1. Introduction
Arsenic is relatively scarce in the natural environment (20th position in elemental abundances on the continental crust), but it is widely distributed. It can be found in detectable amounts in nearly all rocks and soils. As in soils relates to the geological substratum and consequently a rather wide range of As levels have been reported in soils around the world with an average of 5–10 mg/kg in uncontaminated soils [
1]. However, As may accumulate in soils due to human activities such as waste discharges of metal processing plants, burning of fossil fuels, mining of As containing ores and agricultural use of arsenical pesticides. In this respect it is important to know that As has different degrees of toxicity depending on the speciation of As, dose of exposure, individual and local tissue susceptibilities [
2]. The oxidation state of As is important since it affects movement, persistence and toxicity in soils and sediments. In oxygen-rich environments and well-drained soils, arsenate (AsO
43−) species dominate, whereas under reducing conditions, such as regularly flooded soils, arsenite (AsO
33−) is the stable oxidation state, but elemental As and arsine (AsH
3) can also be present in strongly reducing environments. As may also exist in organometallic forms such as monomethylarsonic acid (MMAA, CH
5AsO
3), dimethylarsinic acid (DMAA, C
2H
7AsO
2) and the more volatile methylarsines. Iron, aluminum and calcium arsenates are, like the corresponding phosphates, virtually insoluble in water and arsenate exhibits a phosphate-like specific adsorption (inner sphere complexation) on oxide and clay surfaces. Arsenites, however, are 10 times more soluble and mobile and are, furthermore, more toxic than arsenates [
3].
Trace elements occur in soils in certain ‘pools’ or ‘sinks’ of different solubility and mobility, of which in general six categories can be differentiated in the solid phase: (i) occupying exchangeable sites as diffuse ion or as outer-sphere complexes; (ii) specific adsorption as inner-sphere complexes; (iii) associated with insoluble organic matter; (iv) (co)precipitated as pure or mixed solids; and finally present in the structure of (v) secondary or (vi) primary minerals [
4]. Trace element speciation analysis in soils must almost always be performed by operational fractionation methods like chemical extractions since direct determination of specific chemical compounds is mostly impossible. Chemical extraction is, however, complicated by the fact that no chemical solution uniquely extracts trace elements out of one of these pools.
Sequential extractions, in which a soil sample is reacted with a series of carefully selected chemical solutions of increasing strengths, were developed to increase extraction selectivity of the distinct geochemical fractions [
5]. Today, sequential extractions are the most frequently applied analytical method for studying the solid phase speciation of environmentally relevant heavy metals [
6]. However, sequential extractions determine only an operational defined speciation (i.e., a fractionation), which depends on different factors like choice and order of extraction agents, the extraction duration, solid/liquid ratio, and sample preparation and conservation [
7,
8].
Sample preparation and conservation are very important, especially for samples taken from reducing environments, since (labile) phases can actually be transformed into other phases during sample preparation. Some significant differences in operational speciation are even encountered when the same scheme is applied by different analysts [
9]. However, lack of precision (especially at concentrations close to the detection limit) and selectivity are the main problems that occur among sequential extractions [
10,
11,
12]. Lack of selectivity is manifested by incomplete dissolution of the intended phase by the extraction agents for example when the chemical system becomes overburdened, in (partial) dissolution of undesired phases resulting in overrating the targeted phase, and/or in readsorption and redistribution phenomena.
Validation tests of sequential extractions have been a matter of debate for a long time and can be accomplished either by treating synthetic and/or pure mineralogical phases or by applying some kind of standard additions [
13,
14,
15,
16,
17].
It has been suggested that extraction efficiency could be improved by monitoring some parameters. For example, incomplete dissolution of acid-soluble minerals due to strong acid/base buffering could be checked by measuring the pH of the solution before and after extraction [
18]. Rauret et al. [
19] suggested, therefore, a multiple successive extraction procedure with monitoring the mandatory parameters (pH, Fe and Mn concentration, and Eh) in metal release to determine the number of extractions required. This should lead, according to these authors, to a more reliable distribution pattern especially when dealing with heavily contaminated samples. Keon et al. [
20] suggested using low sediment-to-extractant ratios (1/100) to ensure that each extractant did not become exhausted (e.g., to prevent that iron oxide dissolution is limited by oxalate concentration).
The coal-burning plant Federico II of Brindisi (Apulia, southern Italy) discussed in this paper is outfitted with a partially-enclosed conveyor belt for the transport of the solid fuel to the coal-fired boiler. Coal sometimes, depending on its geographical origin, can be characterized by a high arsenic content that could have been transferred to the areas close to the power plant both by scattering of coal grains and/or by particles formed by combustion. This contingence was taken into account with a previous public agency funded investigation aiming to evaluate the environmental impact of the power plant on the surrounding area. Reported As concentration values were generally within the accepted safety limits, except for a single sampling point, where this earlier investigation detected extremely high values of As concentration (>1500 mg kg
−1,
Table 1).
This work, therefore, describes arsenic sequential extraction analysis performed on the 30 soil samples, deriving from 5 cores extracted in the circumscribed area where the earlier investigation detected anomalous values of As concentration, in order to confirm or deny the environmental significance of the data previously collected.
Chemical analyses were performed in order to obtain the following information: (1) As availability in leaching test; (2) As sequential extraction by using different solvents; (3) As speciation analysis.
Different sequential extraction schemes have been designed for the analysis of metals in soils and sediments [
10,
21]. In this case, the speciation aims at understanding the distribution of a metal over the various sedimentary substrates such as carbonates, iron and manganese oxyhydroxides, organic matter, sulphides, silicates, etc. Under particular conditions some of these substrates will dissolve (for example the oxyhydroxides under reducing conditions and the carbonates under acidic conditions) or release adsorbed metals (for example when the electrolytic strength of the solution is increased). It is thus possible, by carefully selecting the composition of the extraction solutions, also called extractants, to destroy selectively specific soil or sediment substrates. Information obtained from sequential extraction procedures can be very interesting, although a prerequisite is that the analytical procedures are well-defined and accepted.
Moreover, in order to evaluate the As availability in soil samples we performed also a classic leaching test in acetic acid (the Istituto di ricerca sulle acque – Consiglio Nazionale delle Ricerche, IRSA-CNR, method): this assay analyzes the behaviour of a solid matrix (both organic and inorganic materials) towards the action of meteoric water. Finally a speciation analysis was performed by combining high-performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry (ICP-MS) techniques.
2. Materials and Methods
2.1. Sample Collection
Thirty soil samples, coming from the agricultural area of Brindisi (Apulia, southern Italy), were collected along the coal conveyor belt and around the coal-burning power plant Federico II in the month of July 2008. In particular, the soil samples belonged to 5 cores extracted in a limited area where a previous investigation had detected very high values of As concentration (>1500 mg kg
−1,
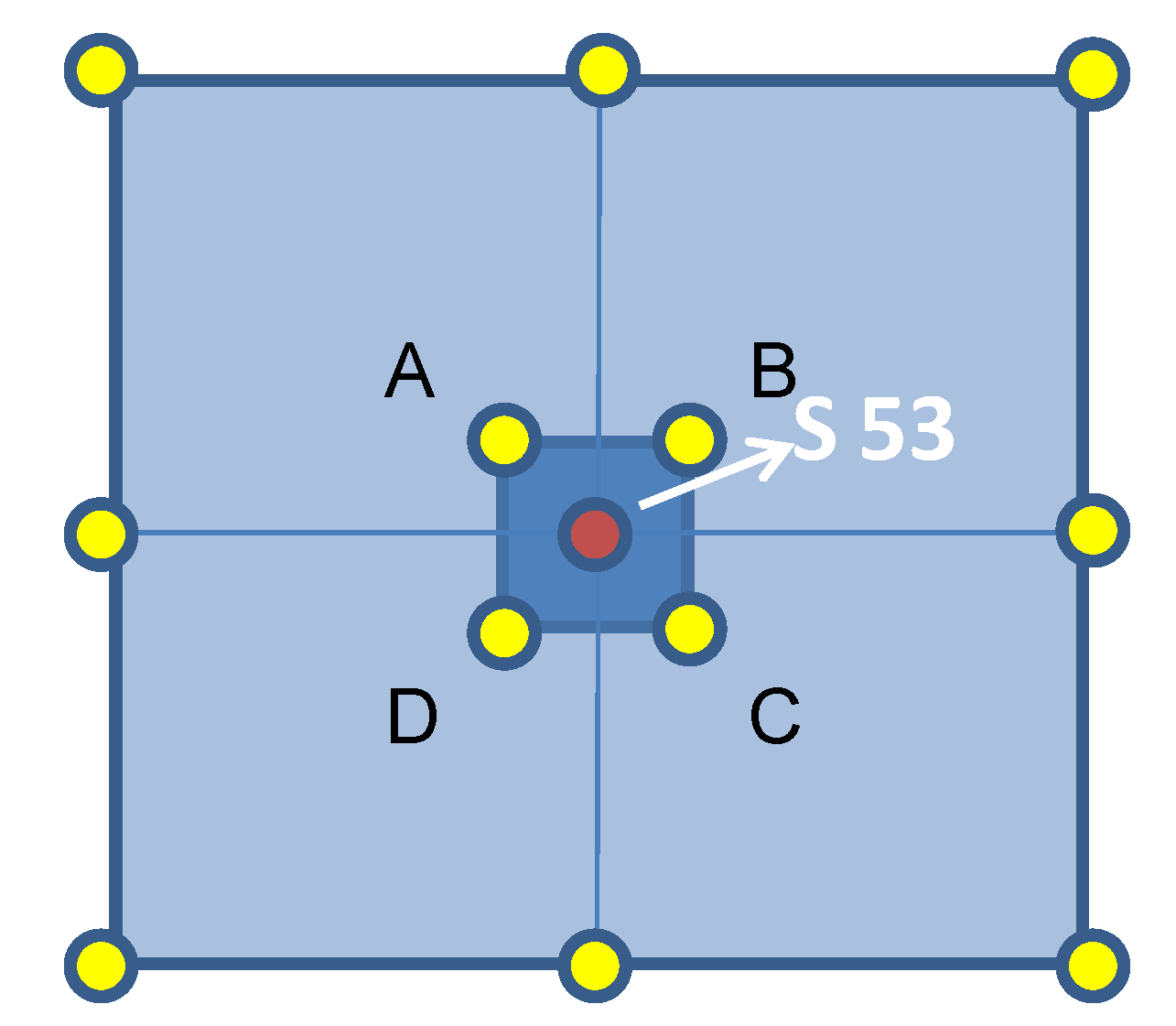
Table 1). Each core was 5 m deep and 6 different soil sample were derived from each one, corresponding to 6 different depth levels: 0.0–1.0 m, 1.0–1.5 m, 1.5–2.0 m, 2.0–3.0 m, 3.0–4.0 m, 4.0–5.0 m. The 5 cores were called: S53/1, corresponding with the sampling point of the past analysis performed by “Sviluppo Italia” in the year 2006, and 4 cores forming its close surroundings, S53/1A, S53/1B, S53/1C, and S53/1D (
Figure 1).
2.2. Sequential Extractions
A sequential extraction procedure, consisting in five steps, was performed [
22]. As the first step, 90 mL of ammonium acetate 1 M (adjusted to pH 5 with acetic acid) were added to 5 g of sediment, previously dried at room temperature. The samples were stirred for 24 h and then centrifuged to separate the extracted solution from the solid residue. Ammonium acetate allowed to extract the water-soluble fraction of metals and metalloids, bound as carbonates and highly exchangeable forms [
23].
After at least two washings with distilled water, at the second step each sample was treated with 90 mL of hydroxylammine chloride 1 M (adjusted to pH 5 with sodium citrate) by stirring for 24 h. In these conditions, an improvement of As solubility, due to the increase of Fe and Mn oxides and hydroxides solubilization, was verified. In fact, there is a significant correlation between Fe solubility and As mobility thus confirming that Fe oxides and hydroxides are the main As adsorption agents [
23,
24]. It has been also reported that As mobility in soils mainly depends on complexation reactions with oxides and hydroxides of Al, Mn, and Fe [
25]. Moreover, it is well known that Fe oxides and hydroxides are dominant in the adsorption process of As in soils: reducing and chelating agents cause the co-dissolution of As and oxides and hydroxides, making this metalloid more soluble and available [
26]. The separation of the extracted solution and the washing of the solid residue were performed as already described in the first step.
At the third step of the sequential extraction procedure 25 mL of HCl 0.1 M were added to the solid residue of the previous step, stirring for 24 h. The forth extraction was performed by adding 25 mL of NaOH to each residue of the third step and stirring for 48 h. The third and the forth step allowed to extract metal fraction bound to the organic matter and the clay minerals. In fact, HCl breaks the bonds between clays and humic substances and NaOH extracts arsenates strongly adsorbed on the clay surface [
26,
27,
28].
The solid residue of the fourth step was washed at least twice and then treated with 25 mL of HNO3 8 M, stirring for 3 h at 85 °C. The extracted solution was separated by centrifugation and solid residue was washed at least twice with distilled water. The fifth step extracted the not easily available As fraction (for example, sulphide), but it was soluble in acid conditions and high temperature.
After the fifth step, the solid residue is stored for the following digestion and spectrometric analysis. So we obtained six different shares for each soil sample: five shares from the five steps of the sequential extraction procedure and one share from the final solid residue.
The As extracted at the fifth step of the sequential extractions and still present in the final solid residue is the so called “occluded As” [
26], i.e., As incorporated in minerals that do not easily release the metalloid, for example some very insoluble sulphides such as arsenopyrite [
27].
The solutions extracted from soil samples during the sequential extraction procedure and the final solid residue were analyzed by ICP-MS spectrometry (Environmental Protection Agency, EPA, method 6020). In particular, the final solid residues of the five step sequential extraction were digested by aqua regia (one volume of HNO3 and three volumes of HCl) mineralization.
The final results were expressed in µg/kg (ppb) of dry weight and were referred to 1 kg of soil, dried at room temperature in order to avoid As, which forms very volatile species, being lost and the measurements being altered.
2.3. Leaching Test
A classic leaching test in acetic acid (IRSA-CNR method, APAT CNR IRSA 3020 Man 29 2003) was performed in order to evaluate the As availability in soil samples: this assay analyzes the behaviour of a solid matrix (both organic and inorganic materials) towards the action of meteoric water. We treated 2.5 g of each soil sample (particles with a size <2 mm dried at room temperature) with 50 mL of water (extraction ratio 1:20) for 24 h (adjusted to pH 5 with diluted acetic acid). The resulting solution, filtered through 0.45 µm filters, were used for As determination by ICP-MS spectrometry.
2.4. Speciation Analysis
A speciation analysis was performed in order to determine As species in the soil samples: As(III), As(V), and organic fractions (MMAA, DMAA). 0.200 g of each sample, dried at room temperature and sifted to eliminate particles with dimensions >2 mm, were suspended in 50 mL of ammonium oxalate 0.2 M and then left in an ultrasonic bath for 10 min. The extracted As was detected by the combination of HPLC (Waters 626 LC System equipped with a ionic exchange Hamilton PRP-X100 column, 250 × 4.6 mm inner diameter), and ICP-MS (Varian ICP-MS 820-MS) techniques.
2.5. Instrumental Details
All determinations of As were performed on a Varian 820 MS inductively coupled plasma mass spectrometer (ICP-MS).
4. Discussion
The impact of environmental pollution due to metals and metalloids in soil depends not only on their total concentration but also on their mobility and availability, which affects their release and interaction with other environmental components, such as water, air, and vegetables. In order to evaluate metal mobility in soil, sequential extraction procedures can be performed, in which a soil sample is treated with solutions with different extractant capacity that provoke the release of metal rates characterized by different solubility and, consequently, mobility. The fractions extracted by weak reagents are more mobile and dangerous from an environmental point of view, because they represent the metal portion which is released more easily from soil to other environmental compartments, such as superficial and ground water, or adsorbed by vegetables which grow in the contaminated site. In soil As is adsorbed mainly by minerals characterized by variable charge: in fact, it was observed by EXAFS methodologies, that AsO
43− ions were adsorbed on the surface of minerals with variable charge forming “inner-sphere complexes”, characterized by very strong covalent bonds [
29]. These bonds can be mono- or bidentate mononuclear and/or bidentate binuclear in different proportions according to the characteristics of the absorbent surfaces. Recent studies showed that AsO
43− ions are adsorbed more efficiently by Mn and Fe oxides than Al oxides, whereas AsO
33− ions are strongly adsorbed by Fe oxides and weakly adsorbed by Al oxides [
30,
31,
32,
33,
34].
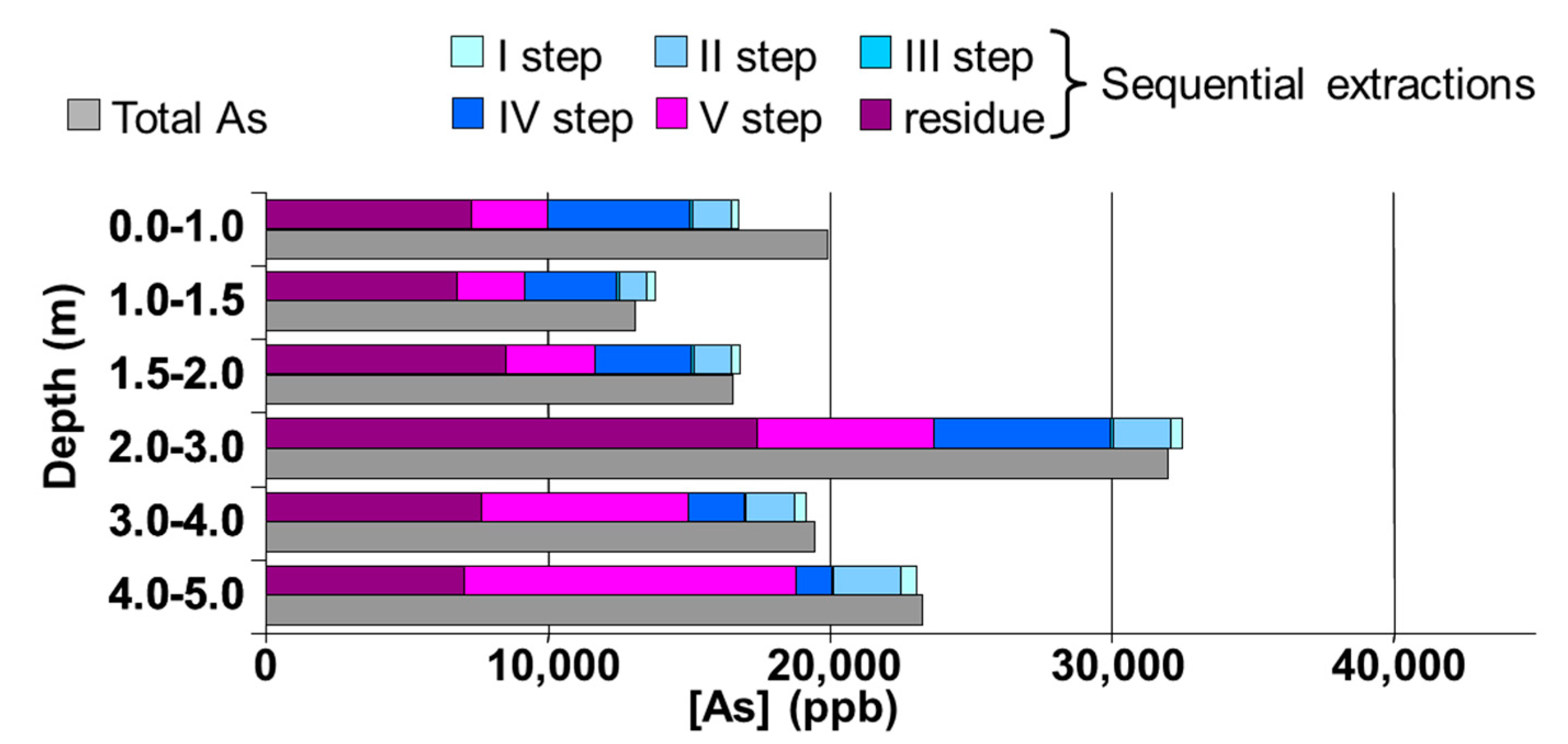
Sixteen of the 30 soil samples taken at the studied area were characterized by an As concentration higher than 20 ppm, the limit set by the 471/99 D.M. in the areas for public parks and private and residential use, and the highest value was 44.51 ppm (S53/1 core, depth 2.0–3.0 m). No soil sample showed an As amount higher than 50 ppm, the limit set by the 471/99 D.M. in the commercial and industrial areas.
As extracted in the first step of the sequential extraction procedures was the less abundant in all the analyzed soil sample. Even if sequential extraction methods can change significantly, the first step is always performed in less drastic conditions and allows to solubilize the most available fraction of analyte and, therefore, the rate of metal or metalloid with the highest potential toxicity. Also As extracted in the second step is significantly bio-available, even if in these specific extraction conditions As species are not so water-soluble like As forms extracted in the first step.
In general, As extracted at the fourth step represented an abundant fraction of total As in all analyzed soil samples. The organic matter has an important role in determining As mobility: the adsorption of As(III) and As(V) on humic acids is influenced by pH and the concentration of the element. Humic acids could be important for As adsorption in relatively acid environments, whereas in alkaline environments As release and solubilization are favored [
35]. Sodium hydroxide, used at the fourth step of the sequential extractions, is able to solubilize the organic matter, as pointed out by the darkening of the extracts from the superficial soil samples, where we expect a higher concentration of humic substances. But the As fraction extracted at the fourth step also includes arsenates bound to Fe-rich minerals and, in general, to clay matrices [
27].
As species extracted at the fifth step and in the final residue (dissolved in aqua regia) form the so called As occluded [
26], As included in minerals such as sulphides. However, we also have to consider that in the superficial levels sulphide oxidation is possible due to the metabolism of the bacterial species present in the soil, and consequently the transformation of As in more available forms [
36]. This hypothesis is not applicable to the deepest levels due to the anaerobic conditions which characterize these depths. The collected data for the soil samples from the S53 sampling point and its close surroundings suggest that As is present mainly in an occluded form.
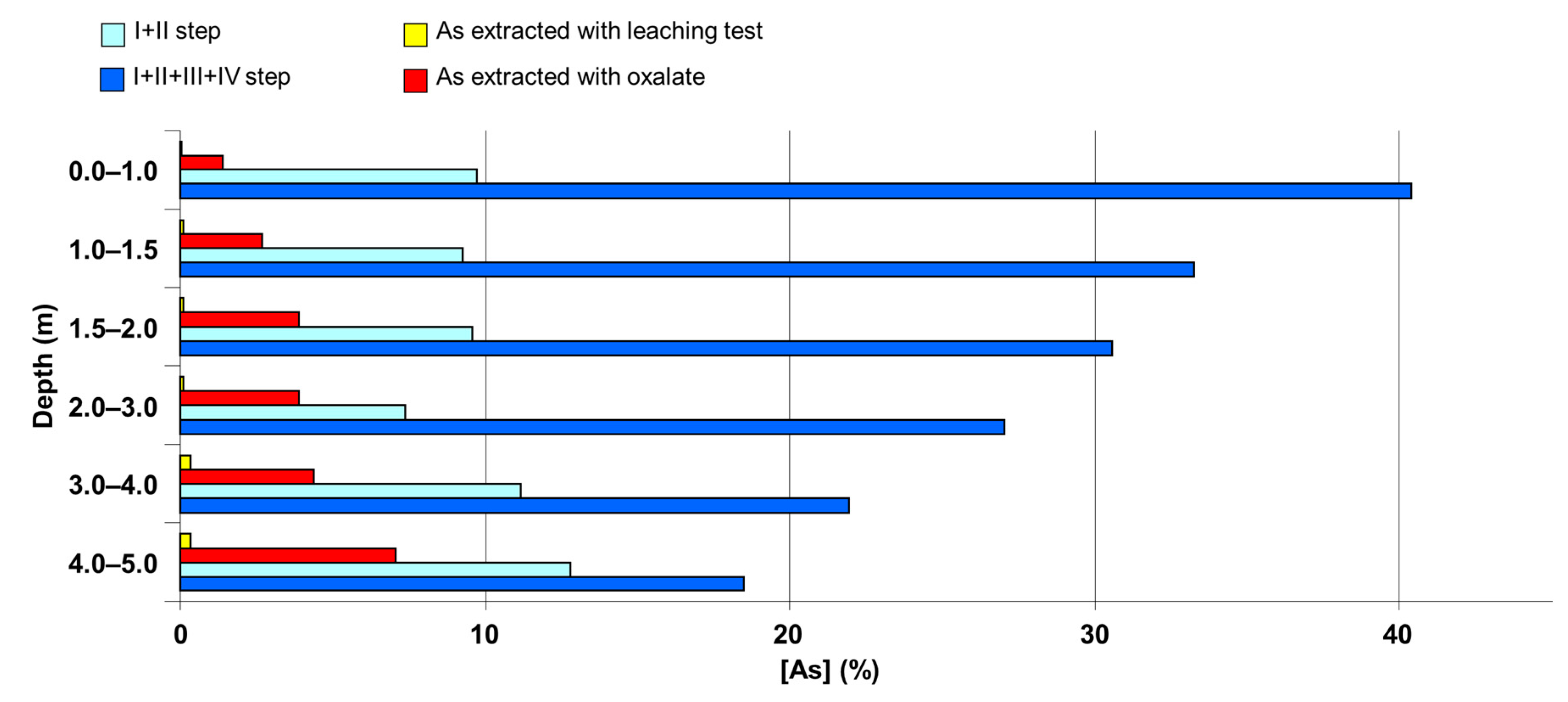
Figure 4 describes the comparison between the As amount extracted by sequential extractions and the As quantity extracted by ammonium oxalate 0.2 M in the chemical speciation procedure. In general, As extracted with ammonium oxalate was always less than the sum of the As fractions extracted in the first and second step of sequential extractions. Moreover, in the superficial levels As extracted by oxalate was very similar to that extracted in the first step of sequential extractions, whereas when increasing the depth of the soil sample As extracted by oxalate became more abundant.
As extraction by ammonium oxalate for the chemical speciation analysis could be considered like a leaching test which allows the release of the highest possible amount of As, without changing his chemical properties. Analyzing As leaching in ammonium oxalate 0.2 M, we noticed that the highest levels of As release was obtained from the deepest soil sample, with a maximum generally between 2 and 3 m of depth (core S53/1, S53/1A e S53/1B). The As amount extracted from soil samples of S53 sampling point and its surroundings was on average 4% of total As. This value was very different from As extracted by ammonium oxalate from other areas located near the site analyzed in this study. For example, in the Sviluppo Industriale e dei Servizi Reali alle Imprese (SISRI) area of Brindisi, As extracted by ammonium oxalate 0.2 M was 62% of total As. Comparing As amounts extracted by acetic acid with As extracted by ammonium oxalate, we observed much higher values of As extracted for the chemical speciation analysis. In fact, As extracted by acetic acid ranges from 6 to 126 ppb, whereas As extracted by ammonium oxalate ranges from 152 to 1962 ppb.
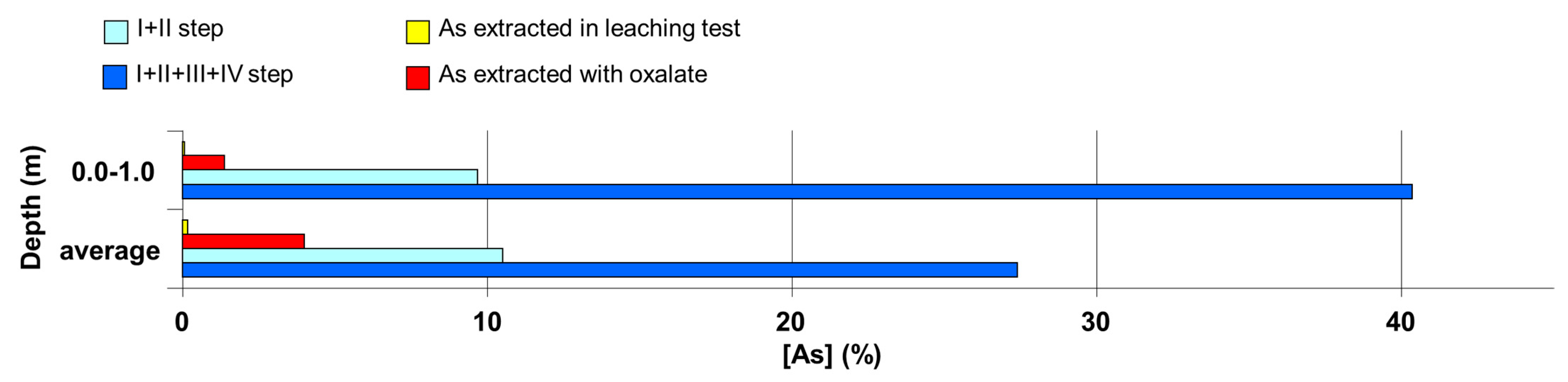
In this regard,
Figure 5 and
Figure 6 show the comparison between the average percentage of As extracted for the leaching test by ammonium oxalate and in the first two steps and the first four steps of sequential extraction procedures for soil samples in the most superficial layers on the one hand, and the corresponding average values of all levels of depth on the other hand. For total extracted As values in the first four steps of the sequential extraction only, the averages found in the upper layer were higher than the averages measured in all the other levels of depth. This is probably due to the increased presence of organic matter in the upper layer, the matrix on which some of the As species extracted by NaOH in the fourth step of the sequential extraction are adsorbed. Further studies concerning the determination of As release due to the removal of the organic substance performed in the third and the fourth step of the sequential extraction and due to the possible use of fertilizers, especially phosphates with an anionic component with high exchange capacity for As oxyanions, are needed.
5. Conclusions
Total As determination in the 30 soil samples deriving from the 5 probes of the S53 sampling point and its surroundings showed that As concentration was lower than the As amount detected by “Sviluppo Italia”. In general, As accumulates in the deepest depth levels. In 16 of the 30 soil samples As concentration exceeds 20 ppm limit, established by D.M. 471/99 for green areas for public use and private and residential use, and in no case did As concentration exceed the limit of 50 ppm, specified by the same decree for commercial and industrial areas.
The As sequential extraction protocol showed that the most abundant As amount was contained in the final residue of the sequential extraction procedure. As extracted in the fifth step and As in the final residue is the so-called occluded As that is embedded in rock formations that do not release the metalloid, including some very insoluble sulphides such as arsenopyrite.
The graphs showed in this work summarize the different scenarios proposed by us for an evaluation at different levels of approximation of environmental risk related to the presence of As, if the total concentration is not taken into account. This is because the environmental impact of pollution by metals and metalloids in soils depends not only on their total concentration but also on their mobility and availability.
The first scenario is derived from considering for risk evaluation only the As extracted by acetic acid in the leaching test. This test allows us to evaluate the amount of extractable As from the leaching effect of rain water. As also described above, the amount of As extracted by acetic acid was very limited, an order of magnitude lower than As extracted by ammonium acetate in the first step of sequential extractions and by ammonium oxalate in the speciation analysis. It was on average 0.16%, both of total As obtained in a unique digestion and total As extracted by the sequential extraction protocol.
A more cautious approach may lead to consideration for risk evaluation the amount of As obtained in the first two steps of sequential extractions. This amounts to an average of 9.8% of total As determined in a single mineralization and to 10.5% of As extracted in the sequential extraction procedures. The second proposed scenario also takes into account the As extracted by ammonium oxalate, used for the analysis of chemical speciation and which consists in the percentage of As extracted in mild conditions that do not change the percentages of As(III), As(V) and organic species possibly present. As extracted by 0.2 M ammonium oxalate is on average 4% of total As extracted in the sequential extractions and 3.9% of total As determined in a single digestion. These values are still below the sum of the percentages of As extracted in the first two steps of sequential extractions.
Another scenario is derived from considering the As extracted in the first four steps of sequential extractions. We should consider that the third and fourth steps, although carried out using “strong” extractants (0.1 M HCl and 0.5 M NaOH) hard to find in natural conditions, mobilize the arsenates bound to the surface of minerals rich in Fe and, in general, to clay and, if present, As bound to organic matrices, which in the more superficial layers of soil can be a substantial fraction.
Our results showed that only for total extracted As values in the first four steps of the sequential extraction are the averages found in the upper layer higher than the averages measured in all the other levels of depth, and we attributed this effect to a greater amount of organic matter in the upper layer, a matrix certainly capable of absorbing some of the As species extracted by NaOH in the fourth step of the sequential extraction.
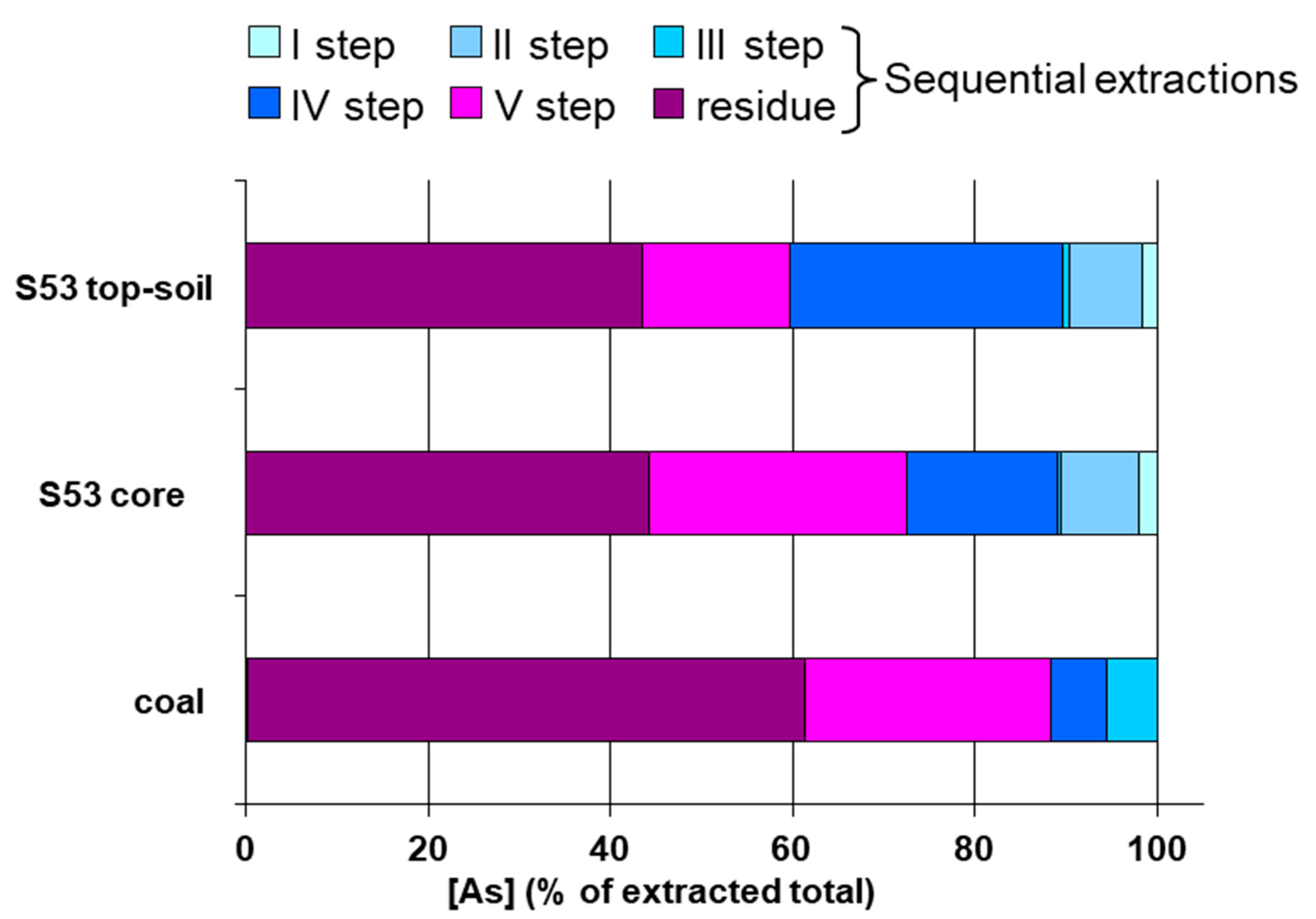
As extracted in the studied area was characterized by different chemical species as compared with As from other areas of the Sito di Interesse Nazionale National Interest Site, SIN) of Brindisi. In fact, in the soil sample collected in the agricultural area of Brindisi, along the coal conveyor belt and around the coal-burning power plant Federico II, As fraction extracted by ammonium oxalate 0.2 M was on average 4.0% and ranged from 0.8% to 8.6%, whereas in the SISRI area As extracted by ammonium oxalate was 62% (35.9–91.7%). However, the percentage of As extracted by ammonium oxalate was always higher than the As amount extracted by ammonium acetate at the first step of sequential extractions.
In the soil samples from S53 sampling point and its close surroundings speciation chemical analysis showed that As(V) was by far the most abundant species, whereas As(III) was only a very little fraction. Organic species were completely absent. The result of studies performed in a very close area (such as the SISRI area) have already shown that As(V) was more than 90% of extracted As, whereas As(III), which represents the most soluble and available species, was less than 10%.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}