Laser-Control of Ultrafast π-Electron Ring Currents in Aromatic Molecules: Roles of Molecular Symmetry and Light Polarization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Laser Control of Ring Currents in a Molecule Having a Single Aromatic Ring
2.1. Basic Idea of Laser-Driven Ring Currents: Molecular Symmetry and Angular Momentum Eigenstates
2.2. Laser-Polarization Effects on Ring Currents within the Frozen-Nuclei Approximation
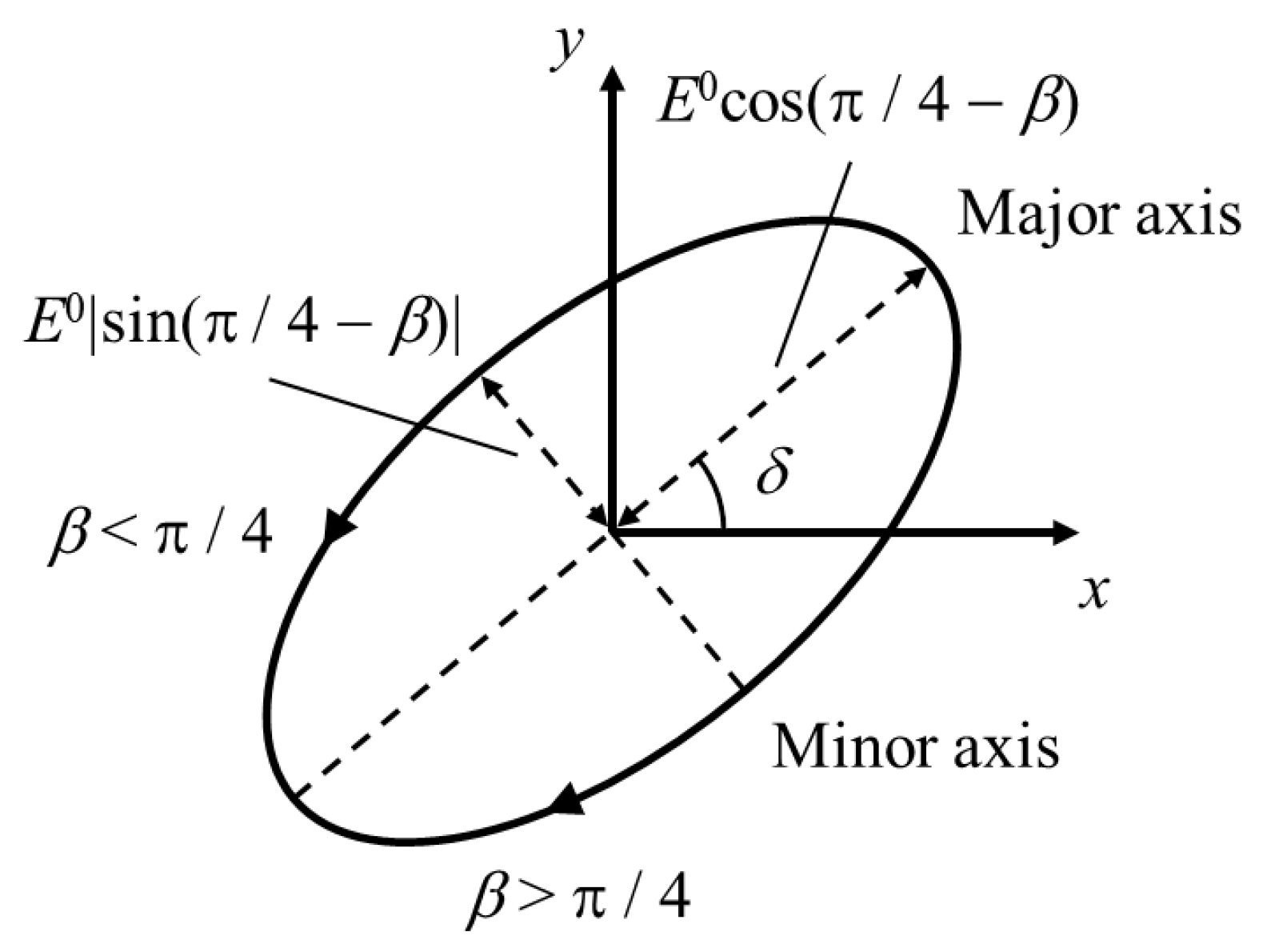
2.2.1. General Form of Polarized Laser Pulses
2.2.2. Three-Level Model Analysis of Optical Excitation
2.2.3. Case of Degenerate Excited States
2.2.4. Case of Quasi-Degenerate Excited States
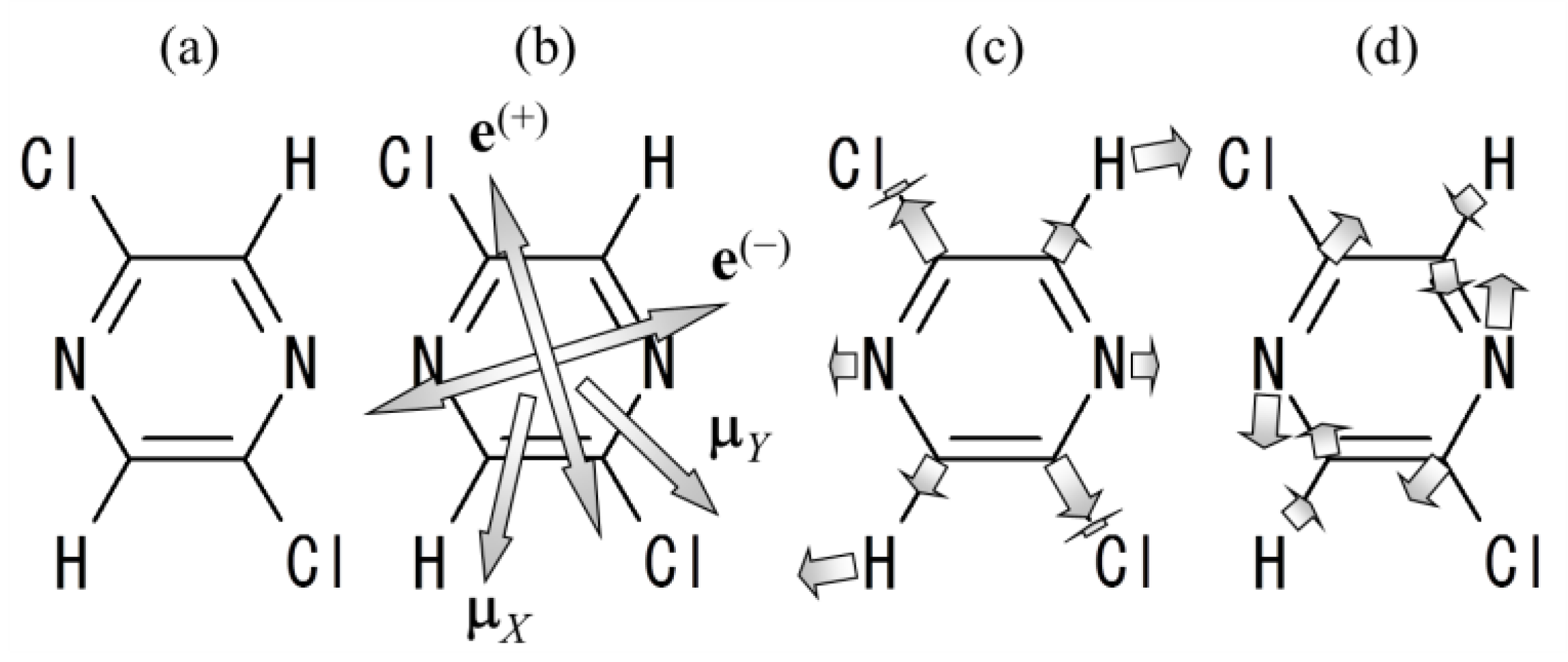
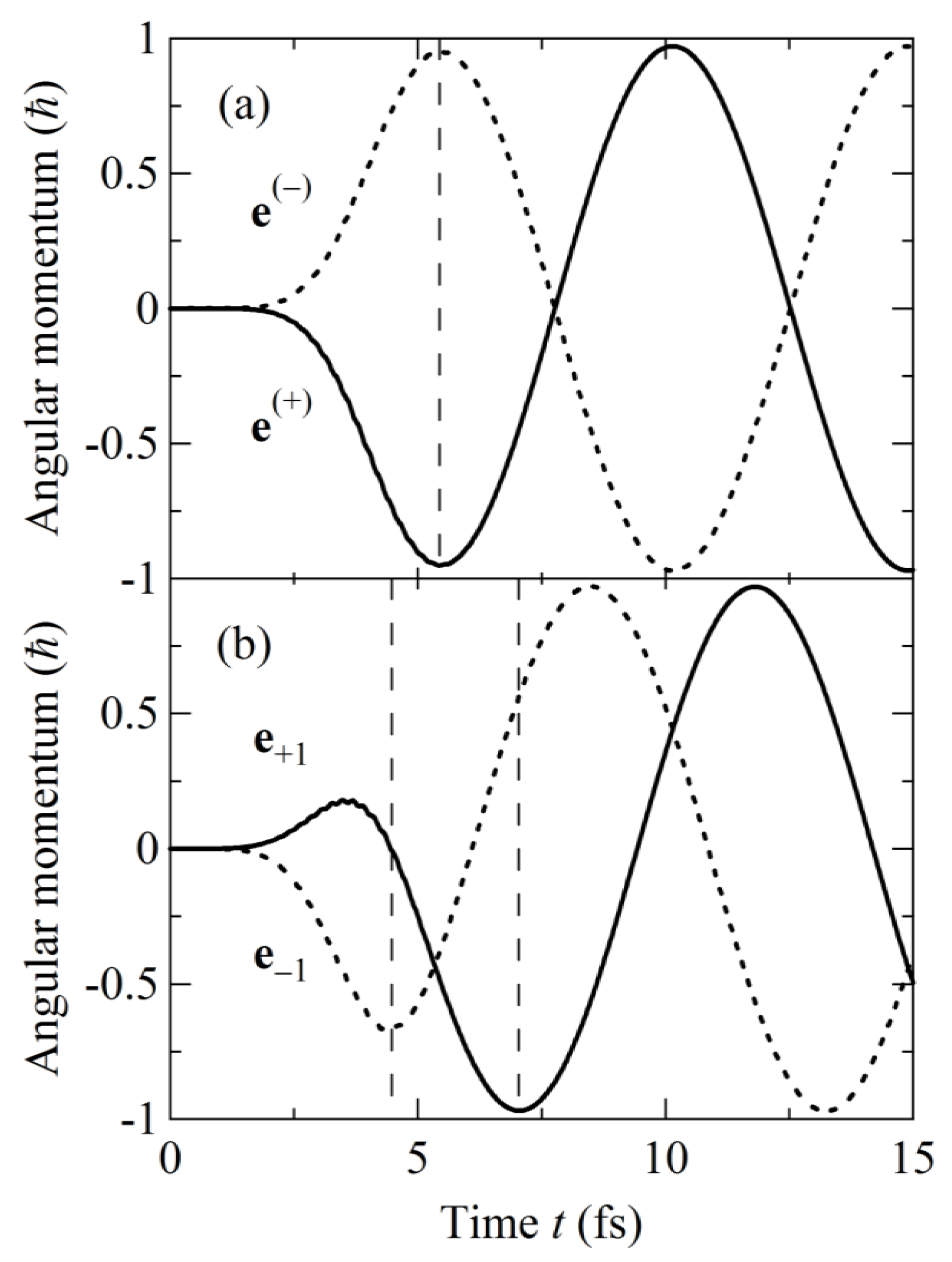
2.2.5. Numerical Demonstration for 2,5-Dichloropyrazine
2.3. Beyond the Frozen-Nuclei Approximation: Nonadiabatically Coupled Vibronic Dynamics
2.3.1. Selection of Effective Vibrational Modes and Method of Nuclear Wave Packet Propagation
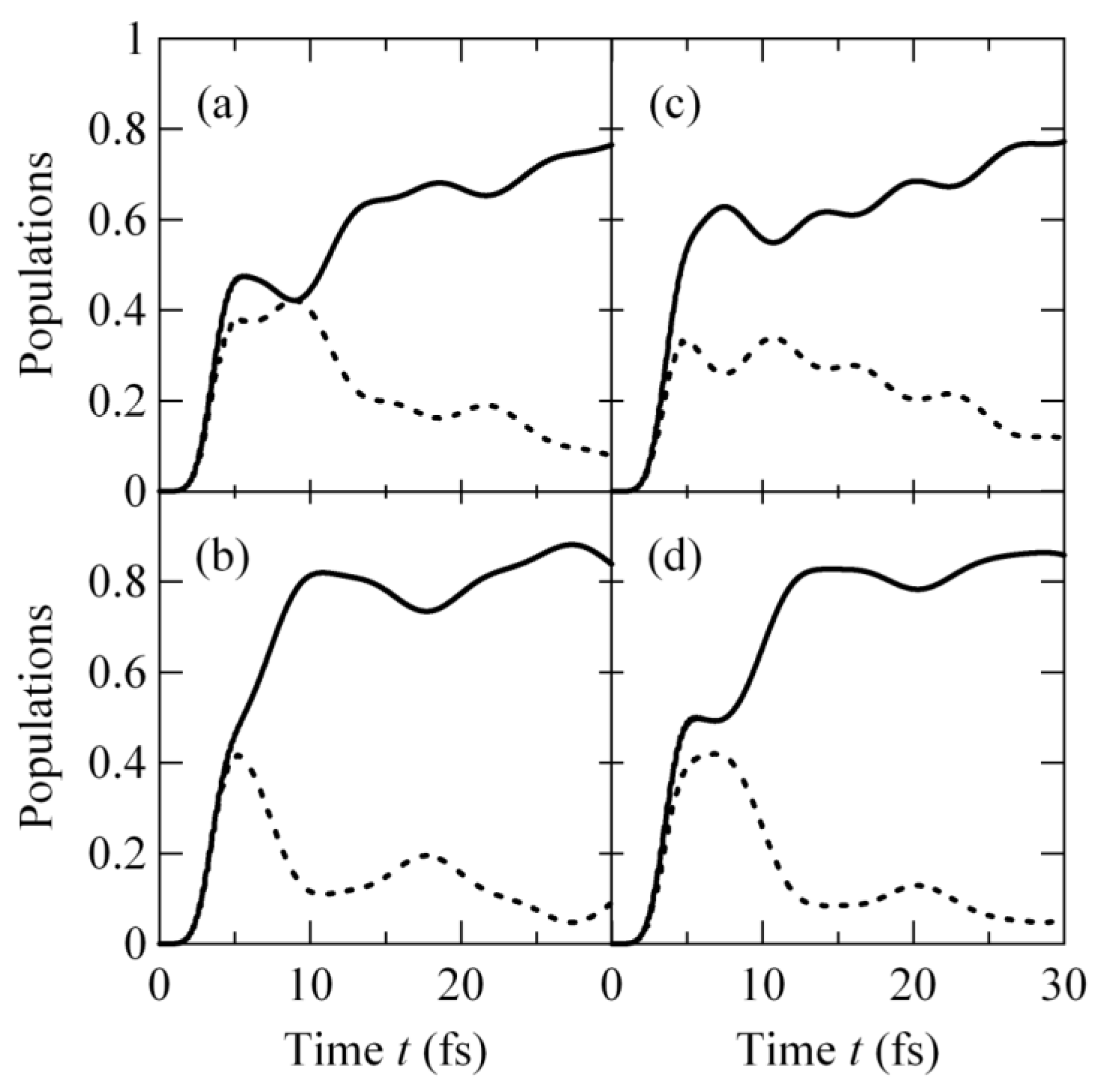
2.3.2. Population Transfer Dynamics
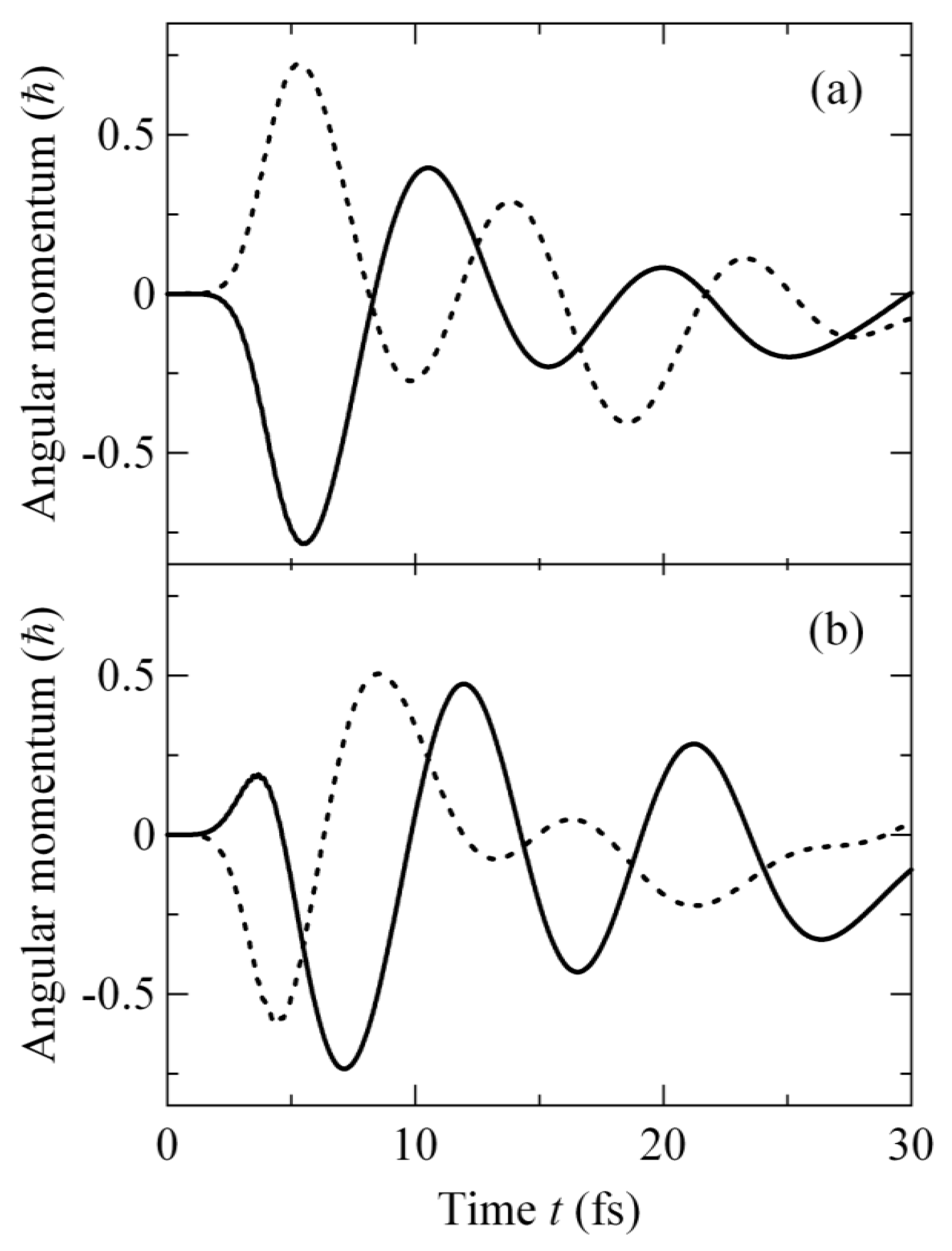
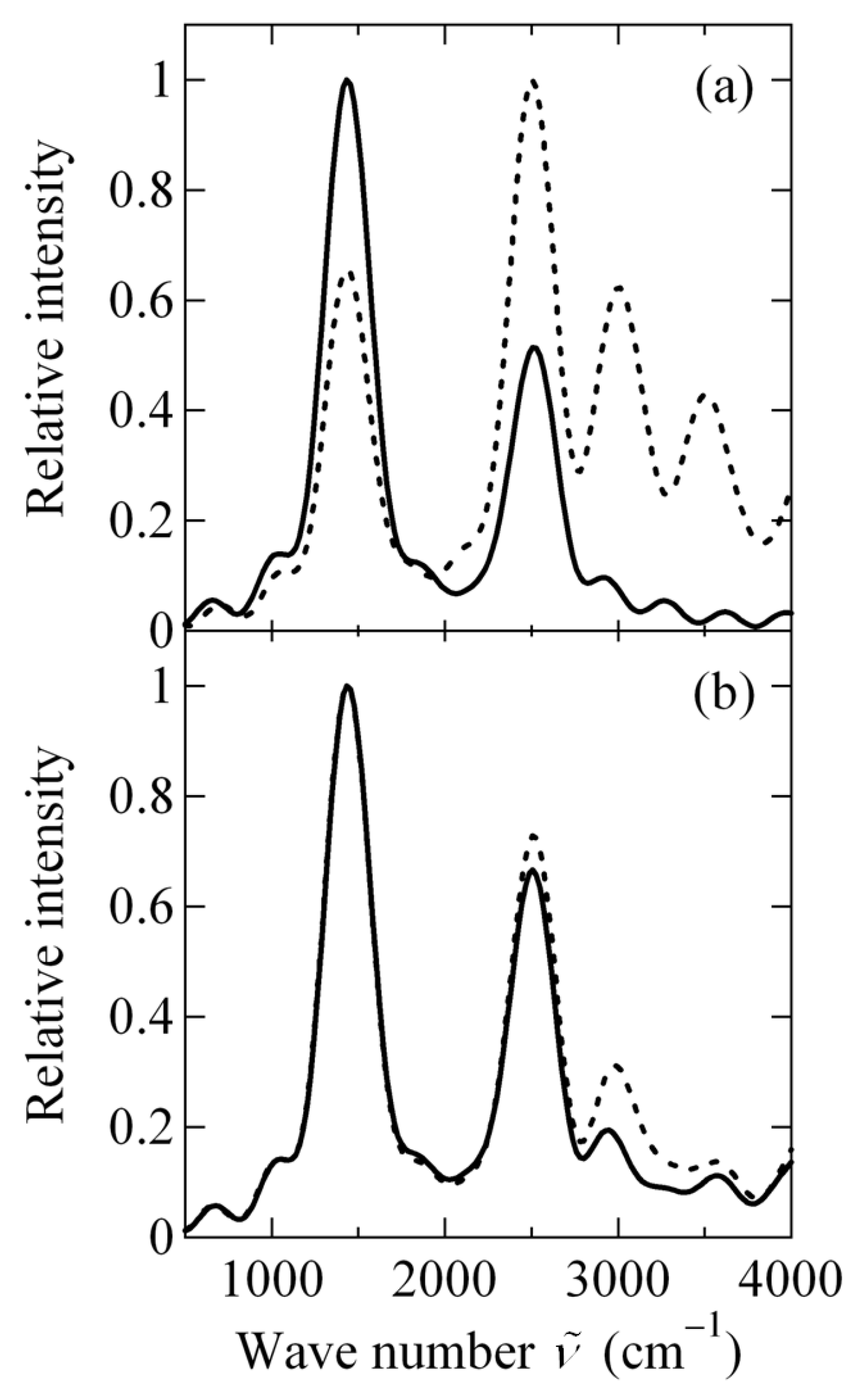
2.3.3. Angular Momentum Expectation Value and Vibrational Spectrum
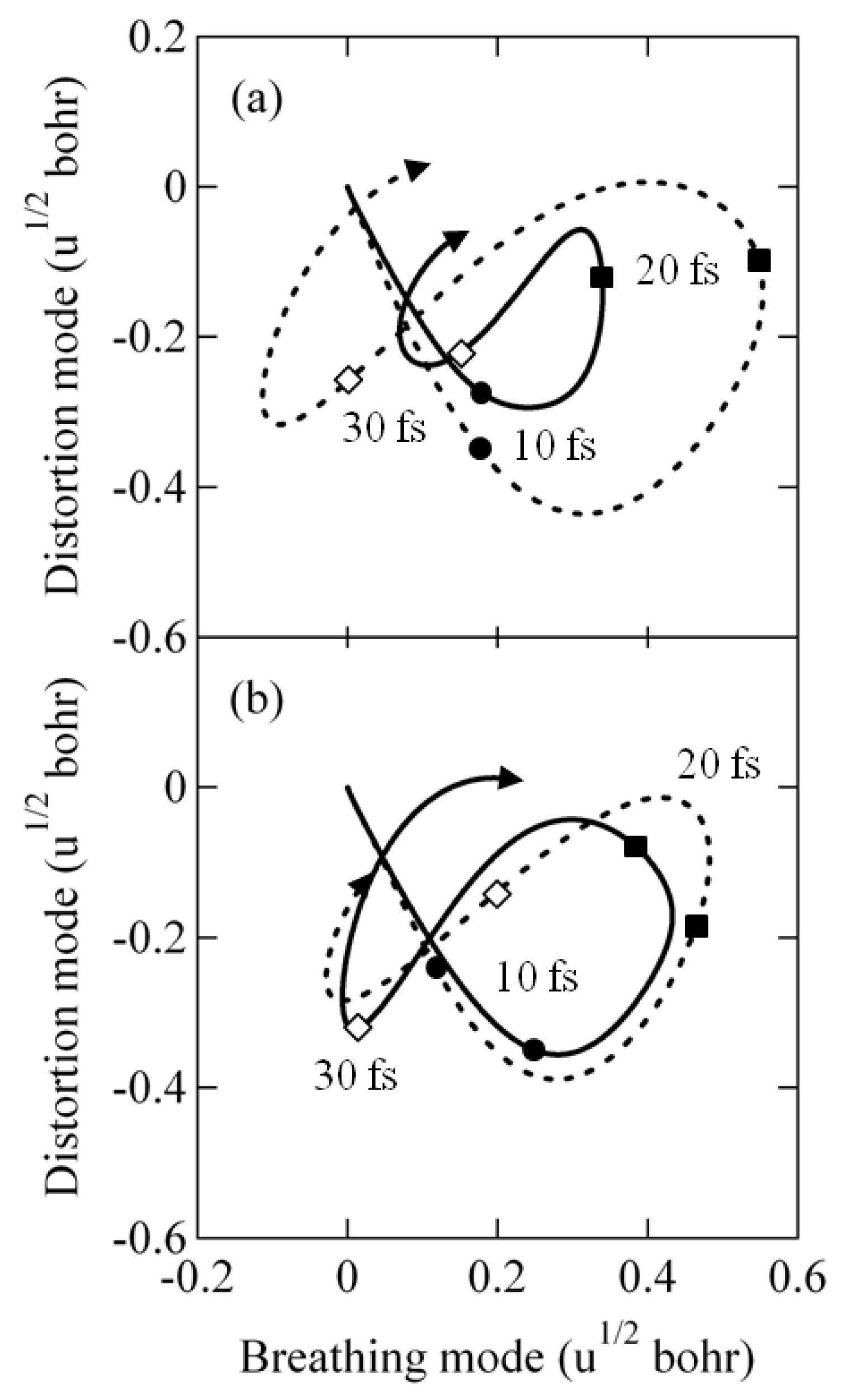
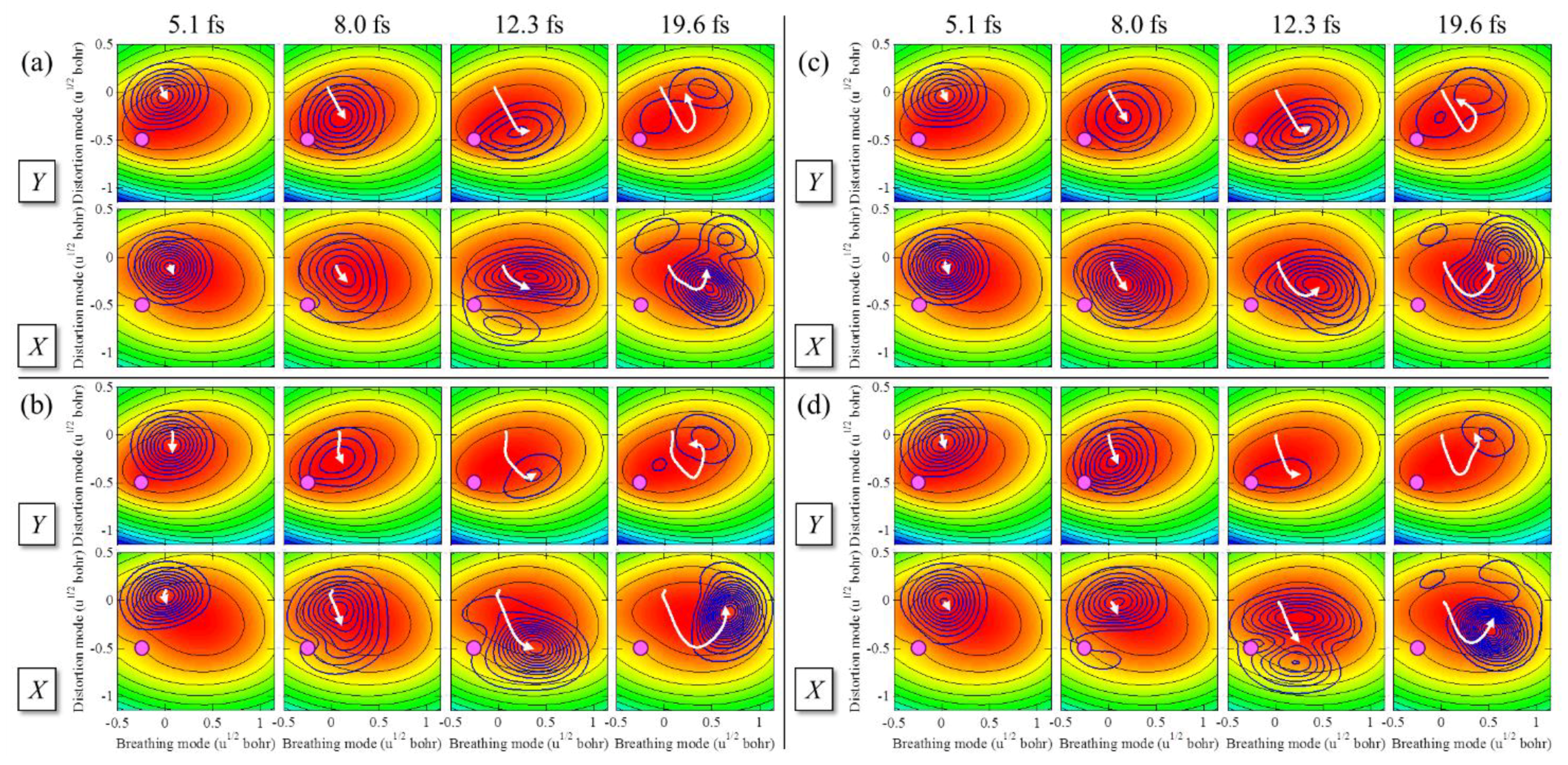
2.3.4. Laser Control of Wave Packet Interference
3. Coherent π-Electron Angular Momentum and Current of a Molecule Having Two Aromatic Rings
3.1. Two Ring Components of π-Electron Angular Momentum and Current
3.1.1. Decomposition of Angular Momentum and Current into Atomic Orbital Components
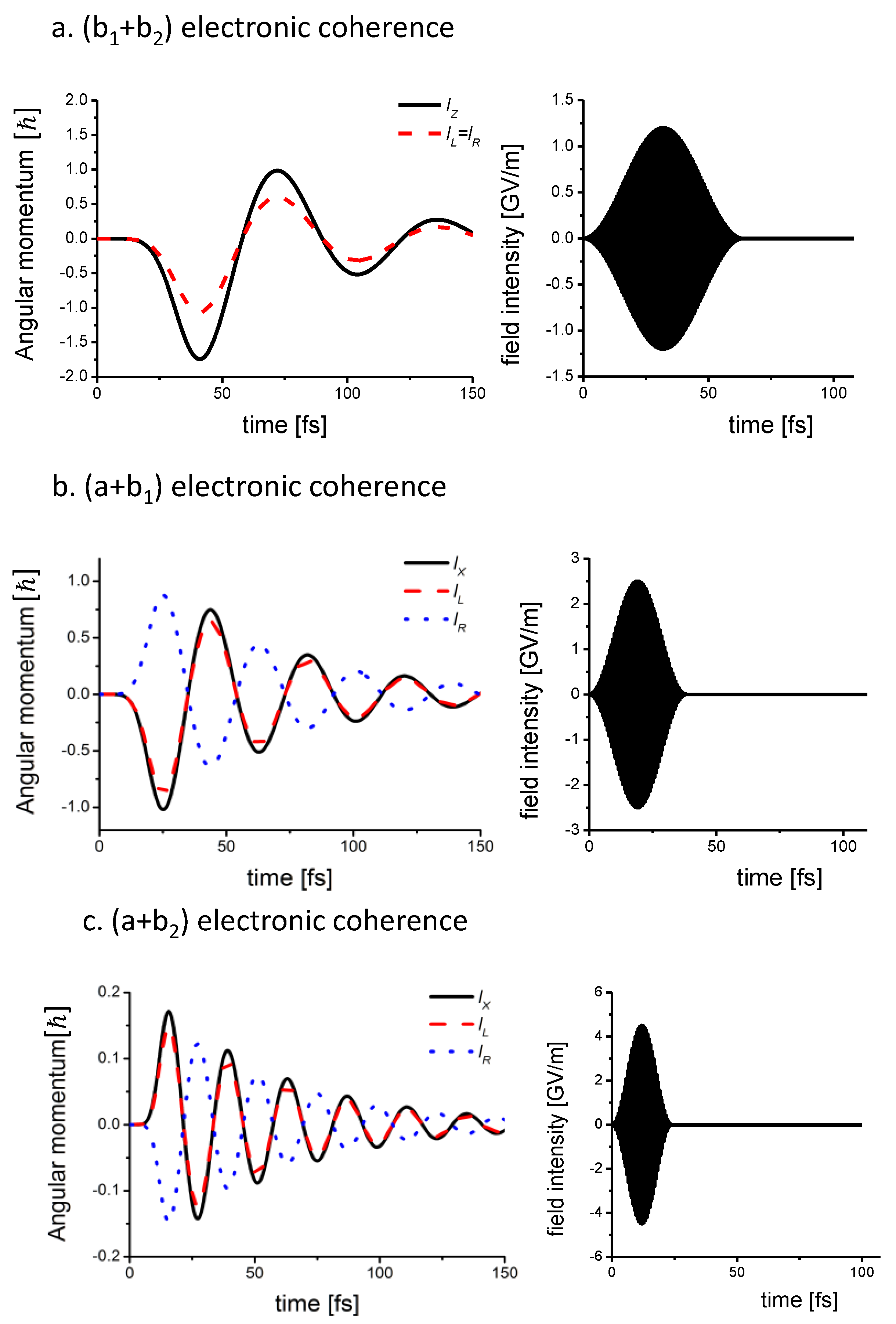
3.1.2. Coherent Angular Momentum
3.1.3. Coherent Ring Current
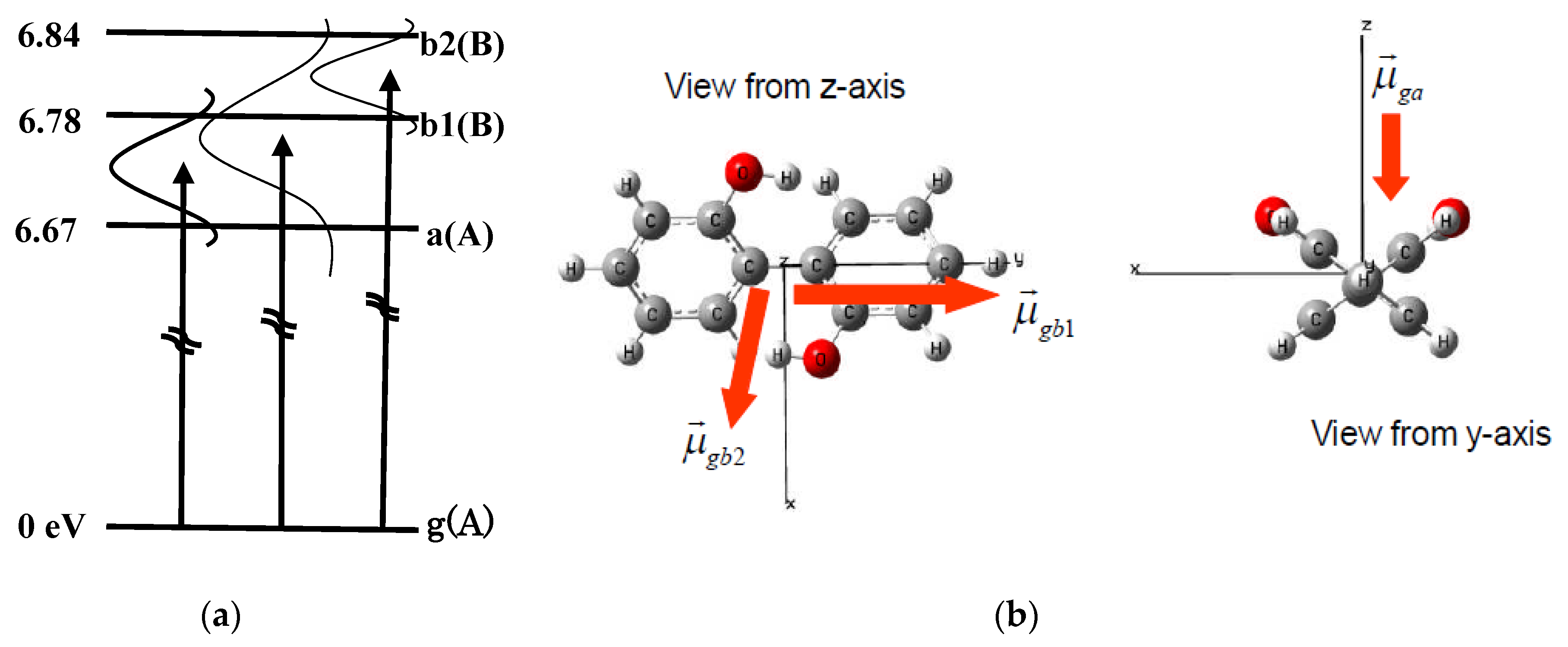
3.1.4. Application to (P)-2,2′-Biphenol
3.1.5. Generation of Two-State Electronic Coherence by Linearly Polarized UV Pulses
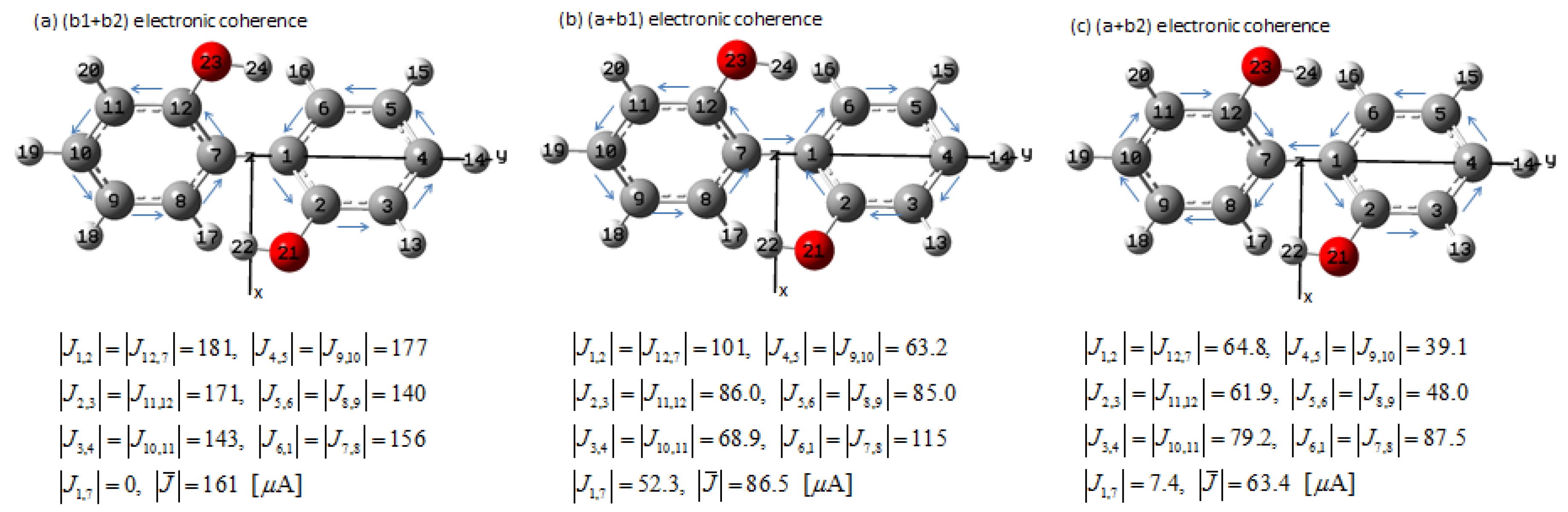
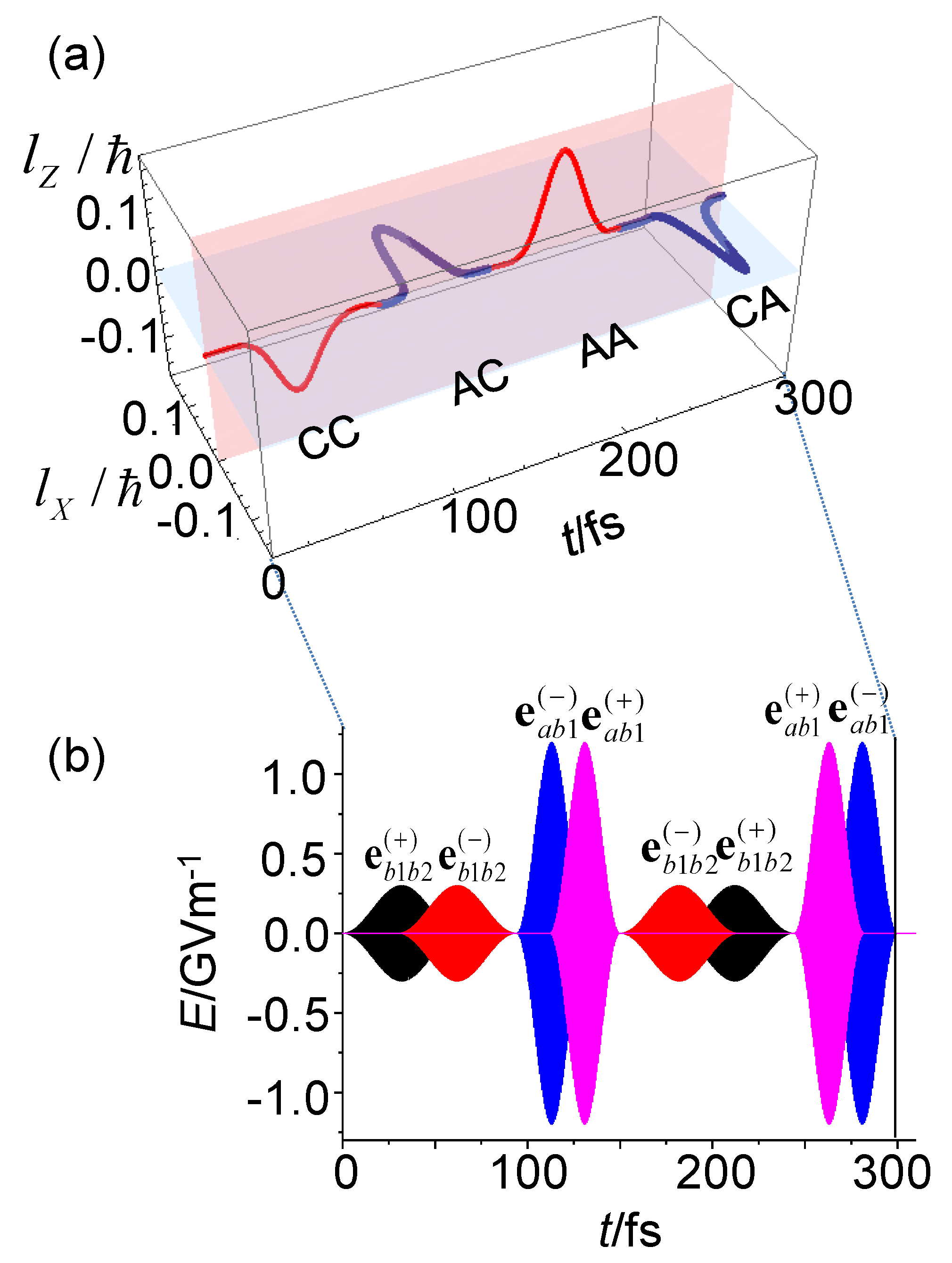
3.1.6. Bond Currents and Time Evolution of Coherent Ring Current
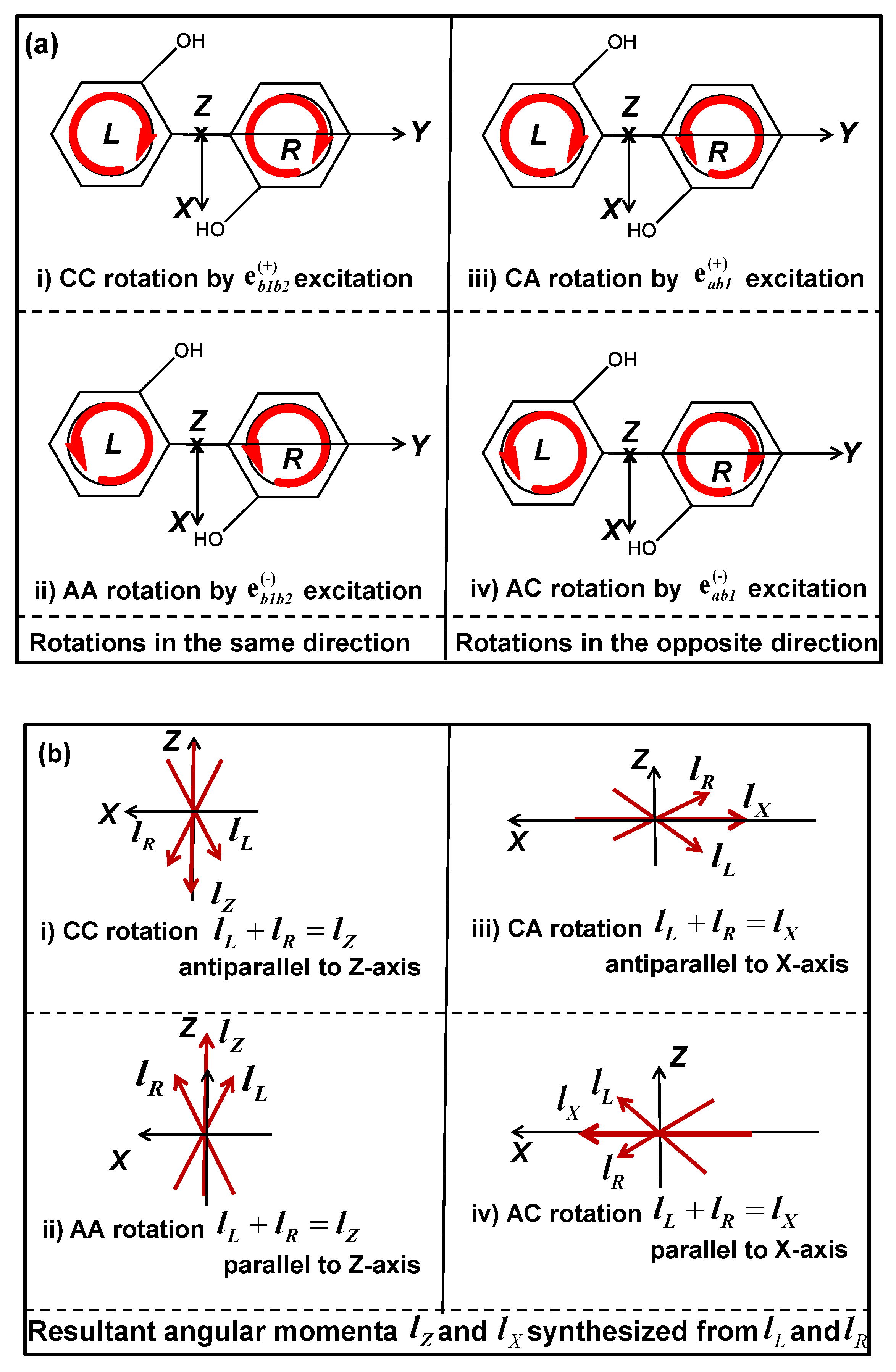
3.2. Quantum Switching of Coherent π-Electron Rotations in a Nonplanar Chiral Aromatic Molecule
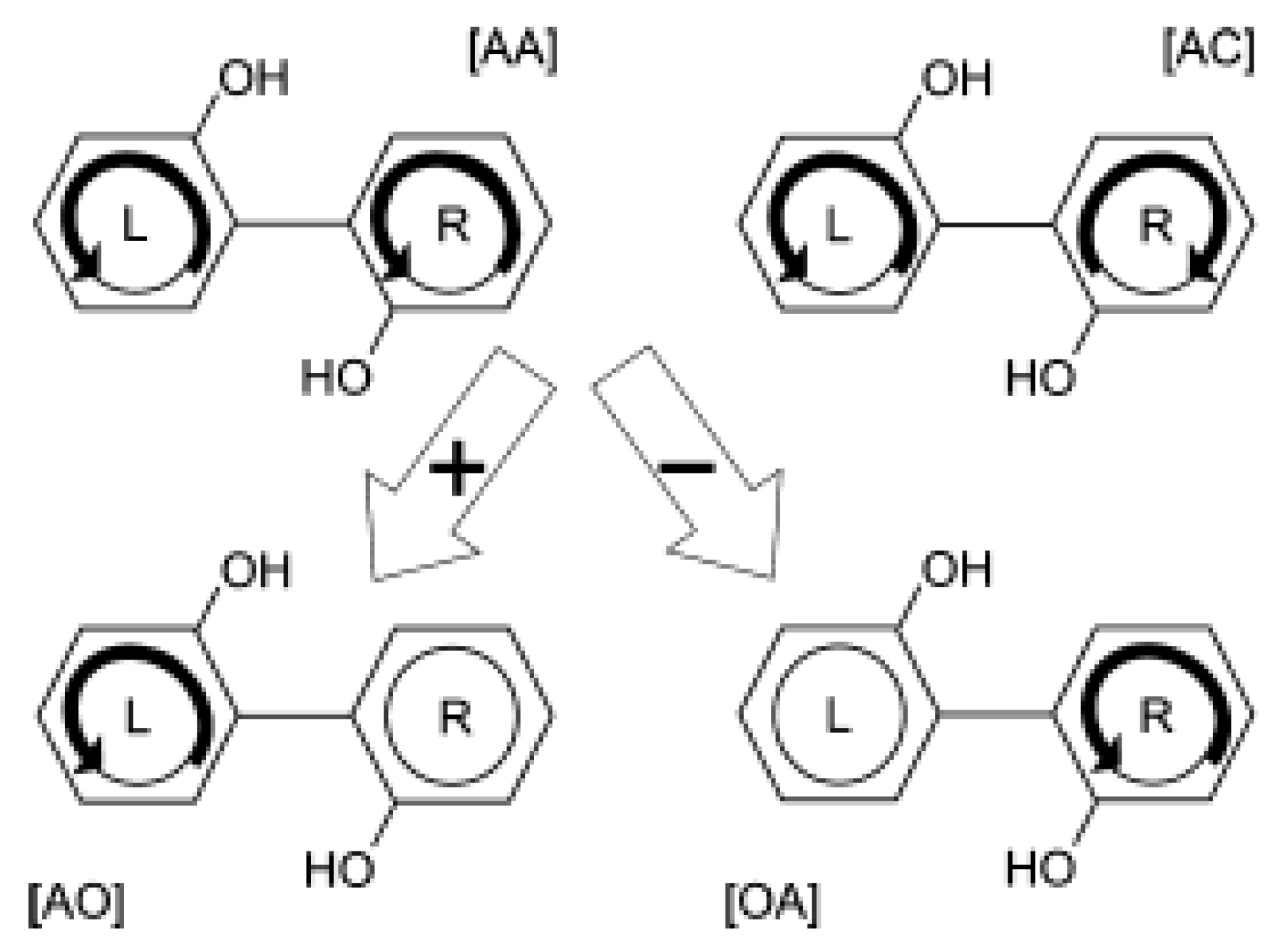
3.2.1. Control Scheme for Angular Momentum Switching
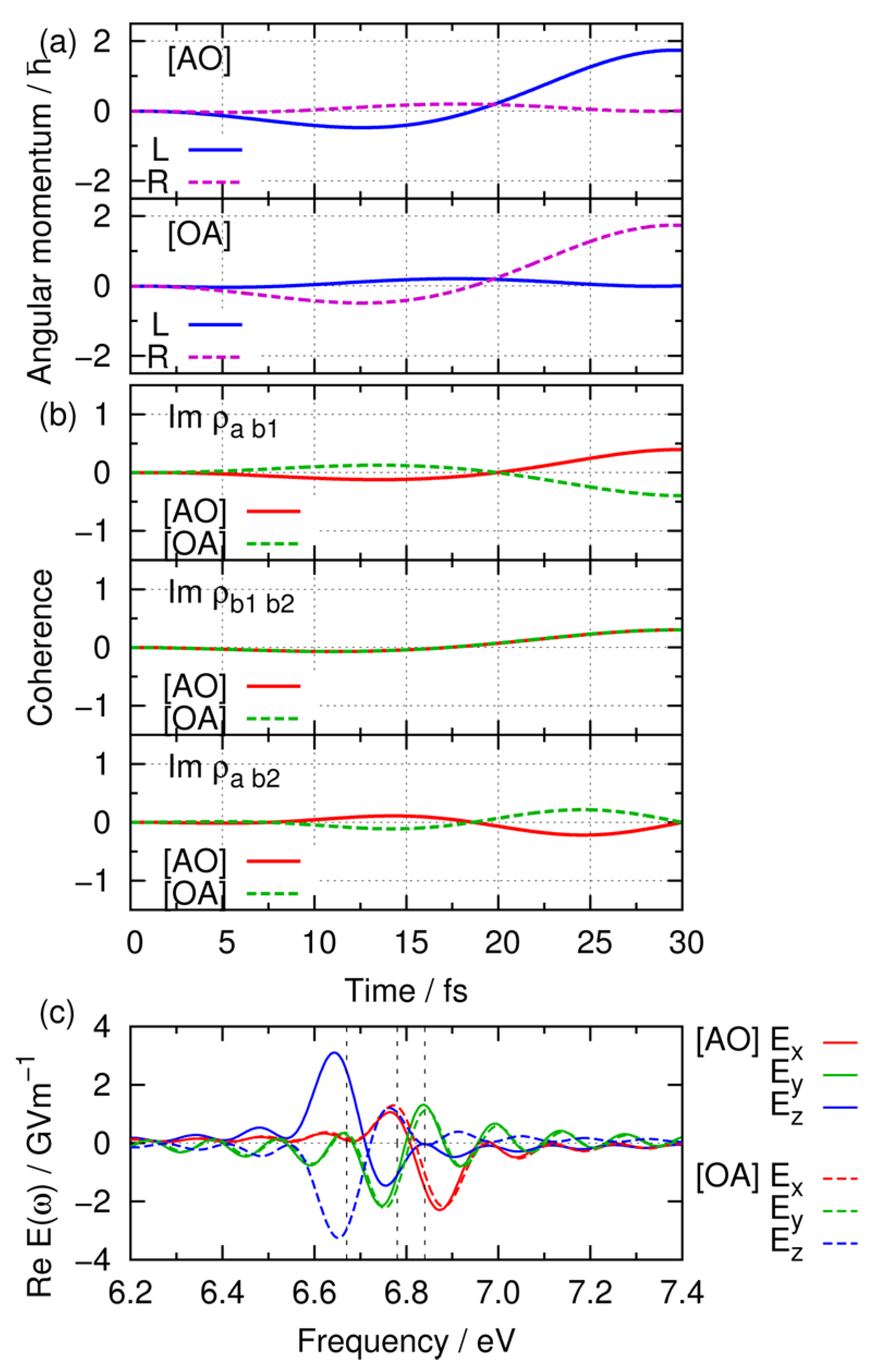
3.2.2. Simulation Results of Angular Momentum Switching
3.3. Quantum Localization of Coherent π-Electron Angular Momentum
3.3.1. Optimal Control Theory
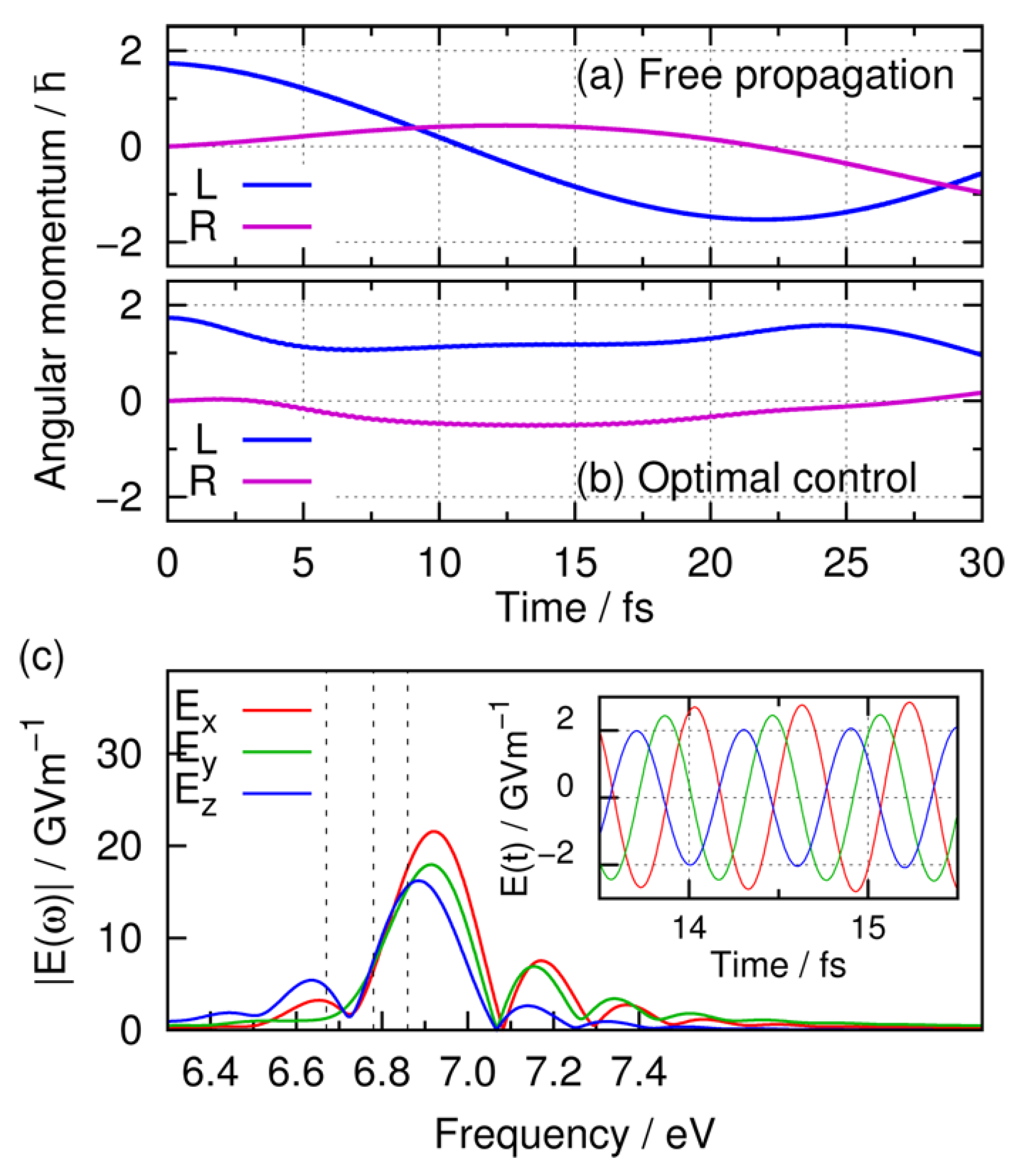
3.3.2. Simulation Results of Angular Momentum Localization
3.3.3. Simulation Results of Maintaining Angular Momentum Localization
4. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Polanyi, J.C.; Zewail, A.H. Direct Observation of the Transition State. Acc. Chem. Res 1995, 28, 119–132. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Ultrafast Dynamics of the Chemical Bond; World Scientific: Singapore, 1994; Volume 1. [Google Scholar]
- Zewail, A.H. Femtochemistry: Ultrafast Dynamics of the Chemical Bond; World Scientific: Singapore, 1994; Volume 2. [Google Scholar]
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond Using Ultrafast Lasers. Angew. Chem. Int. Ed. 2000, 39, 2587–2631. [Google Scholar] [CrossRef]
- Agostini, P. Observation of a Train of Attosecond Pulses from High Harmonic Generation. Science 2001, 292, 1689–1692. [Google Scholar]
- Hentschel, M.; Kienberger, R.; Spielmann, C.; Reider, G.A.; Milosevic, N.; Brabec, T.; Corkum, P.; Heinzmann, U.; Drescher, M.; Krausz, F. Attosecond metrology. Nature 2001, 414, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Tzallas, P.; Charalambidis, D.; Papadogiannis, N.A.; Witte, K.; Tsakiris, G.D. Direct observation of attosecond light bunching. Nature 2003, 426, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Mairesse, Y.; de Bohan, A.; Frasinski, L.J.; Merdji, H.; Dinu, L.C.; Monchicourt, P.; Breger, P.; Kovačev, M.; Taïeb, R.; Carré, B.; et al. Attosecond Synchronization of High-Harmonic Soft X-rays. Science 2003, 302, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Kienberger, R.; Goulielmakis, E.; Uiberacker, M.; Baltuska, A.; Yakovlev, V.; Bammer, F.; Scrinzi, A.; Westerwalbesloh, T.; Kleineberg, U.; Heinzmann, U.; et al. Atomic transient recorder. Nature 2004, 427, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Sekikawa, T.; Kosuge, A.; Kanai, T.; Watanabe, S. Nonlinear optics in the extreme ultraviolet. Nature 2004, 432, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Nabekawa, Y.; Shimizu, T.; Okino, T.; Furusawa, K.; Hasegawa, H.; Yamanouchi, K.; Midorikawa, K. Interferometric Autocorrelation of an Attosecond Pulse Train in the Single-Cycle Regime. Phys. Rev. Lett. 2006, 97, 153904. [Google Scholar] [CrossRef] [PubMed]
- Sansone, G.; Benedetti, E.; Calegari, F.; Vozzi, C.; Avaldi, L.; Flammini, R.; Poletto, L.; Villoresi, P.; Altucci, C.; Velotta, R.; et al. Isolated Single-Cycle Attosecond Pulses. Science 2006, 314, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Goulielmakis, E.; Schultze, M.; Hofstetter, M.; Yakovlev, N.S.; Gagnon, J.; Uiberacker, M.; Aquila, A.L.; Gullikson, E.M.; Attwood, D.T.; Kienberger, R.; et al. Single-Cycle Nonlinear Optics. Science 2008, 320, 1614–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krausz, F.; Ivanov, M. Attosecond physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Lépine, F.; Sansone, G.; Vrakking, M.J.J. Molecular applications of attosecond laser pulses. Chem. Phys. Lett. 2013, 578, 1–14. [Google Scholar] [CrossRef]
- McPherson, A.; Gibson, G.; Jara, H.; Johann, U.; Luk, T.S.; McIntyre, I.A.; Boyer, K.; Rhodes, C.K. Studies of multiphoton production of vacuum-ultraviolet radiation in the rare gases. J. Opt. Soc. Am. B 1987, 4, 595–601. [Google Scholar] [CrossRef]
- Ferray, M.; L’Huillier, A.; Li, X.F.; Lompré, L.A.; Mainfray, G.; Manus, C. Multiple-harmonic conversion of 1064 nm radiation in rare gases. J. Phys. B 1988, 21, L31–L35. [Google Scholar] [CrossRef]
- Sarukura, N.; Hata, K.; Adachi, T.; Nodomi, R.; Watanabe, M.; Watanabe, S. Coherent soft-x-ray generation by the harmonics of an ultrahigh-power KrF laser. Phys. Rev. A 1991, 43, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Rundquist, A.; Wang, H.; Murnane, M.M.; Kapteyn, H.C. Generation of Coherent Soft X Rays at 2.7 nm Using High Harmonics. Phys. Rev. Lett. 1997, 79, 2967–2970. [Google Scholar] [CrossRef]
- Spielmann, C.; Burnett, N.H.; Sartania, S.; Koppitsch, R.; Schnürer, M.; Kan, C.; Lenzner, M.; Wobrauschek, P.; Krausz, F. Generation of Coherent X-rays in the Water Window Using 5-Femtosecond Laser Pulses. Science 1997, 278, 661–664. [Google Scholar] [CrossRef]
- Seres, J.; Seres, E.; Verhoef, A.J.; Tempea, G.; Streli, C.; Wobrauschek, P.; Yakovlev, V.; Scrinzi, A.; Spielmann, C.; Krausz, F. Source of coherent kiloelectronvolt X-rays. Nature 2005, 433, 596. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Klamroth, T.; Saalfrank, P. Time-dependent configuration-interaction calculations of laser-pulse-driven many-electron dynamics: Controlled dipole switching in lithium cyanide. J. Chem. Phys. 2005, 123, 074105. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Klamroth, T.; Saalfrank, P. Molecular response properties from explicitly time-dependent configuration interaction methods. J. Chem. Phys. 2007, 127, 034107. [Google Scholar] [CrossRef] [PubMed]
- Remacle, F.; Kienberger, R.; Krausz, F.; Levine, R.D. On the feasibility of an ultrafast purely electronic reorganization in lithium hydride. Chem. Phys. 2007, 338, 342–347. [Google Scholar] [CrossRef]
- Remacle, F.; Nest, M.; Levine, R.D. Laser Steered Ultrafast Quantum Dynamics of Electrons in LiH. Phys. Rev. Lett. 2007, 99, 183902. [Google Scholar] [CrossRef] [PubMed]
- Nest, M.; Remacle, F.; Levine, R.D. Pump and probe ultrafast electron dynamics in LiH: A computational study. New J. Phys. 2008, 10, 025019. [Google Scholar] [CrossRef]
- Barth, I.; Manz, J.; Serrano-Andrés, L. Quantum simulations of toroidal electric ring currents and magnetic fields in linear molecules induced by circularly polarized laser pulses. Chem. Phys. 2008, 347, 263–271. [Google Scholar] [CrossRef]
- Barth, I.; Serrano-Andrés, L.; Seideman, T. Nonadiabatic orientation, toroidal current, and induced magnetic field in BeO molecules. J. Chem. Phys. 2008, 129, 164303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, I.; Serrano-Andrés, L.; Seideman, T. Erratum: “Nonadiabatic orientation, toroidal current, and induced magnetic field in BeO molecules” [J. Chem. Phys. 129, 164303 (2008)]. J. Chem. Phys. 2009, 130, 109901. [Google Scholar] [CrossRef] [Green Version]
- Yonehara, T.; Takatsuka, K. Characterization of electron-deficient chemical bonding of diborane with attosecond electron wavepacket dynamics and laser response. Chem. Phys. 2009, 366, 115–128. [Google Scholar] [CrossRef]
- Takatsuka, K.; Yonehara, T. Exploring dynamical electron theory beyond the Born–Oppenheimer framework: From chemical reactivity to non-adiabatically coupled electronic and nuclear wavepackets on-the-fly under laser field. Phys. Chem. Chem. Phys. 2011, 13, 4987–5016. [Google Scholar] [CrossRef] [PubMed]
- Mignolet, B.; Gijsbertsen, A.; Vrakking, M.J.J.; Levine, R.D.; Remacle, F. Stereocontrol of attosecond time-scale electron dynamics in ABCU using ultrafast laser pulses: A computational study. Phys. Chem. Chem. Phys. 2011, 13, 8331–8344. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, I.S.; Nest, M. Correlated Electron Dynamics: How Aromaticity Can Be Controlled. J. Am. Chem. Soc. 2011, 133, 20230–20236. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Liu, C.; Manz, J.; Paulus, B.; Pérez-Torres, J.F.; Pohl, V.; Tremblay, J.C. Multidirectional Angular Electronic Flux during Adiabatic Attosecond Charge Migration in Excited Benzene. J. Phys. Chem. A 2016, 120, 5360–5369. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Manz, J.; Paulus, B.; Pohl, V.; Tremblay, J.C.; Yang, Y. Quantum control of electronic fluxes during adiabatic attosecond charge migration in degenerate superposition states of benzene. Chem. Phys. 2017, 482, 146–159. [Google Scholar] [CrossRef]
- Hermann, G.; Liu, C.; Manz, J.; Paulus, B.; Pohl, V.; Tremblay, J.C. Attosecond angular flux of partial charges on the carbon atoms of benzene in non-aromatic excited state. Chem. Phys. Lett. 2017, 683, 553–558. [Google Scholar] [CrossRef]
- Cederbaum, L.S.; Zobeley, J. Ultrafast charge migration by electron correlation. Chem. Phys. Lett. 1999, 307, 205–210. [Google Scholar] [CrossRef]
- Breidbach, J.; Cederbaum, L.S. Migration of holes: Formalism, mechanisms, and illustrative applications. J. Chem. Phys. 2003, 118, 3983–3996. [Google Scholar] [CrossRef]
- Bandrauk, A.D.; Chelkowski, S.; Nguyen, H.S. Attosecond localization of electrons in molecules. Int. J. Quantum Chem. 2004, 100, 834–844. [Google Scholar] [CrossRef]
- Hennig, H.; Breidbach, J.; Cederbaum, L.S. Electron Correlation as the Driving Force for Charge Transfer: Charge Migration Following Ionization in N-Methyl Acetamide. J. Phys. Chem. A 2005, 109, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Breidbach, J.; Cederbaum, L.S. Universal Attosecond Response to the Removal of an Electron. Phys. Rev. Lett. 2005, 94, 033901. [Google Scholar] [CrossRef] [PubMed]
- Kuleff, A.I.; Breidbach, J.; Cederbaum, L.S. Multielectron wave-packet propagation: General theory and application. J. Chem. Phys. 2005, 123, 044111. [Google Scholar] [CrossRef] [PubMed]
- Remacle, F.; Levine, R.D. An electronic time scale in chemistry. Proc. Natl. Acad. Sci. USA 2006, 103, 6793–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleff, A.I.; Cederbaum, L.S. Charge migration in different conformers of glycine: The role of nuclear geometry. Chem. Phys. 2007, 338, 320–328. [Google Scholar] [CrossRef]
- Lünnemann, S.; Kuleff, A.I.; Cederbaum, L.S. Charge migration following ionization in systems with chromophore-donor and amine-acceptor sites. J. Chem. Phys. 2008, 129, 104305. [Google Scholar] [CrossRef] [PubMed]
- Kuleff, A.I.; Cederbaum, L.S. Radiation Generated by the Ultrafast Migration of a Positive Charge Following the Ionization of a Molecular System. Phys. Rev. Lett. 2011, 106, 053001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calegari, F.; Ayuso, D.; Trabattoni, A.; Belshaw, L.; De Camillis, S.; Anumula, S.; Frassetto, F.; Poletto, L.; Palacios, A.; Decleva, P.; et al. Ultrafast electron dynamics in phenylalanine initiated by attosecond pulses. Science 2014, 346, 336–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K. To catch and smash charge on the hop. Science 2015, 350, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Kraus, P.M.; Mignolet, B.; Baykusheva, D.; Rupenyan, A.; Horný, L.; Penka, E.F.; Grassi, G.; Tolstikhin, O.I.; Schneider, J.; Jensen, F.; et al. Measurement and laser control of attosecond charge migration in ionized iodoacetylene. Science 2015, 350, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobusada, K.; Yabana, K. Photoinduced electric currents in ring-shaped molecules by circularly polarized laser pulses. Phys. Rev. A 2007, 75, 032518. [Google Scholar] [CrossRef]
- Barth, I.; Manz, J. Periodic Electron Circulation Induced by Circularly Polarized Laser Pulses: Quantum Model Simulations for Mg Porphyrin. Angew. Chem. Int. Ed. 2006, 45, 2962–2965. [Google Scholar] [CrossRef] [PubMed]
- Barth, I.; Manz, J.; Shigeta, Y.; Yagi, K. Unidirectional Electronic Ring Current Driven by a Few Cycle Circularly Polarized Laser Pulse: Quantum Model Simulations for Mg–Porphyrin. J. Am. Chem. Soc. 2006, 128, 7043–7049. [Google Scholar] [CrossRef] [PubMed]
- Barth, I.; Manz, J. Quantum Switching of Magnetic Fields by Circularly Polarized Re-Optimized π Laser Pulses: From One-Electron Atomic Ions to Molecules. In Progress in Ultrafast Intense Laser Science; Yamanouchi, K., Gerber, G., Bandrauk, A.D., Eds.; Springer: Berlin, Germany, 2010; Volume 6, pp. 21–44. [Google Scholar]
- Kanno, M.; Kono, H.; Fujimura, Y. Control of π-Electron Rotations in Chiral Aromatic Molecules Using Intense Laser Pulses. In Progress in Ultrafast Intense Laser Science; Yamanouchi, K., Charalambidis, D., Normand, D., Eds.; Springer: Berlin, Germany, 2011; Volume 7, pp. 53–78. [Google Scholar]
- Kanno, M.; Ono, Y.; Kono, H.; Fujimura, Y. Laser-Polarization Effects on Coherent Vibronic Excitation of Molecules with Quasi-Degenerate Electronic States. J. Phys. Chem. A 2012, 116, 11260–11272. [Google Scholar] [CrossRef] [PubMed]
- Kanno, M.; Kono, H.; Lin, S.H.; Fujimura, Y. Laser-Induced Electronic and Nuclear Coherent Motions in Chiral Aromatic Molecules. In Quantum Systems in Chemistry and Physics: Progress in Methods and Applications; Nishikawa, K., Maruani, J., Brändas, E.J., Delgado-Barrio, G., Piecuch, P., Eds.; Progress in Theoretical Chemistry and Physics; Springer: Amsterdam, The Netherlands, 2012; Volume 26, pp. 121–148. [Google Scholar]
- Kanno, M.; Ono, Y.; Kono, H.; Fujimura, Y. Nonadiabatically Coupled π-Electron Rotation and Molecular Vibration in Aromatic Molecules Excited by Polarized UV/Vis Laser Pulses. Chin. J. Phys. 2014, 52, 617–651. [Google Scholar]
- Tannor, D.J. Introduction to Quantum Mechanics: A Time-Dependent Perspective; University Science Books: Mill Valley, CA, USA, 2007; pp. 479–482. [Google Scholar]
- Kanno, M.; Kono, H.; Fujimura, Y. Control of π-electron rotation in chiral aromatic molecules by nonhelical laser pulses. Angew. Chem. Int. Ed. 2006, 45, 7995–7998. [Google Scholar] [CrossRef] [PubMed]
- Kanno, M.; Hoki, K.; Kono, H.; Fujimura, Y. Quantum optimal control of electron ring currents in chiral aromatic molecules. J. Chem. Phys. 2007, 127, 204314. [Google Scholar] [CrossRef] [PubMed]
- Kanno, M.; Kono, H.; Fujimura, Y.; Lin, S.H. Nonadiabatic Response Model of Laser-Induced Ultrafast π-Electron Rotations in Chiral Aromatic Molecules. Phys. Rev. Lett. 2010, 104, 108302. [Google Scholar] [CrossRef] [PubMed]
- Selle, R.; Nuernberger, P.; Langhojer, F.; Dimler, F.; Fechner, S.; Gerber, G.; Brixner, T. Generation of polarization-shaped ultraviolet femtosecond pulses. Opt. Lett. 2008, 33, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, P.; Selle, R.; Langhojer, F.; Dimler, F.; Fechner, S.; Gerber, G.; Brixner, T. Polarization-shaped femtosecond laser pulses in the ultraviolet. J. Opt. A 2009, 11, 085202. [Google Scholar] [CrossRef]
- Seidel, M.T.; Zhang, Z.; Yan, S.; Tan, H.-S. Ultraviolet polarization pulse shaping using sum-frequency generation. J. Opt. Soc. Am. B 2011, 28, 1146–1151. [Google Scholar] [CrossRef]
- Seidel, M.T.; Zhang, Z.; Yan, S.; Wells, K.L.; Tan, H.-S. Characterization of polarization shaped ultraviolet femtosecond laser pulses. J. Opt. Soc. Am. B 2011, 28, 2718–2725. [Google Scholar] [CrossRef]
- Yuan, K.-J.; Bandrauk, A.D. Circularly polarized attosecond pulses from molecular high-order harmonic generation by ultrashort intense bichromatic circularly and linearly polarized laser pulses. J. Phys. B 2012, 45, 074001. [Google Scholar] [CrossRef]
- Seideman, T. Revival Structure of Aligned Rotational Wave Packets. Phys. Rev. Lett. 1999, 83, 4971–4974. [Google Scholar] [CrossRef]
- Stapelfeldt, H.; Seideman, T. Aligning molecules with strong laser pulses. Rev. Mod. Phys. 2003, 75, 543–557. [Google Scholar] [CrossRef]
- Levine, I.N. Quantum Chemistry, 6th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2009; pp. 471–635. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Lindh, R.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Rauhut, G.; Amos, R.D.; Bernhardsson, A.; et al. MOLPRO, version 2006.1; Cardiff, UK, 2006.
- Hampel, C.; Peterson, K.; Werner, H.-J. A comparison of the efficiency and accuracy of the quadratic configuration interaction (QCISD), coupled cluster (CCSD), and Brueckner coupled cluster (BCCD) methods. Chem. Phys. Lett. 1992, 190, 1–12. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J. A second order multiconfiguration SCF procedure with optimum convergence. J. Chem. Phys. 1985, 82, 5053–5063. [Google Scholar] [CrossRef]
- Knowles, P.J.; Werner, H.-J. An efficient second-order MCSCF method for long configuration expansions. Chem. Phys. Lett. 1985, 115, 259–267. [Google Scholar] [CrossRef]
- Handbook of Mathematical Functions: With Formulas, Graphs, and Mathematical Tables, 9th ed.; Abramowitz, M.; Stegun, I.A. (Eds.) Dover: New York, NY, USA, 1970; p. 896. [Google Scholar]
- Werner, H.-J. Third-order multireference perturbation theory The CASPT3 method. Mol. Phys. 1996, 89, 645–661. [Google Scholar] [CrossRef]
- Celani, P.; Werner, H.-J. Multireference perturbation theory for large restricted and selected active space reference wave functions. J. Chem. Phys. 2000, 112, 5546–5557. [Google Scholar] [CrossRef]
- Baer, M. Beyond Born-Oppenheimer; Wiley: Hoboken, NJ, USA, 2006; pp. 26–57. [Google Scholar]
- Sarkar, B.; Adhikari, S. A rigorous approach to the formulation of extended Born-Oppenheimer equation for a three-state system. Int. J. Quantum Chem. 2009, 109, 650–667. [Google Scholar] [CrossRef]
- Simah, D.; Hartke, B.; Werner, H.-J. Photodissociation dynamics of H2S on new coupled ab initio potential energy surfaces. J. Chem. Phys. 1999, 111, 4523–4534. [Google Scholar] [CrossRef]
- Ohtsuki, Y.; Nakagami, K.; Fujimura, Y. Quantum Control of Molecular Dynamics. In Advances in Multi-Photon Processes and Spectroscopy; Lin, S.H., Villaeys, A.A., Fujimura, Y., Eds.; World Scientific: Singapore, 2001; Volume 13, pp. 1–127. [Google Scholar]
- Gross, P.; Neuhauser, D.; Rabitz, H. Optimal control of curve-crossing systems. J. Chem. Phys. 1992, 96, 2834–2845. [Google Scholar] [CrossRef]
- Born, M.; Oppenheimer, J.R. Zur Quantentheorie der Molekeln. Ann. Phys. 1927, 84, 457–484. [Google Scholar] [CrossRef]
- Tannor, D.J. Introduction to Quantum Mechanics: A Time-Dependent Perspective; University Science Books: Mill Valley, CA, USA, 2007; pp. 81–86. [Google Scholar]
- Fujimura, Y.; Sakai, H. Electronic and Nuclear Dynamics in Molecular Systems; World Scientific: Singapore, 2011; pp. 117–132. [Google Scholar]
- Mineo, H.; Yamaki, M.; Teranishi, Y.; Hayashi, M.; Lin, S.H.; Fujimura, Y. Quantum Switching of π-Electron Rotations in a Nonplanar Chiral Molecule by Using Linearly Polarized UV Laser Pulses. J. Am. Chem. Soc. 2012, 134, 14279–14282. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Lin, S.H.; Fujimura, Y. Coherent π-electron dynamics of (P)-2,2′-biphenol induced by ultrashort linearly polarized UV pulses: Angular momentum and ring current. J. Chem. Phys. 2013, 138, 074304. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y.; Kono, H.; Nakajima, T.; Lin, S.H. A theoretical study of resonance Raman scattering from molecules. III. Resonance Raman scattering and resonance fluorescence. J. Chem. Phys. 1981, 75, 99–106. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Baskin, J.S.; Felker, P.M.; Zewail, A.H. Doppler-free time-resolved polarization spectroscopy of large molecules: Measurement of excited state rotational constants. J. Chem. Phys. 1986, 84, 4708–4710. [Google Scholar] [CrossRef]
- Yamaki, M.; Mineo, H.; Teranishi, Y.; Hayashi, M.; Fujimura, Y.; Nakamura, H.; Lin, S.H. Quantum Localization of Coherent π-Electron Angular Momentum in (P)-2,2′-Biphenol. J. Phys. Chem. Lett. 2014, 5, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Botina, J.; Rabitz, H. Rapidly convergent iteration methods for quantum optimal control of population. J. Chem. Phys. 1998, 108, 1953–1963. [Google Scholar] [CrossRef]
- Ohtsuki, Y.; Zhu, W.; Rabitz, H. Monotonically convergent algorithm for quantum optimal control with dissipation. J. Chem. Phys. 1999, 110, 9825–9832. [Google Scholar] [CrossRef]
- Umeda, H.; Fujimura, Y. Quantum control of chemical reaction dynamics in a classical way. J. Chem. Phys. 2000, 113, 3510–3518. [Google Scholar] [CrossRef]
- Mineo, H.; Lin, S.H.; Fujimura, Y. Vibrational effects on UV/Vis laser-driven π-electron ring currents in aromatic ring molecules. Chem. Phys. 2014, 442, 103–110. [Google Scholar] [CrossRef]
- Mineo, H.; Lin, S.H.; Fujimura, Y.; Xu, J.; Xu, R.X.; Yan, Y.J. Non-Markovian response of ultrafast coherent electronic ring currents in chiral aromatic molecules in a condensed phase. J. Chem. Phys. 2013, 139, 214306. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Kanno, M.; Kono, H.; Chao, S.D.; Lin, S.H.; Fujimura, Y. Ultrafast coherent dynamics of nonadiabatically coupled quasi-degenerate excited states in molecules: Population and vibrational coherence transfers. Chem. Phys. 2012, 392, 136–142. [Google Scholar] [CrossRef]
- Yamaki, M.; Mineo, H.; Teranishi, Y.; Lin, S.H.; Fujimura, Y. Quantum Control of Coherent π-Electron Dynamics in Chiral Aromatic Molecules. J. Chin. Chem. Soc. 2016, 63, 87–92. [Google Scholar] [CrossRef]
- Yamaki, M.; Teranishi, Y.; Nakamura, H.; Lin, S.H.; Fujimura, Y. The generation of stationary π-electron rotations in chiral aromatic ring molecules possessing non-degenerate excited states. Phys. Chem. Chem. Phys. 2016, 18, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Yamaki, M.; Kim, G.-S.; Teranishi, Y.; Lin, S.H.; Fujimura, Y. Induction of unidirectional π-electron rotations in low-symmetry aromatic ring molecules using two linearly polarized stationary lasers. Phys. Chem. Chem. Phys. 2016, 18, 26786–26795. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.E. The Larger Acenes: Versatile Organic Semiconductors. Angew. Chem. Int. Ed. 2008, 47, 452–483. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Fujimura, Y. Quantum Design of π-Electron Ring Currents in Polycyclic Aromatic Hydrocarbons: Parallel and Antiparallel Ring Currents in Naphthalene. J. Phys. Chem. Lett. 2017, 8, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Fujimura, Y. Quantum control of coherent π-electron ring currents in polycyclic aromatic hydrocarbons. J. Chem. Phys. 2017, 147, 224301. [Google Scholar] [CrossRef] [PubMed]
- Hertel, I.V.; Shchatsinin, I.; Laarmann, T.; Zhavoronkov, N.; Ritze, H.-H.; Schulz, C.P. Fragmentation and Ionization Dynamics of C60 in Elliptically Polarized Femtosecond Laser Fields. Phys. Rev. Lett. 2009, 102, 023003. [Google Scholar] [CrossRef] [PubMed]
- Shchatsinin, I.; Ritze, H.-H.; Schulz, C.P.; Hertel, I.V. Multiphoton excitation and ionization by elliptically polarized, intense short laser pulses: Recognizing multielectron dynamics and doorway states in C60 vs. Xe. Phys. Rev. A 2009, 79, 053414. [Google Scholar] [CrossRef]
- Kanno, M.; Inada, N.; Kono, H. Single-active-electron analysis of laser-polarization effects on atomic/molecular multiphoton excitation. J. Chem. Phys. 2017, 147, 154310. [Google Scholar] [CrossRef] [PubMed]
















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, M.; Kono, H.; Fujimura, Y. Laser-Control of Ultrafast π-Electron Ring Currents in Aromatic Molecules: Roles of Molecular Symmetry and Light Polarization. Appl. Sci. 2018, 8, 2347. https://0-doi-org.brum.beds.ac.uk/10.3390/app8122347
Kanno M, Kono H, Fujimura Y. Laser-Control of Ultrafast π-Electron Ring Currents in Aromatic Molecules: Roles of Molecular Symmetry and Light Polarization. Applied Sciences. 2018; 8(12):2347. https://0-doi-org.brum.beds.ac.uk/10.3390/app8122347
Chicago/Turabian StyleKanno, Manabu, Hirohiko Kono, and Yuichi Fujimura. 2018. "Laser-Control of Ultrafast π-Electron Ring Currents in Aromatic Molecules: Roles of Molecular Symmetry and Light Polarization" Applied Sciences 8, no. 12: 2347. https://0-doi-org.brum.beds.ac.uk/10.3390/app8122347