instance, Liu et al. used ATP binding aptamers to control the mechanical strength of their DNA hydrogels. They showed that in the presence of ATP their hydrogels exhibit a G’ of 380 Pa and a G’ of 204 Pa in the absence of ATP [33]. Moreover, they demonstrated the ATP concentration dependence of their system that allows for fine-tuning of the hydrogel’s mechanical properties.
3.2. Photoresponsive
The photomodulation of hydrogel properties is desirable as light offers a simplistic and noninvasive manner of altering a hydrogel’s structure, thus averting diffusion requirements and potential nonspecific interactions within a dynamic cell environment. The photomodulation of DNA interactions has been previously explored without regards for DNA hydrogels synthesis, as this could be a powerful technology for gene regulation. Prior studies yielded a variety of approaches to photomodulation [78,79], though many are irreversible, as they are designed for single stimulus activation or for the inhibition of genes. A few reversible processes have, however, been developed [80,81,82,83], most notably with regards to DNA hydrogels, such as the Azo-benzene moiety and the synthetic base, 3-cyanovinylcarbazole nucleoside, CNVK. The Azo-benzene moiety undergoes cis-to-trans isomerization when exposed to visible light (>400 nm) and trans-to-cis when exposed to UV light between 300–400 nm. The cis isomer caused a decrease in melting temperature, due to increased steric hindrance, from 24.8 to 15.9 °C in an 8 bp poly-T dsDNA strand [80] Azo-benzene-modified DNA crosslinkers were used as in acrylamide based gels to induce the release of fluorescein, gold nanoparticles, and horseradish peroxidase with UV-light [38]. The synthetic base CNVK offers rapid conformational switching inducing DNA hybridization with a complementary pyrimidine base, Cytosine or Thymine, within 1 s of UV (366 nm) irradiation [81,83]. Inversely, a minute of irradiation with 312 nm UV-light will split the base-pairing by inducing conformational switching to the cis state. Kandatsu et al. utilized CNVK to form a pure DNA hydrogel of tetravalent DNAns [84]. The DNAns’ sticky ends consisted of 4, 6, or 8 nts with CNVK base at the end. Gel–sol transition was monitored using a steel ball which sinks in the sol state or is suspended in the gel state. The sticky ends of 4 and 6 nt showed a reversible gel–sol transition, while an 8 nt sticky end did not, demonstrating the importance of working within the relevant shift of linker Tm induced by these photoswitchable bases.
Additionally, the previously discussed covalent crosslinker of DNA, EGDE, has been shown to degrade under exposure to light irradiation with a wavelength comprised of between 360–400 nm causing the degradation of EGDE crosslinked DNA hydrogels [85]. This approach was utilized to control the release of p53 gene, doxorubicin (DOX), epirubicin (EPIR), and paclitaxel (PTX) in EGDE crosslinked nanogels as a therapeutic technology [86]. While many of these existing studies on the photomodulation of DNA hydrogels are focused on the gel–sol transition and induced solute release, it is reasonable to imagine these same concepts being applied to only a fraction of DNA crosslinks or within the internal DNAns structures to produce photomodular mechanical properties within DNA hydrogels. As well, photocontrolled crosslinking of DNA hydrogels allows them to be photographically patterned as demonstrated by Shimomura et al. who used quencher-modified DNA strands to block the sticky ends of trivalent DNAns such that they could be photothermally released and permit gelation within a localized region (Figure 2b) [87]. However, given the focus on solute release, these studies do not investigate the mechanical properties for their formed hydrogels and further studies must be undertaken to unveil the potential of such photoresponsive DNA crosslinkers to modulate mechanical properties in DNA hydrogels.
3.3. Enzyme Responsive
Leveraging the sequence specificity of DNA to design crosslinkers allows the insertion of specific restriction sites for efficient enzymatic digestion by restriction endonuclease (Figure 2c). For instance, Xing et al. used this principle to design linkers that can be orthogonally cut by restriction enzymes, BamHI and EcoRI, thus inducing the gel-to-sol transition in the presence of the specific enzyme [88]. However, despite being a very efficient mechanism to control the mechanical properties and the dissolution of hydrogels, this strategy is not reversible and is in general used for cargo release from DNA-based hydrogels [89]. More recently English et al. described the use of the CRISPR-Cas9 system to modulate the physical properties of a polyacrylamide DNA crosslinked hydrogel [90]. In this paper they demonstrated the possibility to trigger degradation of the hydrogel by using ssDNA linkers containing Cas12a recognition and cleavage sequence. As previously discussed in this section, this strategy is irreversible and would be used to control release of molecules from the hydrogels and to control hydrogel degradation.
3.4. Thermal Responsive
The use of hybridized DNA as a crosslinker inherently makes hydrogels thermally responsive and has been demonstrated in various studies to produce changes in the dynamic moduli [36,53,88,91]. Li et al. fabricated peptide based DNA hydrogels with identical crosslinking point density but with three different DNA crosslinkers to study the effect of the crosslinker Tm on the rheological properties of the hydrogel [92]. The three linkers have melting temperatures of 20.8, 39.6, and 53.5 °C, which corresponded to linker lengths of 8 (1 mismatched bp), 8, and 12 bp. The crosslinker with a mismatch did not form a gel above room temperature while the 8 and 12 bp crosslinkers yielded a storage modulus of 4500 and 5300 Pa and gelling temperatures of 42.5 and 53.5 °C, respectively. Furthermore, when cyclically tested for thermo-reversibility between 25 and 60 °C, the 12 bp crosslinker maintained its storage modulus when returning to the gel state [92]. Xing et al. similarly varied the sticky ends but used a 3-arm DNAns, in contrast to the previously mentioned peptide-DNA hydrogel, and observed that the lower melting temperature of the sticky ends significantly increased the frequency dependence of the storage moduli though each variation shared a storage modulus of 1700 Pa at a frequency of 3 Hz [88]. The DNAns gel showed similar thermo-reversibility as demonstrated by Li et al. and showed gelling temperatures of 38, 48, and 58 °C for sticky ends of 8 (1 mismatched bp), 8, and 12 bp. The previously mentioned pH responsive hydrogel by Zhou et al. which utilized an i-motif to induce stiffening at low pH also demonstrated an increase in the gelling temperature from 39 to 45 °C [31]. These examples demonstrate the unique thermal responsiveness of DNA hydrogels and the ability to exploit these interactions for fine-tuning of the mechanical properties (Figure 2d).
3.5. Strand Displacement
The strand displacement method relies on the replacement of a hybridized strand with a new strand (fuel strand) containing a greater number of complementary bases to the target strand (Figure 2e). The fuel strand hybridizes to an exposed “toe-hold” binding site and gradually displaces the previously hybridized strand. This phenomenon offers an interesting tool given the sequence-programmability and dynamic modulation over a few hours [93,94,95]. Fern et al. have shown that DNA crosslinked polyacrylamide gels could be expanded significantly by incorporating hairpin strands to displace the existing crosslinks and could be programmed to expand to a controlled limit in response to a stimulus such as a DNA strand or ATP, a mechanism that can be adapted to programmably alter the mechanical properties of a hydrogel [95]. Bomboi et al. designed a tetravalent DNAns gel in which toehold regions were programmed into the sticky ends to enable short 6 nt oligonucleotides to displace the linker from the sticky end at low temperatures, causing the gel to melt in response to both cooling or heating, thus producing a binary temperature dependence of the mechanical properties [61]. Yue et al. also used strand displacement of the sticky end linkage between trivalent DNAns to reversibly modulate the mechanical properties between three states with effective Young’s moduli of ~4, 13, and 23 kPa [47].
4. DNA–Polymer Hybrids
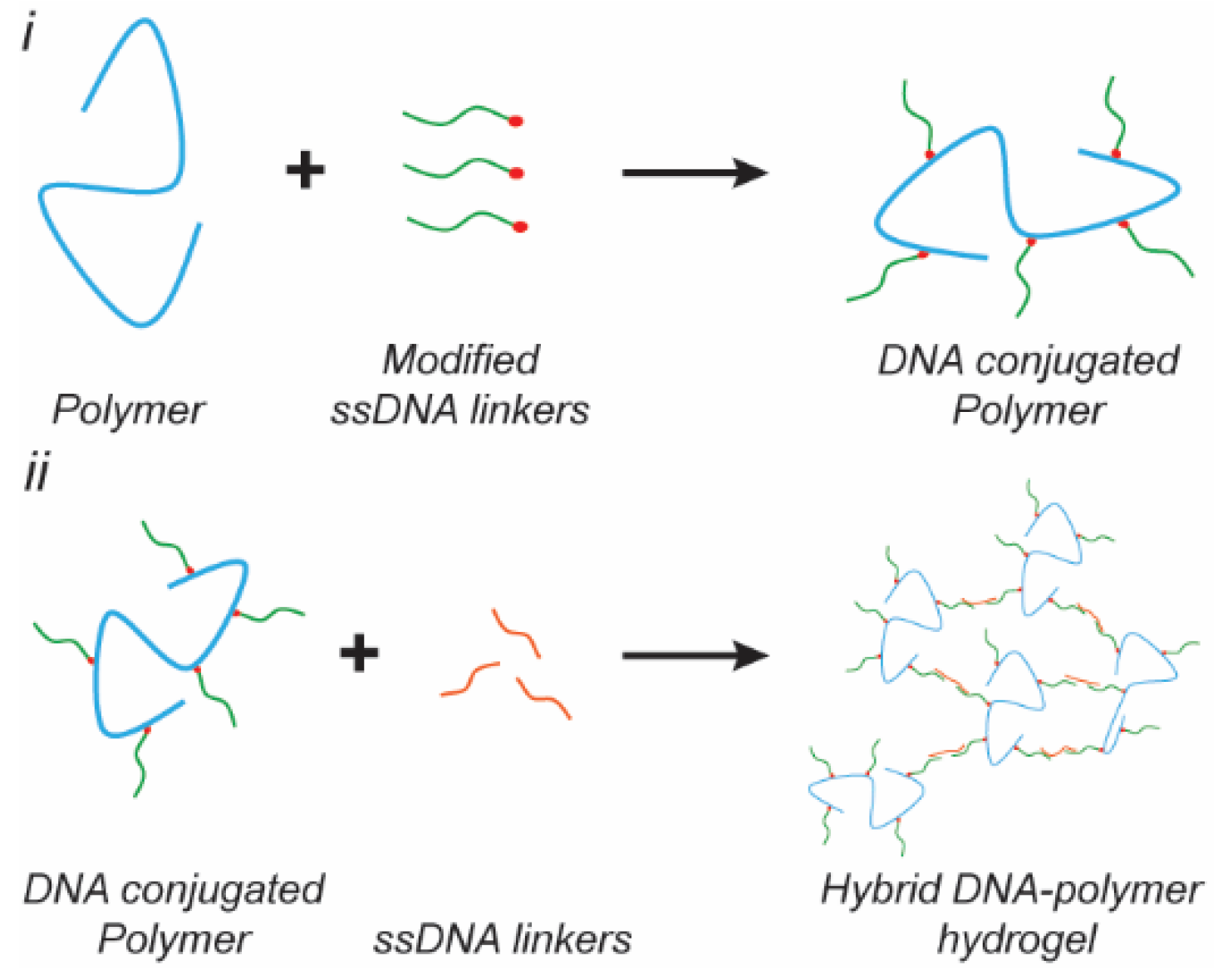
Beyond pure DNA hydrogels, DNA can be used as a crosslinker to tune the mechanical properties and responsiveness of polymer hydrogels. In this section, we categorize DNA–polymer hybrid hydrogels based on the type of the constituent polymer, synthetic- or peptide-based. DNA–polymer hybrid hydrogels offer the advantage of reduced DNA content, which results in a lower material cost, as well as enabling the incorporation of alternative functional motifs to enable cell adhesion, hydrogel degradation [96], or alternative mechanisms for the dynamic tuning of the mechanical properties [92].
4.1. Synthetic Polymer Based Hybrids
Polyacrylamide (PAAm) was one of the first polymers to be crosslinked via anchored DNA hybridization [91]. Acrydite modified DNA oligonucleotides are used to covalently anchor DNA crosslinkers to the PAAm chain and a free ssDNA strand is typically added to hybridize across two anchored ssDNA sites (Figure 3). Lin et al. used this approach on a PAAm based hydrogel and included an available toehold region on the free ssDNA crosslinker to enable strand displacement mediated rupture of the hybridized crosslink. Additionally, the DNA crosslinker only replaces a defined fraction of the covalent crosslinking points which enables a broad range of stiffnesses to be achieved, from tens of Pa to ~10 kPa of the elastic modulus from [97,98]. Furthermore, the concentration of hybridized linker strands could be altered in situ by the addition of the fuel strand [97,98]. In addition to using complementary DNA as a crosslinker, peptide nucleic acid (PNA) oligonucleotides have been used. PNA are polymer mimics of DNA with a pseudopeptidic backbone that replaces the sugar-phosphate backbone of DNA [99]. Chu et al. designed a PNA/DNA/polymer hybrid hydrogel for which PNA and/or DNA was conjugated onto the polymer backbone, N-(2-hydroxypropyl)methacrylamide (HPMA), and ssDNA was used as a crosslinker to yield two different conformations of the crosslinker, a double helix via Watson–Crick base pairing or a triple helix formation via Hoogsteen base pairing [14]. The triple-helix forming gel showed a significant decrease in pore size to a few microns as compared to the pore size of ~100 microns for the double helix crosslinked version.
4.2. Peptide Based Hybrids
In addition to synthetic polymers, polypeptide chains have also been functionalized with DNA linkers to create hybrid hydrogel systems. Wu et al. used a human serum albumin (HSA) derived polypeptide copolymer (PcP) functionalized with ssDNA and a dimer of trivalent DNAns was used as the crosslinker to form a hydrogel with tunable mechanical properties [96]. Additionally, by varying solid content of the PcP from 1% to 5%, they were able to change the storage modulus from ~1 Pa to 4200 Pa [96]. Li et al. used a similar approach to crosslink poly(L-glutamic acid240-co-ɣ-propargyl-L-glutamate20) (p(LGA240-co-PLG20), Mw 34,060, PDI 1.4) with dimeric trivalent DNAns which yielded a storage modulus of ~0.4 to 400 Pa at 1–5% w/v [16]. Moreover, the inclusion of DNA nanostructures as the crosslinker in this hydrogel added self-healing and shear-thinning properties, which are required for bioprinting. Interestingly, these hydrogels were crosslinked with similar DNA nanostructures though exhibiting an order of magnitude difference in their storage modulus at similar concentrations, which is due to the helical structure of the HSA-derived-PcP. This demonstrates the potential of biopolymers to provide further functionality for tuning the mechanical properties in conjunction with DNA-based crosslinkers. Additionally, the previously mentioned peptide, p(LGA240-co-PLG20), further demonstrated pH-induced α-helix formation of the peptide backbone when crosslinked with linear dsDNA, which increased the storage modulus from 1750 to 2000 Pa when the pH was lowered from 8 to 5 [92]. Desphande et al. functionalized oligo(ethylene glycol)-functionalized polyisocyanopeptide (PIC) with ssDNA to create a hydrogel with stress-stiffening properties. By varying the ratio of crosslinking DNA to anchored DNA sites from 0.1 to 0.9 the storage modulus was tuned from ~1 Pa to 30 Pa [100]. However, increasing the ratio to 1.2 decreased the storage modulus to ~23 Pa due to saturation of the anchored ssDNA by excessive crosslinker. The importance of effective crosslinks was further corroborated by an increase in the storage modulus from 50 to 130 Pa after heating and cooling with a ratio of 0.9, indicating network reorganization and concurrently enhanced crosslinking.
5. Dissecting the Mechanical Properties of DNA Hydrogels
5.1. Characterization Methods
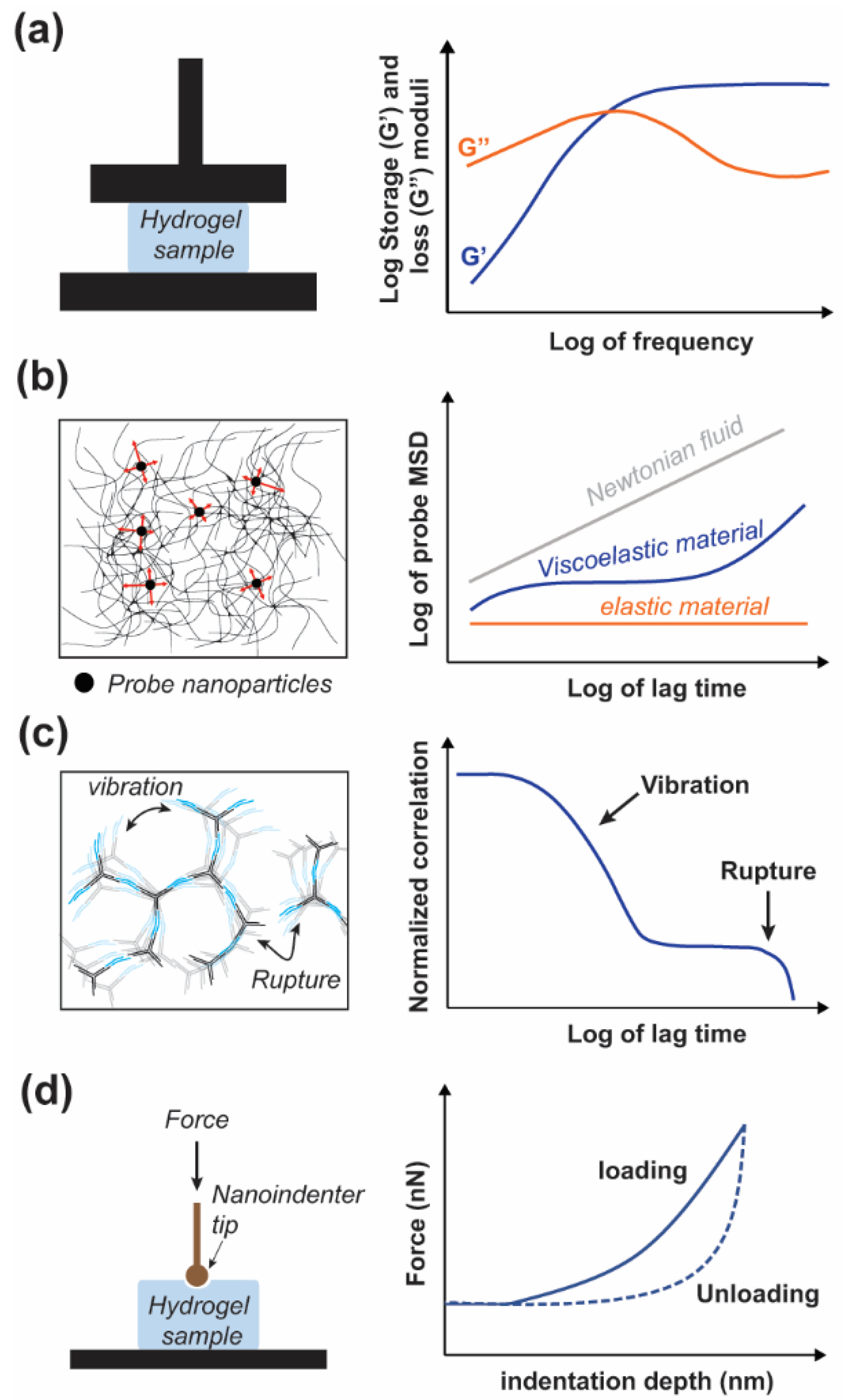
There are a variety of methods to characterize the mechanical properties of hydrogels; generally categorized as compression, indentation, and rheology. Conventional rheological measurements are conducted via bulk rheology (Figure 4a), which includes parallel plate [33,54,64] and parallel cone and plate [51].
Given the innate thermo-reversibility of DNA hydrogels, temperature-controlled rheology is often of interest. Bulk rheology is advantageous because it is a well-established technique; however, it is limited to low frequencies (<100 Hz), often requires a large sample volume, and cannot probe the heterogeneity of the sample. The required large sample volume, typically in the milliliter range, is an important factor for the use of bulk rheology to characterize pure DNA hydrogels due to current material cost. In contrast to bulk rheology, microrheology utilizes a much smaller sample volume, typically less than 100 μL. Microrheology uses the motion of embedded probe particles caused by either thermal fluctuation (passive) or externally applied forces (active) to determine the rheological properties of the sample (Figure 4b). Compared to bulk rheology, microrheology offers access to a broader frequency range (10−1–106 Hz) and the potential to study sample heterogeneity [101,102,103]. The measurement techniques for passive microrheology include dynamic light scattering (DLS), diffusive wave spectroscopy (DWS) and particle tracking microscopy (PTM) [62,101,104,105]. Since the relaxation time for a hydrogel might be longer than the timeframe allowed by DLS or DWS (~10 s) PTM or bulk rheology can be used in conjunction to access the lower frequency regime (<0.1 Hz) [106]. DLS measurements are also dependent on sufficient scattering from the probe particles to mask the scattering signal from the sample, though they typically require low probe concentrations. For samples of greater opacity DWS is a viable alternative, although it requires a much higher density of probe particles which may interfere with the properties of the hydrogel. However, DWS is also more sensitive at higher frequencies compared to DLS [101,103,104]. Alternatively, DLS can be used without probe particles to study the network vibrations and reorganization at multiple length-scales (Figure 4c) [107].
Among the other techniques available to characterize soft materials, nanoindentation represents one of the most convenient methods for the direct measurements of mechanical properties of hydrogels at different scales [108]. This technique relies on the application of a controlled force (load) on the surface of a soft biomaterials with a nanoprobe to induce local deformation (Figure 4d). The depth of indentation or deformation as well as the response time for a given load are monitored and used to identify mechanical properties of the sample. Nanoindenter instrument and atomic force microscope (AFM) are the two types of instruments that can be used to perform nanoindentation on these soft biomaterials [108,109]. In addition, AFM enables the combination of nanoscale imaging and mechanical characterization of biomaterials that are used to scaffold cells for biomedical applications such as tissue engineering. While this technique has been used with various types of soft biomaterials and polymer hydrogels, only a few studies report measurements done on DNA-based hydrogels. For example, Yue et al. used a nanoindenter instrument to measure the stiffness (Young’s modulus) of various DNA-based hydrogels with a tunable Young’s moduli ranging from ~3.75 to ~23.45 kPa [47].
5.2. Modeling/Simulations
Computer simulation of DNA hydrogels at the atomic scale is currently beyond the limits of computational power, thus coarse grained models such as oxDNA [110,111] have been developed. oxDNA represent nucleotides as single oriented rigid bodies, accounts for electrostatic interaction via the Debye–Huckel approximation, and accounts for sequence-dependent interactions in accordance with the SantaLucia model [112]. Small gel volumes (~100 nm box length) of tetravalent DNAns have been simulated using oxDNA into realistic times of a few μs [113,114]. Rovigatti et al. used oxDNA simulations of tetravalent DNAns gels to study the temperature and concentration-dependent formation dynamics to yield an accurate phase diagram [113]. Furthermore, using oxDNA simulation of DNAns gels, Rovigatti et al. found that DNAns gels will never crystallize owing to their arm flexibility, thus reducing the free energy of the gel state below that of the crystalline state [114,115]. However, these simulations were run at high salt concentration of 0.5 M, prior to the inclusion of variable salt-dependence within the oxDNA model in 2015 [111]. The oxDNA model utilizes a cutoff distance of three times the Debye–Huckel Length and at lower, physiological, salt concentrations the electrostatic potential becomes long-range and significantly increases the computational cost due to the increased number of interactions. The oxDNA model of a single DNAns at low-salt concentrations was, however, shown to match with experimental results measured by small-angle neutron scattering (SANS) [116]. In 2018, Xing et al. developed a coarse-grained simulation for DNA hydrogels by representing a trivalent DNAns with seven rigid bodies, a center and two bodies per arm, with patchy ends representing interunit hybridization [117]. This model was used to study the temperature and concentration-dependent degree of association, structural organization, and to dissect the rheological response, which will be discussed further in the next section. More recently, Stoev et al. used a similar model in conjunction with oxDNA simulations of a single DNAns and microrheology experiments to study the role of linker flexibility in DNA hydrogels [66]. Using oxDNA the probable angle for DNA sticky ends with rigid or flexible linkers was determined. This angle was incorporated as the range of motion for the patchy ends within the coarse-grained model and the rheological behavior acquired through simulation was validated with microrheological results, demonstrating a significant increase in the crossover frequency when flexible linkers are present, causing the gel to behave as a fluid at low frequencies [66].
5.3. Dynamic Properties of DNA Hydrogels
DNA hydrogels, particularly DNAns hydrogels, present a unique system with a high-degree of control which can be utilized to mimic other complex materials for further study. Specifically, Biffi et al. observed a two-step decay in the correlation function during probe-free DLS measurements, which resembles a property seen in glassy materials [118]. This two-step decay correlates with a fast and slow time constant representing the vibrations of the system and network reorganization, respectively (Figure 4c). A later study utilizing multiangle DLS, Biffi et al. found the slow relaxation time to be length-scale independent above the minimum wavevector used, 8 μm−1, which corresponds to a length-scale ~780 nm comprising tens of DNAns at ~14 nm in length [119]. This length-independent relaxation time suggests that the relaxation time is also weakly dependent on concentration but follows an Arrhenius equation with regards to temperature dependence [119]. A later study by Nava et al. explored the lower wavevector regime down to 0.2 μm−1, and as large as 30 μm−1 corresponding to length-scales of 31 and 0.2 μm, respectively. They found that at larger length-scales the slow-relaxation time becomes dependent on the length-scale by an exponent of two, attributed to diffusive processes [107]. Interestingly, the point of transition between dependence and independence of length scale is temperature-dependent, with lower temperatures resulting in a larger length-scale for transition in agreement with a larger degree of association at lower temperatures. Furthermore, Nava et al. replicated the length-scale independent region with a simulation composed of a linear chain of beads and springs for which there were two different spring constants. Each spring dynamically changes between stiffnesses in order to replicate the local changes in bulk modulus due to localized network reorganization in DNAns gels [107]. The formation of clusters during DNAns gel formation was studied by Fernandez-Castanon et al. using DLS and variable ratios of tetravalent DNAns to linker. They found that at low temperatures a percolating network was formed regardless of the ratio; however, the ratio affected the formation of clusters at higher temperatures and thus could be used as a factor to program the formation of the gel [120]. As previously mentioned, Conrad et al. studied the effect of DNAns valency on DNA hydrogels at different concentrations ranging from 200–800 μM of DNAns. At ~500 μM of DNAns the plateau modulus increased from 100 to 4000 Pa with valency from 3 to 6, and although a higher valency implies a higher DNA content per structure, they show that a valency of 6 still results in a plateau modulus an order of magnitude greater than for the equivalent volume fraction of trivalent DNAns [63]. Additionally, while there was a small change in the crossover frequency at different valencies, the temperature dependence was identical for each valency and followed an Arrhenius equation; further validating the notion that the hybridization energy of the interunit linkage is responsible for the timescale of network reorganization [63]. Xing et al. used their previously mentioned coarse-grained model to extract the contributions of the DNA arm rigidity, angular constraint, and interunit hybridization to the storage modulus at different frequencies [117]. They found that the arm rigidity was responsible for over 90% of the storage modulus and largely independent of frequency, while the angular constraints contributed to only a few percent of the total storage modulus and was similarly frequency independent. Interestingly, the contribution due to interunit hybridization was two orders of magnitude less than the total plateau modulus at low frequencies, however, it increased by almost an order of magnitude at higher frequencies. This is indicative of the bonds breaking at longer timescales and providing a greater degree of structural support at high frequencies [117]. While DNAns will inherently separate during sticky end rupture, Romano and Sciortino utilized a cyclic strand-displacement cascade which prevents the full rupture of the interunit linkage by maintaining a dynamic strand-displacement reaction during bond-switching [121].
5.4. Nanoscale Mechanical Properties
The nanoscale mechanical properties of DNA [122] are known to influence the bulk mechanical properties of the DNA hydrogels assembled [50,123]. Indeed, DNA has inherent nanoscale mechanical properties that depend on its structure and assembly [22,26]. It has an elastic modulus comprised between 0.3 and 1 GPa and a persistence length of 50 nm (~150 bp) in a duplex form [124]. In addition, the presence of nicks, bulges, single stranded DNA, or multiple helix bundles also effects the nanoscale mechanical properties of double stranded DNA linkers and motifs [23,24,25], which subsequently effects the overall mechanical properties of the DNA hydrogels formed with these motifs [123]. An illustration of the importance of these nanoscale features has been given by the study from Um et al. in 2006, which demonstrated that the crosslinking of DNA motifs with ligase to avoid the presence of nicks can drastically increase the mechanical strength of DNA-based hydrogels [50]. Furthermore, hydration around the DNA strands increases stiffness [125], and salt interactions determine the length-scale of this hydration layer resulting in significant swelling of DNA fibers in the absence of salt [60]. Salts act as counterions to stabilize the negative charge of the DNA backbone and govern hole transfer along dsDNA, while the conformational state is responsible for electronic coupling through π-orbital base stacking [126,127,128,129] This mechano-electronic coupling has been utilized to develop a FRET-based nanosensor for mercury [130] and demonstrated an order of magnitude increase in conductivity for A-form DNA relative to its B-form [131]. An alternative mechanoelectric DNA-based probe was developed for potassium detection based on the conformational transition of DNA into a G-quadruplex [132]. Inversely, DNA extension can be programmed to occur above a certain force threshold (<100 pN) and FRET reporter probes have been used to further study mechanotransduction at single cell-ECM and cell-cell adhesion sites [133,134,135,136,137].
6. Mimicking Biomaterials for Biomedical Applications
The inherent biocompatibility of DNA-based biomaterials and the control over the mechanical properties provide DNA-based hydrogels with several advantages over other biomaterials, such as hydrogels assembled from ECM-derived proteins, polysaccharides, and natural or synthetic polymers [138,139]. These are commonly used to control cellular behaviors and create ECM mimics or scaffolds for biomedical application such as regenerative medicine, tissue engineering, sustained drug delivery [140], immunomodulation [41,141], and wound healing [142]. Indeed, the nanoscale precision for biomolecules’ conjugation and the ease of DNA motifs’ functionalization have enabled the assembly of hybrid biomaterials conjugated with several bioactive signals such as ECM-cell adhesion peptides and growth factors [143,144], or antigens, and immunomodulatory motifs [41,42], which can facilitate cell interactions and induce control of cell behaviors [145]. For example, Baig et al. designed a multiligand DNA origami for the pathway-specific modulation of neocortical neurons while monitoring local membrane tension with a DNA-based FRET tension sensor [136]. Alternatively, in a study by Aldaye et al. the authors designed DNA-based ribbons to assemble a fiberlike ECM mimicking biomaterial that was later functionalized with ECM proteins—containing the RGD peptide—in order to promote cell attachment [46]. Another strategy was developed by Finke et al. to assemble a printable peptide-DNA hybrid biomaterial generated using PCR with branched primers that can serve as a coating layer to promote cell attachment on the surface [146]. These materials show that cells can adhere, grow, and proliferate on DNA-based hydrogels. DNA-based biomaterials also appear to be well-suited for the sustained release of drugs or antigens. Nishikawa et al. notably used injectable DNA-based hydrogels loaded with the ovalbumin model antigen and CpG motifs to induce a strong immune response in mice [43]. In another study, Komura et al. developed a hybrid DNA/RNA hydrogel that can stimulate Toll-like receptors (TLR) by releasing GU-rich RNA sequences and inducing a strong immune response [40]. Several studies have also highlighted the use of DNA-based materials or hybrid DNA materials with controlled and tunable mechanical and rheological properties to mimic those of the ECM and influence cell behavior. For instance, using DNA oligonucleotides as a linker for a polyacrylamide gel functionalized with ECM proteins, Jiang et al. have notably demonstrated that they could control the gel stiffness from a few hundred Pa to 30 kPa by varying the length and density of the crosslinker strands [147]. They showed that temporally altered mechanical properties influenced the length and number of dendrites of neurons cultured on their hydrogels. Basu et al. have shown that incorporating silicate nanodisks into DNA-based hydrogels could enhance toughness and elasticity and serve as good ECM mimics for tissue repair with sustained drug delivery capability [148]. These works highlight the potential of DNA-based hydrogels for functional tissue engineering with a broad array of stimuli-responsiveness [149].
7. Conclusions
Herein, we have reviewed various designs for DNA hydrogels with regards to their mechanical properties, responsiveness, characterization, dynamics, and use as functional biomaterials. Notably there are a wide variety of design implementations to form DNA hydrogels and correlating their nanoscale design into programmable macroscale mechanical properties remains a vital challenge. Given the broad design space and the various characterization methods, it becomes difficult to establish standard design principles for utilizing DNA hydrogels as biomaterials with specific mechanical properties. In order to facilitate the progression towards mechanically programmable DNA-based biomaterials, we identify important factors to consider for mechanical design of DNA hydrogels including structural rigidity, linker affinity or Tm, valency, linker flexibility, and steric availability of crosslinking points. Furthermore, total DNA content should be considered in the design of DNA hydrogels as large-scale synthesis costs currently remain high. As discussed earlier, DNA hydrogels can be produced by large quantities of biologically derived dsDNA; however, similar mechanical properties were achieved at lower DNA content by forming hydrogels with DNA nanostructures. Future studies may unveil structural motifs that can yield improved mechanical properties at lower DNA concentrations, thus lowering the cost of DNA hydrogels. Currently, to avert this cost and utilize the programmable nature of DNA for mechanically tunable biomaterials, hybrid DNA hydrogels have been developed. These hybrid hydrogels offer the distinct advantage of enabling a broader range of accessible mechanical properties for DNA-based biomaterials, whereas all-DNA hydrogels are currently limited to relatively soft mechanical properties, which may limit their application potential, although the innate thermal-responsiveness and thixotropic properties of DNA hydrogels yield them considerable potential with regards to bioprinting and injectable therapeutics. For in vivo applications of DNA hydrogels further work is required to improve the stability under physiological conditions [150]. Similarly, these developments in stability will be critical to maintaining the designed mechanical properties for in vivo applications. However, for drug releasing in vivo applications the mechanical properties may only be significant for the injection of the hydrogel and in vitro applications are currently viable when cells are cultured in the absence of serum. Thus, the primary challenges to implementing mechanically programmable DNA hydrogels can be surmised as cost, stability, and established design principles. These challenges are currently shared with the DNA origami field as a whole, and as such, significant efforts are being made to overcome these challenges, specifically the challenges of cost and stability. However, DNA origami design principles have developed an appreciable level of standardization through the development of automated and manual design software, whereas standard design principles for DNA hydrogels do not yet exist. By compiling various designs from a mechanical point of view we begin to move towards identifying these standard design principles. Given the variety of conditions, designs, and characterization methods, future studies will need to analyze the present variety of DNA hydrogel designs and undertake a systemic interrogation of each design parameter tuned within a similar system to establish these design principles.
Author Contributions
All authors wrote, commented, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seeman, N.C. DNA in a Material World. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. An Overview of Structural DNA Nanotechnology. Mol. Biotechnol. 2007, 37, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and Opportunities for Structural DNA Nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, Y.; Su, M.; Ko, S.H.; Ye, T.; Leng, Y.; Sun, X.; Ribbe, A.E.; Jiang, W.; Mao, C. DNA Self-Assembly: From 2D to 3D. Faraday Discuss. 2009, 143, 221–233. [Google Scholar] [CrossRef]
- Han, D.; Pal, S.; Nangreave, J.; Deng, Z.; Liu, Y.; Yan, H. DNA Origami with Complex Curvatures in Three-Dimensional Space. Science 2011, 332, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneziano, R.; Ratanalert, S.; Zhang, K.; Zhang, F.; Yan, H.; Chiu, W.; Bathe, M. Designer Nanoscale DNA Assemblies Programmed from the Top Down. Science 2016, 352, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Zhang, F.; Liu, Y.; Yan, H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117, 12584–12640. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Y.; Liu, H.; Ribbe, A.E.; Mao, C. Self-Assembly of Hexagonal DNA Two-Dimensional (2D) Arrays. J. Am. Chem. Soc. 2005, 127, 12202–12203. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Mohammed, A.; Gardell, J.; Masich, S.; Czeizler, E.; Orponen, P.; Högberg, B. DNA Rendering of Polyhedral Meshes at the Nanoscale. Nature 2015, 523, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Meyer, T.A.; Pan, V.; Dutta, P.K.; Ke, Y. The Beauty and Utility of DNA Origami. Chem 2017, 2, 359–382. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Castro, C.; Choi, J.H. Structural DNA Nanotechnology: Artificial Nanostructures for Biomedical Research. Annu. Rev. Biomed. Eng. 2018, 20, 375–401. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, M.; Shi, Z.; Castro, C.E.; Arya, G. Dynamic DNA Nanotechnology: Toward Functional Nanoscale Devices. Nanoscale Horiz. 2020, 5, 182–201. [Google Scholar] [CrossRef]
- Chu, T.-W.; Feng, J.; Yang, J.; Kopeček, J. Hybrid Polymeric Hydrogels via Peptide Nucleic Acid (PNA)/DNA Complexation. J. Control. Release 2015, 220, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Lu, C.-H.; Guo, W.; Aleman-Garcia, M.A.; Ren, J.; Willner, I. A Shape Memory Acrylamide/DNA Hydrogel Exhibiting Switchable Dual PH-Responsiveness. Adv. Funct. Mater. 2015, 25, 6867–6874. [Google Scholar] [CrossRef]
- Li, C.; Chen, P.; Shao, Y.; Zhou, X.; Wu, Y.; Yang, Z.; Li, Z.; Weil, T.; Liu, D. A Writable Polypeptide–DNA Hydrogel with Rationally Designed Multi-Modification Sites. Small 2015, 11, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Rowland, M.J.; Shao, Y.; Cao, T.; Chen, C.; Jia, H.; Zhou, X.; Yang, Z.; Scherman, O.A.; Liu, D. Responsive Double Network Hydrogels of Interpenetrating DNA and CB[8] Host–Guest Supramolecular Systems. Adv. Mater. 2015, 27, 3298–3304. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; LaBean, T.H.; Feng, L.; Reif, J.H. Directed Nucleation Assembly of DNA Tile Complexes for Barcode-Patterned Lattices. Proc. Natl. Acad. Sci. USA 2003, 100, 8103–8108. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ye, T.; Su, M.; Zhang, C.; Ribbe, A.E.; Jiang, W.; Mao, C. Hierarchical Self-Assembly of DNA into Symmetric Supramolecular Polyhedra. Nature 2008, 452, 198–201. [Google Scholar] [CrossRef]
- Ong, L.L.; Hanikel, N.; Yaghi, O.K.; Grun, C.; Strauss, M.T.; Bron, P.; Lai-Kee-Him, J.; Schueder, F.; Wang, B.; Wang, P.; et al. Programmable Self-Assembly of Three-Dimensional Nanostructures from 104 Unique Components. Nature 2017, 552, 72–77. [Google Scholar] [CrossRef]
- Jun, H.; Wang, X.; Bricker, W.P.; Bathe, M. Automated Sequence Design of 2D Wireframe DNA Origami with Honeycomb Edges. Nat. Commun. 2019, 10, 5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Marras, A.E.; Su, H.-J.; Castro, C.E. DNA Origami Compliant Nanostructures with Tunable Mechanical Properties. ACS Nano 2014, 8, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.E.; Su, H.-J.; Marras, A.E.; Zhou, L.; Johnson, J. Mechanical Design of DNA Nanostructures. Nanoscale 2015, 7, 5913–5921. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.-J.; Lee, C.; Lee, J.G.; Yagyu, H.; Tabata, O.; Kim, D.-N. Investigating the Sequence-Dependent Mechanical Properties of DNA Nicks for Applications in Twisted DNA Nanostructure Design. Nucleic Acids Res. 2019, 47, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.-H.; Chen, E.; Veneziano, R.; Gaitanaros, S.; Chen, Y. Stretching DNA Origami: Effect of Nicks and Holliday Junctions on the Axial Stiffness. Nucleic Acids Res. 2020, 48, 12407–12414. [Google Scholar] [CrossRef]
- Saran, R.; Wang, Y.; Li, I.T.S. Mechanical Flexibility of DNA: A Quintessential Tool for DNA Nanotechnology. Sensors 2020, 20, 7019. [Google Scholar] [CrossRef]
- Saccà, B.; Meyer, R.; Erkelenz, M.; Kiko, K.; Arndt, A.; Schroeder, H.; Rabe, K.S.; Niemeyer, C.M. Orthogonal Protein Decoration of DNA Origami. Angew. Chem. Int. Ed. Engl. 2010, 49, 9378–9383. [Google Scholar] [CrossRef]
- Wang, P.; Gaitanaros, S.; Lee, S.; Bathe, M.; Shih, W.M.; Ke, Y. Programming Self-Assembly of DNA Origami Honeycomb Two-Dimensional Lattices and Plasmonic Metamaterials. J. Am. Chem. Soc. 2016, 138, 7733–7740. [Google Scholar] [CrossRef]
- Aghebat Rafat, A.; Sagredo, S.; Thalhammer, M.; Simmel, F.C. Barcoded DNA Origami Structures for Multiplexed Optimization and Enrichment of DNA-Based Protein-Binding Cavities. Nat. Chem. 2020, 12, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, X.; Liu, P.; Wang, F.; Ariyama, H.; Ando, T.; Lin, J.; Wang, L.; Hu, J.; Li, B.; et al. Capturing Transient Antibody Conformations with DNA Origami Epitopes. Nat. Commun. 2020, 11, 3114. [Google Scholar] [CrossRef]
- Zhou, X.; Li, C.; Shao, Y.; Chen, C.; Yang, Z.; Liu, D. Reversibly Tuning the Mechanical Properties of a DNA Hydrogel by a DNA Nanomotor. Chem. Commun. 2016, 52, 10668–10671. [Google Scholar] [CrossRef] [PubMed]
- Bohidar, H.; Pandey, P.; Rawat, K.; Vk, A.; Kohlbrecher, J. Structural Hierarchy in DNA Hydrogels. J. Appl. Biotechnol. Bioeng. 2017, 2, 144–150. [Google Scholar]
- Liu, H.; Cao, T.; Xu, Y.; Dong, Y.; Liu, D. Tuning the Mechanical Properties of a DNA Hydrogel in Three Phases Based on ATP Aptamer. Int. J. Mol. Sci. 2018, 19, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gačanin, J.; Synatschke, C.V.; Weil, T. Biomedical Applications of DNA-Based Hydrogels. Adv. Funct. Mater. 2020, 30, 1906253. [Google Scholar] [CrossRef] [Green Version]
- Gačanin, J.; Kovtun, A.; Fischer, S.; Schwager, V.; Quambusch, J.; Kuan, S.L.; Liu, W.; Boldt, F.; Li, C.; Yang, Z.; et al. Spatiotemporally Controlled Release of Rho-Inhibiting C3 Toxin from a Protein–DNA Hybrid Hydrogel for Targeted Inhibition of Osteoclast Formation and Activity. Adv. Healthc. Mater. 2017, 6, 1700392. [Google Scholar] [CrossRef]
- Lyu, D.; Chen, S.; Guo, W. Liposome Crosslinked Polyacrylamide/DNA Hydrogel: A Smart Controlled-Release System for Small Molecular Payloads. Small 2018, 14, 1704039. [Google Scholar] [CrossRef]
- Liwinska, W.; Stanislawska, I.; Lyp, M.; Stojek, Z.; Zabost, E. Switchable Conformational Changes of DNA Nanogel Shells Containing Disulfide–DNA Hybrids for Controlled Drug Release and Efficient Anticancer Action. RSC Adv. 2019, 9, 13736–13748. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Liu, H.; Zhang, X.; Yan, J.; Zhu, Z.; Peng, L.; Yang, H.; Kim, Y.; Tan, W. Photoresponsive DNA-Cross-Linked Hydrogels for Controllable Release and Cancer Therapy. Langmuir 2011, 27, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Prasad Thelu, H.V.; Albert, S.K.; Golla, M.; Krishnan, N.; Ram, D.; Murty Srinivasula, S.; Varghese, R. Size Controllable DNA Nanogels from the Self-Assembly of DNA Nanostructures through Multivalent Host–Guest Interactions. Nanoscale 2018, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Komura, F.; Okuzumi, K.; Takahashi, Y.; Takakura, Y.; Nishikawa, M. Development of RNA/DNA Hydrogel Targeting Toll-Like Receptor 7/8 for Sustained RNA Release and Potent Immune Activation. Molecules 2020, 25, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Ohtsuki, S.; Araie, Y.; Umeki, Y.; Endo, M.; Emura, T.; Hidaka, K.; Sugiyama, H.; Takahashi, Y.; Takakura, Y.; et al. Self-Assembling DNA Hydrogel-Based Delivery of Immunoinhibitory Nucleic Acids to Immune Cells. Nanomedicine 2016, 12, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ishii-Mizuno, Y.; Umeki, Y.; Onuki, Y.; Watanabe, H.; Takahashi, Y.; Takakura, Y.; Nishikawa, M. Improved Sustained Release of Antigen from Immunostimulatory DNA Hydrogel by Electrostatic Interaction with Chitosan. Int. J. Pharm. 2017, 516, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ogawa, K.; Umeki, Y.; Mohri, K.; Kawasaki, Y.; Watanabe, H.; Takahashi, N.; Kusuki, E.; Takahashi, R.; Takahashi, Y.; et al. Injectable, Self-Gelling, Biodegradable, and Immunomodulatory DNA Hydrogel for Antigen Delivery. J. Control. Release 2014, 180, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonášová, E.P.; Stokke, B.T. Bioresponsive DNA-Co-Polymer Hydrogels for Fabrication of Sensors. Curr. Opin. Colloid Interface Sci. 2016, 26, 1–8. [Google Scholar] [CrossRef]
- Li, F.; Lyu, D.; Liu, S.; Guo, W. DNA Hydrogels and Microgels for Biosensing and Biomedical Applications. Adv. Mater. 2019, 32, 1806538. [Google Scholar] [CrossRef]
- Aldaye, F.A.; Senapedis, W.T.; Silver, P.A.; Way, J.C. A Structurally Tunable DNA-Based Extracellular Matrix. J. Am. Chem Soc. 2010, 132, 14727–14729. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Wang, S.; Wulf, V.; Willner, I. Stiffness-Switchable DNA-Based Constitutional Dynamic Network Hydrogels for Self-Healing and Matrix-Guided Controlled Chemical Processes. Nat. Commun. 2019, 10, 4774. [Google Scholar] [CrossRef] [Green Version]
- Merindol, R.; Delechiave, G.; Heinen, L.; Catalani, L.H.; Walther, A. Modular Design of Programmable Mechanofluorescent DNA Hydrogels. Nat. Commun. 2019, 10, 528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.; Xing, Y.; Chen, P.; Yang, Y.; Sun, Y.; Zhou, D.; Xu, L.; Fan, Q.; Liu, D. A PH-Triggered, Fast-Responding DNA Hydrogel. Angew. Chem. Int. Ed. 2009, 48, 7660–7663. [Google Scholar] [CrossRef]
- Um, S.H.; Lee, J.B.; Park, N.; Kwon, S.Y.; Umbach, C.C.; Luo, D. Enzyme-Catalysed Assembly of DNA Hydrogel. Nat. Mater. 2006, 5, 797–801. [Google Scholar] [CrossRef]
- Lee, J.B.; Peng, S.; Yang, D.; Roh, Y.H.; Funabashi, H.; Park, N.; Rice, E.J.; Chen, L.; Long, R.; Wu, M.; et al. A Mechanical Metamaterial Made from a DNA Hydrogel. Nat. Nanotechnol. 2012, 7, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yao, C.; Kou, X.; Tang, J.; Luo, D.; Yang, D. A Fluorescent Biofunctional DNA Hydrogel Prepared by Enzymatic Polymerization. Adv. Healthc. Mater. 2018, 7, 1700998. [Google Scholar] [CrossRef]
- Topuz, F.; Okay, O. Rheological Behavior of Responsive DNA Hydrogels. Macromolecules 2008, 41, 8847–8854. [Google Scholar] [CrossRef]
- Karacan, P.; Cakmak, H.; Okay, O. Swelling Behavior of Physical and Chemical DNA Hydrogels. J. Appl. Polym. Sci. 2013, 128, 3330–3337. [Google Scholar] [CrossRef]
- Basu, S.; Pacelli, S.; Paul, A. Self-Healing DNA-Based Injectable Hydrogels with Reversible Covalent Linkages for Controlled Drug Delivery. Acta Biomater. 2020, 105, 159–169. [Google Scholar] [CrossRef]
- Kosuri, S.; Church, G.M. Large-Scale de Novo DNA Synthesis: Technologies and Applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, F.H.T.; Goossens, E.P.M.; Tessari, M.; Heus, H.A. Enzymatic Preparation of Multimilligram Amounts of Pure Single-Stranded DNA Samples for Material and Analytical Sciences. Anal. Biochem. 2015, 475, 68–73. [Google Scholar] [CrossRef] [PubMed]
- van Emmerik, C.L.; Gachulincova, I.; Lobbia, V.R.; Daniëls, M.A.; Heus, H.A.; Soufi, A.; Nelissen, F.H.T.; van Ingen, H. Ramified Rolling Circle Amplification for Synthesis of Nucleosomal DNA Sequences. Anal. Biochem. 2020, 588, 113469. [Google Scholar] [CrossRef]
- Bush, J.; Singh, S.; Vargas, M.; Oktay, E.; Hu, C.-H.; Veneziano, R. Synthesis of DNA Origami Scaffolds: Current and Emerging Strategies. Molecules 2020, 25, 3386. [Google Scholar] [CrossRef]
- Lee, C.K.; Shin, S.R.; Lee, S.H.; Jeon, J.-H.; So, I.; Kang, T.M.; Kim, S.I.; Mun, J.Y.; Han, S.-S.; Spinks, G.M.; et al. DNA Hydrogel Fiber with Self-Entanglement Prepared by Using an Ionic Liquid. Angew. Chem. Int. Ed. 2008, 47, 2470–2474. [Google Scholar] [CrossRef]
- Bomboi, F.; Romano, F.; Leo, M.; Fernandez-Castanon, J.; Cerbino, R.; Bellini, T.; Bordi, F.; Filetici, P.; Sciortino, F. Re-Entrant DNA Gels. Nat. Commun. 2016, 7, 13191. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Caciagli, A.; Cao, T.; Stoev, I.; Zupkauskas, M.; O’Neill, T.; Wenzel, T.; Lamboll, R.; Liu, D.; Eiser, E. Microrheology of DNA Hydrogels. Proc. Natl. Acad. Sci. USA 2018, 115, 8137–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, N.; Kennedy, T.; Fygenson, D.K.; Saleh, O.A. Increasing Valence Pushes DNA Nanostar Networks to the Isostatic Point. Proc. Natl. Acad. Sci. USA 2019, 116, 7238–7243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Pan, V.; Vivek, S.; Weeks, E.R.; Ke, Y. Programmable DNA Hydrogels Assembled from Multidomain DNA Strands. ChemBioChem 2016, 17, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Wen, H.; Niu, L.; Su, C.; Liu, C.; Zhao, J.; Mao, C.; Liang, D. Effects of Chain Flexibility on the Properties of DNA Hydrogels. Soft Matter 2016, 12, 5537–5541. [Google Scholar] [CrossRef]
- Stoev, I.D.; Cao, T.; Caciagli, A.; Yu, J.; Ness, C.; Liu, R.; Ghosh, R.; O’Neill, T.; Liu, D.; Eiser, E. On the Role of Flexibility in Linker-Mediated DNA Hydrogels. Soft Matter 2020, 16, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Zhang, Y.; Usdin, K.; Lobachev, K.S. When Secondary Comes First–The Importance of Non-Canonical DNA Structures. Biochimie 2013, 95, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.; Kaushik, S.; Roy, K.; Singh, A.; Mahendru, S.; Kumar, M.; Chaudhary, S.; Ahmed, S.; Kukreti, S. A Bouquet of DNA Structures: Emerging Diversity. Biochem. Biophys. Rep. 2016, 5, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making Sense of G-Quadruplex and i-Motif Functions in Oncogene Promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [Green Version]
- Raiber, E.-A.; Kranaster, R.; Lam, E.; Nikan, M.; Balasubramanian, S. A Non-Canonical DNA Structure Is a Binding Motif for the Transcription Factor SP1 in Vitro. Nucleic Acids Res. 2012, 40, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Brazier, J.A.; Sugimoto, N. Topological Impact of Noncanonical DNA Structures on Klenow Fragment of DNA Polymerase. Proc. Natl. Acad. Sci. USA 2017, 114, 9605–9610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Assi, H.; Garavís, M.; González, C.; Damha, M.J. I-Motif DNA: Structural Features and Significance to Cell Biology. Nucleic Acids Res. 2018, 46, 8038–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikova, S.; Curtis, E.A. Structure and Function of Multimeric G-Quadruplexes. Molecules 2019, 24, 3074. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, M.; Xing, Y.; Li, Y.; Joedecke, C.-C.; Jin, J.; Yang, Z.; Liu, D. Study of PH-Induced Folding and Unfolding Kinetics of the DNA i-Motif by Stopped-Flow Circular Dichroism. Langmuir 2012, 28, 17743–17748. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.R.; Rusling, D.A. Triplex-Forming Oligonucleotides: A Third Strand for DNA Nanotechnology. Nucleic Acids Res. 2018, 46, 1021–1037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, Y.; Ito, Y.; Fujimoto, K. Interstrand Photocrosslinking of DNA via P-Carbamoylvinyl Phenol Nucleoside. Bioorg. Med. Chem. Lett. 2005, 15, 1299–1301. [Google Scholar] [CrossRef]
- Liu, Q.; Deiters, A. Optochemical Control of Deoxyoligonucleotide Function via a Nucleobase-Caging Approach. Acc. Chem. Res. 2014, 47, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, H.; Ito, T.; Yoshida, T.; Liang, X.; Komiyama, M. Photoregulation of the Formation and Dissociation of a DNA Duplex by Using the Cis–Trans Isomerization of Azobenzene. Angew. Chem. Int. Ed. 1999, 38, 2393–2395. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Fujimoto, K. Ultrafast Reversible Photo-Cross-Linking Reaction: Toward in Situ DNA Manipulation. Org. Lett. 2008, 10, 3227–3230. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yoshino, H.; Ami, T.; Yoshimura, Y.; Saito, I. A Light-Controlled Reversible DNA Photoligation via Carbazole-Tethered 5-Carboxyvinyluracil. Org. Lett. 2008, 10, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Yamada, A.; Yoshimura, Y.; Tsukaguchi, T.; Sakamoto, T. Details of the Ultrafast DNA Photo-Cross-Linking Reaction of 3-Cyanovinylcarbazole Nucleoside: Cis–Trans Isomeric Effect and the Application for SNP-Based Genotyping. J. Am. Chem. Soc. 2013, 135, 16161–16167. [Google Scholar] [CrossRef] [PubMed]
- Kandatsu, D.; Cervantes-Salguero, K.; Kawamata, I.; Hamada, S.; Nomura, S.M.; Fujimoto, K.; Murata, S. Reversible Gel–Sol Transition of a Photo-Responsive DNA Gel. ChemBioChem 2016, 17, 1118–1121. [Google Scholar] [CrossRef]
- Costa, D.; Valente, A.J.M.; Miguel, M.G.; Lindman, B. Light Triggered Release of Solutes from Covalent DNA Gels. Colloids Surf. A Physicochem. Eng. Asp. 2011, 391, 80–87. [Google Scholar] [CrossRef]
- Costa, D.; Valente, A.J.M.; Queiroz, J. Plasmid DNA Nanogels as Photoresponsive Materials for Multifunctional Bio-Applications. J. Biotechnol. 2015, 202, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, S.; Nishimura, T.; Ogura, Y.; Tanida, J. Photothermal Fabrication of Microscale Patterned DNA Hydrogels. R. Soc. Open Sci. 2018, 5, 171779. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Cheng, E.; Yang, Y.; Chen, P.; Zhang, T.; Sun, Y.; Yang, Z.; Liu, D. Self-Assembled DNA Hydrogels with Designable Thermal and Enzymatic Responsiveness. Adv. Mater. 2011, 23, 1117–1121. [Google Scholar] [CrossRef]
- Ko, O.; Han, S.; Lee, J.B. Selective Release of DNA Nanostructures from DNA Hydrogel. J. Ind. Eng. Chem. 2020, 84, 46–51. [Google Scholar] [CrossRef]
- English, M.A.; Soenksen, L.R.; Gayet, R.V.; de Puig, H.; Angenent-Mari, N.M.; Mao, A.S.; Nguyen, P.Q.; Collins, J.J. Programmable CRISPR-Responsive Smart Materials. Science 2019, 365, 780–785. [Google Scholar] [CrossRef]
- Nagahara, S.; Matsuda, T. Hydrogel Formation via Hybridization of Oligonucleotides Derivatized in Water-Soluble Vinyl Polymers. Polym. Gels Netw. 1996, 4, 111–127. [Google Scholar] [CrossRef]
- Li, C.; Zhou, X.; Shao, Y.; Chen, P.; Xing, Y.; Yang, Z.; Li, Z.; Liu, D. A Supramolecular Hydrogel with Identical Cross-Linking Point Density but Distinctive Rheological Properties. Mater. Chem. Front. 2017, 1, 654–659. [Google Scholar] [CrossRef]
- Fern, J.; Scalise, D.; Cangialosi, A.; Howie, D.; Potters, L.; Schulman, R. DNA Strand-Displacement Timer Circuits. ACS Synth. Biol. 2017, 6, 190–193. [Google Scholar] [CrossRef]
- Cangialosi, A.; Yoon, C.; Liu, J.; Huang, Q.; Guo, J.; Nguyen, T.D.; Gracias, D.H.; Schulman, R. DNA Sequence–Directed Shape Change of Photopatterned Hydrogels via High-Degree Swelling. Science 2017, 357, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Fern, J.; Schulman, R. Modular DNA Strand-Displacement Controllers for Directing Material Expansion. Nat. Commun. 2018, 9, 3766. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, C.; Boldt, F.; Wang, Y.; Kuan, S.L.; Tran, T.T.; Mikhalevich, V.; Förtsch, C.; Barth, H.; Yang, Z.; et al. Programmable Protein–DNA Hybrid Hydrogels for the Immobilization and Release of Functional Proteins. Chem. Commun. 2014, 50, 14620–14622. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Yurke, B.; Langrana, N.A. Mechanical Properties of a Reversible, DNA-Crosslinked Polyacrylamide Hydrogel. J. Biomech. Eng. 2004, 126, 104–110. [Google Scholar] [CrossRef]
- Lin, D.C.; Yurke, B.; Langrana, N.A.; Mills, A.P. A Polyacrylamide Gel With Reversible DNA Crosslinks. Am. Soc. Mech. Eng. Digit. Collect. 2008, 36509, 105–106. [Google Scholar]
- Gupta, A.; Mishra, A.; Puri, N. Peptide Nucleic Acids: Advanced Tools for Biomedical Applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.R.; Hammink, R.; Das, R.K.; Nelissen, F.H.T.; Blank, K.G.; Rowan, A.E.; Heus, H.A. DNA-Responsive Polyisocyanopeptide Hydrogels with Stress-Stiffening Capacity. Adv. Funct. Mater. 2016, 26, 9075–9082. [Google Scholar] [CrossRef]
- Cicuta, P.; Donald, A.M. Microrheology: A Review of the Method and Applications. Soft Matter 2007, 3, 1449–1455. [Google Scholar] [CrossRef]
- Schultz, K.M.; Furst, E.M. Microrheology of Biomaterial Hydrogelators. Soft Matter 2012, 8, 6198–6205. [Google Scholar] [CrossRef]
- Mansel, B.W.; Keen, S.; Patty, P.J.; Hemar, Y.; Williams, M.A.K. A Practical Review of Microrheological Techniques. Rheol. New Concepts Appl. Methods 2013. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wu, C. Rheological Study of Soft Matters: A Review of Microrheology and Microrheometers. Macromol. Chem. Phys. 2018, 219, 1700307. [Google Scholar] [CrossRef]
- Cai, P.C.; Krajina, B.A.; Kratochvil, M.J.; Zou, L.; Zhu, A.; Burgener, E.B.; Bollyky, P.L.; Milla, C.E.; Webber, M.J.; Spakowitz, A.J.; et al. Dynamic Light Scattering Microrheology for Soft and Living Materials. Soft Matter 2020. [Google Scholar] [CrossRef]
- Fernandez-Castanon, J.; Bianchi, S.; Saglimbeni, F.; Leonardo, R.D.; Sciortino, F. Microrheology of DNA Hydrogel Gelling and Melting on Cooling. Soft Matter 2018, 14, 6431–6438. [Google Scholar] [CrossRef] [Green Version]
- Nava, G.; Rossi, M.; Biffi, S.; Sciortino, F.; Bellini, T. Fluctuating Elasticity Mode in Transient Molecular Networks. Phys. Rev. Lett. 2017, 119, 078002. [Google Scholar] [CrossRef]
- Qian, L.; Zhao, H. Nanoindentation of Soft Biological Materials. Micromachines 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, R. Nanomechanical Mapping of Soft Materials with the Atomic Force Microscope: Methods, Theory and Applications. Chem. Soc. Rev. 2020, 49, 5850–5884. [Google Scholar] [CrossRef]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.K.; Louis, A.A. Sequence-Dependent Thermodynamics of a Coarse-Grained DNA Model. J. Chem Phys. 2012, 137, 135101. [Google Scholar] [CrossRef] [PubMed]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing Improved Structural Properties and Salt Dependence into a Coarse-Grained Model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SantaLucia, J.; Allawi, H.T.; Seneviratne, P.A. Improved Nearest-Neighbor Parameters for Predicting DNA Duplex Stability. Biochemistry 1996, 35, 3555–3562. [Google Scholar] [CrossRef]
- Rovigatti, L.; Bomboi, F.; Sciortino, F. Accurate Phase Diagram of Tetravalent DNA Nanostars. J. Chem. Phys. 2014, 140, 154903. [Google Scholar] [CrossRef] [Green Version]
- Rovigatti, L.; Smallenburg, F.; Romano, F.; Sciortino, F. Gels of DNA Nanostars Never Crystallize. ACS Nano 2014, 8, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Smallenburg, F.; Sciortino, F. Liquids More Stable than Crystals in Particles with Limited Valence and Flexible Bonds. Nat. Phys. 2013, 9, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Castanon, J.; Bomboi, F.; Rovigatti, L.; Zanatta, M.; Paciaroni, A.; Comez, L.; Porcar, L.; Jafta, C.J.; Fadda, G.C.; Bellini, T.; et al. Small-Angle Neutron Scattering and Molecular Dynamics Structural Study of Gelling DNA Nanostars. J. Chem. Phys. 2016, 145, 084910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Z.; Ness, C.; Frenkel, D.; Eiser, E. Structural and Linear Elastic Properties of DNA Hydrogels by Coarse-Grained Simulation. Macromolecules 2019, 52, 504–512. [Google Scholar] [CrossRef] [Green Version]
- Biffi, S.; Cerbino, R.; Bomboi, F.; Paraboschi, E.M.; Asselta, R.; Sciortino, F.; Bellini, T. Phase Behavior and Critical Activated Dynamics of Limited-Valence DNA Nanostars. Proc. Natl. Acad. Sci. USA 2013, 110, 15633–15637. [Google Scholar] [CrossRef] [Green Version]
- Biffi, S.; Cerbino, R.; Nava, G.; Bomboi, F.; Sciortino, F.; Bellini, T. Equilibrium Gels of Low-Valence DNA Nanostars: A Colloidal Model for Strong Glass Formers. Soft Matter 2015, 11, 3132–3138. [Google Scholar] [CrossRef]
- Fernandez-Castanon, J.; Bomboi, F.; Sciortino, F. Binding Branched and Linear DNA Structures: From Isolated Clusters to Fully Bonded Gels. J. Chem. Phys. 2018, 148, 025103. [Google Scholar] [CrossRef]
- Romano, F.; Sciortino, F. Switching Bonds in a DNA Gel: An All-DNA Vitrimer. Phys. Rev. Lett. 2015, 114, 078104. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, C.; DeSantis, P.; Scipioni, A. Nanoscale Mechanical and Dynamical Properties of DNA Single Molecules. Biophys. Chem. 2005, 113, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.-A.; Bauleth-Ramos, T.; Santos, H.A. DNA Hydrogel Assemblies: Bridging Synthesis Principles to Biomedical Applications. Adv. Ther. 2018, 1, 1800042. [Google Scholar] [CrossRef]
- Bloom, K.S. Beyond the Code: The Mechanical Properties of DNA as They Relate to Mitosis. Chromosoma 2008, 117, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, C.M.; Ramos, D.; Mendieta-Moreno, J.I.; Fierro, J.L.G.; Mendieta, J.; Tamayo, J.; Calleja, M. Effect of Water-DNA Interactions on Elastic Properties of DNA Self-Assembled Monolayers. Sci. Rep. 2017, 7, 536. [Google Scholar] [CrossRef] [Green Version]
- Maragakis, P.; Barnett, R.L.; Kaxiras, E.; Elstner, M.; Frauenheim, T. Electronic Structure of Overstretched DNA. Phys. Rev. B 2002, 66, 241104. [Google Scholar] [CrossRef]
- Kubař, T.; Elstner, M. What Governs the Charge Transfer in DNA? The Role of DNA Conformation and Environment. J. Phys. Chem. B 2008, 112, 8788–8798. [Google Scholar] [CrossRef]
- Kundu, S.; Karmakar, S.N. Conformation Dependent Electronic Transport in a DNA Double-Helix. AIP Adv. 2015, 5, 107122. [Google Scholar] [CrossRef] [Green Version]
- Salazar, S.V.; Mujica, V.; Medina, E. Spin-Orbit Coupling Modulation in DNA by Mechanical Deformations. CHIMIA Int. J. Chem. 2018, 72, 411–417. [Google Scholar] [CrossRef]
- Thomas, J.M.; Yu, H.-Z.; Sen, D. A Mechano-Electronic DNA Switch. J. Am. Chem. Soc. 2012, 134, 13738–13748. [Google Scholar] [CrossRef]
- Artés, J.M.; Li, Y.; Qi, J.; Anantram, M.P.; Hihath, J. Conformational Gating of DNA Conductance. Nat. Commun. 2015, 6, 8870. [Google Scholar] [CrossRef]
- Wu, Z.-S.; Chen, C.-R.; Shen, G.-L.; Yu, R.-Q. Reversible Electronic Nanoswitch Based on DNA G-Quadruplex Conformation: A Platform for Single-Step, Reagentless Potassium Detection. Biomaterials 2008, 29, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, C.; Zhu, C.; Salaita, K. DNA-Based Digital Tension Probes Reveal Integrin Forces during Early Cell Adhesion. Nat. Commun. 2014, 5, 5167. [Google Scholar] [CrossRef]
- Ma, R.; Kellner, A.V.; Ma, V.P.-Y.; Su, H.; Deal, B.R.; Brockman, J.M.; Salaita, K. DNA Probes That Store Mechanical Information Reveal Transient Piconewton Forces Applied by T Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 16949–16954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, N.; Xie, T.; Bagheri, Y.; Liang, C.; Keshri, P.; Sun, Y.; You, M. Quantifying Tensile Forces at Cell–Cell Junctions with a DNA-Based Fluorescent Probe. Chem. Sci. 2020, 11, 8558–8566. [Google Scholar] [CrossRef]
- Baig, M.M.F.A.; Lai, W.-F.; Akhtar, M.F.; Saleem, A.; Ahmed, S.A.; Xia, X.-H. DNA Nanotechnology as a Tool to Develop Molecular Tension Probes for Bio-Sensing and Bio-Imaging Applications: An up-to-Date Review. Nano-Struct. Nano-Objects 2020, 23, 100523. [Google Scholar] [CrossRef]
- Glazier, R.; Shinde, P.; Ogasawara, H.; Salaita, K. Spectroscopic Analysis of a Library of DNA Tension Probes for Mapping Cellular Forces at Fluid Interfaces. ACS Appl. Mater. Interfaces 2021, 13, 2145–2164. [Google Scholar] [CrossRef]
- Hinderer, S.; Layland, S.L.; Schenke-Layland, K. ECM and ECM-like Materials—Biomaterials for Applications in Regenerative Medicine and Cancer Therapy. Adv. Drug Deliv. Rev. 2016, 97, 260–269. [Google Scholar] [CrossRef]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D Extracellular Matrix Mimics: Fundamental Concepts and Role of Materials Chemistry to Influence Stem Cell Fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef]
- Nishikawa, M.; Mizuno, Y.; Mohri, K.; Matsuoka, N.; Rattanakiat, S.; Takahashi, Y.; Funabashi, H.; Luo, D.; Takakura, Y. Biodegradable CpG DNA Hydrogels for Sustained Delivery of Doxorubicin and Immunostimulatory Signals in Tumor-Bearing Mice. Biomaterials 2011, 32, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.; Kojima, T.; Song, Y.; Takayama, S. DNA-Based Biomaterials for Immunoengineering. Adv. Healthc. Mater. 2019, 8, 1801243. [Google Scholar] [CrossRef]
- Lattuada, E.; Leo, M.; Caprara, D.; Salvatori, L.; Stoppacciaro, A.; Sciortino, F.; Filetici, P. DNA-GEL, Novel Nanomaterial for Biomedical Applications and Delivery of Bioactive Molecules. Front. Pharm. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Patel, K.; Perez-Garrido, S.; Marshall, J.F.; Palma, M. DNA Origami Nanoarrays for Multivalent Investigations of Cancer Cell Spreading with Nanoscale Spatial Resolution and Single-Molecule Control. ACS Nano 2019, 13, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, W.; Huang, D.; Reynolds, P.; Hammond, L.; Ward, M.; Gadegaard, N.; Marshall, J.F.; Iskratsch, T.; Palma, M. Probing the Nanoscale Organisation and Multivalency of Cell Surface Receptors: DNA Origami Nanoarrays for Cellular Studies with Single-Molecule Control. Faraday Discuss. 2019, 219, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Chi, Q.; Yang, Z.; Xu, K.; Wang, C.; Liang, H. DNA Nanostructure as an Efficient Drug Delivery Platform for Immunotherapy. Front. Pharm. 2020, 10. [Google Scholar] [CrossRef]
- Finke, A.; Bußkamp, H.; Manea, M.; Marx, A. Designer Extracellular Matrix Based on DNA–Peptide Networks Generated by Polymerase Chain Reaction. Angew. Chem. Int. Ed. 2016, 55, 10136–10140. [Google Scholar] [CrossRef]
- Jiang, F.X.; Yurke, B.; Firestein, B.L.; Langrana, N.A. Neurite Outgrowth on a DNA Crosslinked Hydrogel with Tunable Stiffnesses. Ann. Biomed. Eng. 2008, 36, 1565. [Google Scholar] [CrossRef]
- Basu, S.; Alkiswani, A.-R.; Pacelli, S.; Paul, A. Nucleic Acid-Based Dual Cross-Linked Hydrogels for in Situ Tissue Repair via Directional Stem Cell Migration. ACS Appl. Mater. Interfaces 2019, 11, 34621–34633. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.S.; Hu, Y.; Willner, I. Stimuli-Responsive DNA-Based Hydrogels: From Basic Principles to Applications. Acc. Chem. Res. 2017, 50, 680–690. [Google Scholar] [CrossRef]
- Bila, H.; Kurisinkal, E.E.; Bastings, M.M.C. Engineering a Stable Future for DNA-Origami as a Biomaterial. Biomater. Sci. 2019, 7, 532–541. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structural DNA hydrogel design. (a) Hydrogel formation by entanglement of RCA-produced concatemeric DNA strands. (b) Hydrogel formed by crosslinking of two RCA-produced ssDNA. (c) Hydrogels formed by hybridization of multivalent DNA nanostars. (d) Hydrogels formed by hybridization of linear sections of dsDNA.
Figure 1.
Structural DNA hydrogel design. (a) Hydrogel formation by entanglement of RCA-produced concatemeric DNA strands. (b) Hydrogel formed by crosslinking of two RCA-produced ssDNA. (c) Hydrogels formed by hybridization of multivalent DNA nanostars. (d) Hydrogels formed by hybridization of linear sections of dsDNA.

Figure 2.
Responsive DNA linkers. (a) G-quadruplex motif, i-motif, and aptamers used to control mechanical properties of DNA hydrogels. (b) Photoresponsive assembly of DNA hydrogels. (c) Enzymatic digestion of DNA hydrogels. (d) Thermoresponsive DNA hydrogels. (e) Strand displacement method used to control DNA hydrogel assembly.
Figure 2.
Responsive DNA linkers. (a) G-quadruplex motif, i-motif, and aptamers used to control mechanical properties of DNA hydrogels. (b) Photoresponsive assembly of DNA hydrogels. (c) Enzymatic digestion of DNA hydrogels. (d) Thermoresponsive DNA hydrogels. (e) Strand displacement method used to control DNA hydrogel assembly.

Figure 3.
Hybrid DNA–polymer hydrogel formation. (i) Polymer chain conjugation with modified ssDNA linkers to form ssDNA conjugated polymer. (ii) ssDNA conjugated polymers crosslinked by complementary ssDNA linkers to form hybrid DNA–polymer hydrogel.
Figure 3.
Hybrid DNA–polymer hydrogel formation. (i) Polymer chain conjugation with modified ssDNA linkers to form ssDNA conjugated polymer. (ii) ssDNA conjugated polymers crosslinked by complementary ssDNA linkers to form hybrid DNA–polymer hydrogel.

Figure 4.
Common characterization methods for DNA hydrogel. (a) Bulk rheology and dynamic moduli. (b) Graphical depiction of microrheology showing constrained probe particle diffusion within the hydrogel network. The probe mean squared displacement (MSD) is used to determine the material behavior. (c) Network vibrations and interparticle hybridization rupture measured by dynamic light scattering (DLS) where a fast and slow decay time are observed and correspond to the timescale for each event, respectively. (d) Probe-based force controlled nanoindentation of DNA-based hydrogel to determine the material behaviors.
Figure 4.
Common characterization methods for DNA hydrogel. (a) Bulk rheology and dynamic moduli. (b) Graphical depiction of microrheology showing constrained probe particle diffusion within the hydrogel network. The probe mean squared displacement (MSD) is used to determine the material behavior. (c) Network vibrations and interparticle hybridization rupture measured by dynamic light scattering (DLS) where a fast and slow decay time are observed and correspond to the timescale for each event, respectively. (d) Probe-based force controlled nanoindentation of DNA-based hydrogel to determine the material behaviors.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bush, J.; Hu, C.-H.; Veneziano, R. Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials. Appl. Sci. 2021, 11, 1885. https://0-doi-org.brum.beds.ac.uk/10.3390/app11041885
AMA Style
Bush J, Hu C-H, Veneziano R. Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials. Applied Sciences. 2021; 11(4):1885. https://0-doi-org.brum.beds.ac.uk/10.3390/app11041885
Chicago/Turabian StyleBush, Joshua, Chih-Hsiang Hu, and Remi Veneziano. 2021. "Mechanical Properties of DNA Hydrogels: Towards Highly Programmable Biomaterials" Applied Sciences 11, no. 4: 1885. https://0-doi-org.brum.beds.ac.uk/10.3390/app11041885
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.