
Applied Nanotechnologies in Anticoagulant Therapy: From Anticoagulants to Coagulation Test Performance of Drug Delivery Systems


 ,
, 
Abstract
:1. Introduction
2. Anticoagulant Therapy
2.1. Conventional Anticoagulant Agents
2.1.1. Unfractionated Heparin (UFH), Low-Molecular-Weight Heparin (LMWH), and Ultra-Low-Molecular-Weight Heparin (ULMWH)

{kind=link}
{kind=link}
{kind=link}
| Agents | Mw a (Da b) | Production Process | Trade Name (Pharma Company) | Anti-Factor Xa:IIa Ratio | References |
|---|---|---|---|---|---|
| LMWH | |||||
| Ardeparin | 6000 | Peroxidative depolymerization | Normiflo© (Pfizer) | 1.9 | [15,22,36] |
| Certoparin | 6000 | Deamine cleavage with isoamyl nitrate degradation | Sandoparin©, Mono-Embolex©, Sandoz© (Novartis) | 2.4 | [15,22,36] |
| Dalteparin | 6000 | Nitrous acid depolymerization | Fragmin© (Pfizer) | 2.7 | [15,22,36] |
| Enoxaparin | 4500 | Benzylation and alkaline depolymerization | Clexane©/Lovenox© (Sanofi-Aventis) | 3.8 | [15,22,37] |
| Nadroparin | 5000 | Nitrous acid depolymerization | Fraxiparin© (Choay/Aspen) | 3.6 | [15,22] |
| Parnaparin | 4500 | Hydrogen peroxide and cupric salt depolymerization | Fluxum© (Wasserman) | 3.0 | [22] |
| Reviparin | 4000 | Nitrous acid depolymerization followed by chromatographic purification | Clivarine© (Abbott) | 3.5 | [15,22] |
| Tinzaparin | 6000 | Heparinase digestion | Innohep© (Novo/Leo) | 1.5 | [15,22,24] |
| ULMWH | |||||
| Bemiparin | 3600 | β-eliminative cleavage through alkaline depolymerization | Hibor©, Ivor©, Zivor© (Sigma Tau) | 8.1 | [15,22,24] |
| Semuloparin | 2400 | β-eliminative cleavage through selective and controlled depolymerization using a phosphazene base | (Sanofi-Aventis) | 80 | [22,28] |
| Synthetic analogues | |||||
| Fondaparinux | 1728 | Chemical synthesis | Arixtra© (GlaxoSmithKline) | ~850 UI anti-Xa/mg | [22,28] |
| Idraparinux | 1728 | Chemical synthesis | (Sanofi-Aventis) | ~1600 UI anti-Xa/mg | [22] |
| Idrabiotaparinux | 2052 | Chemical synthesis | (Sanofi-Aventis) | ~1600 UI anti-Xa/mg | [22] |
2.1.2. Vitamin K Antagonists (VKAs)
2.2. Novel Anticoagulant Drugs
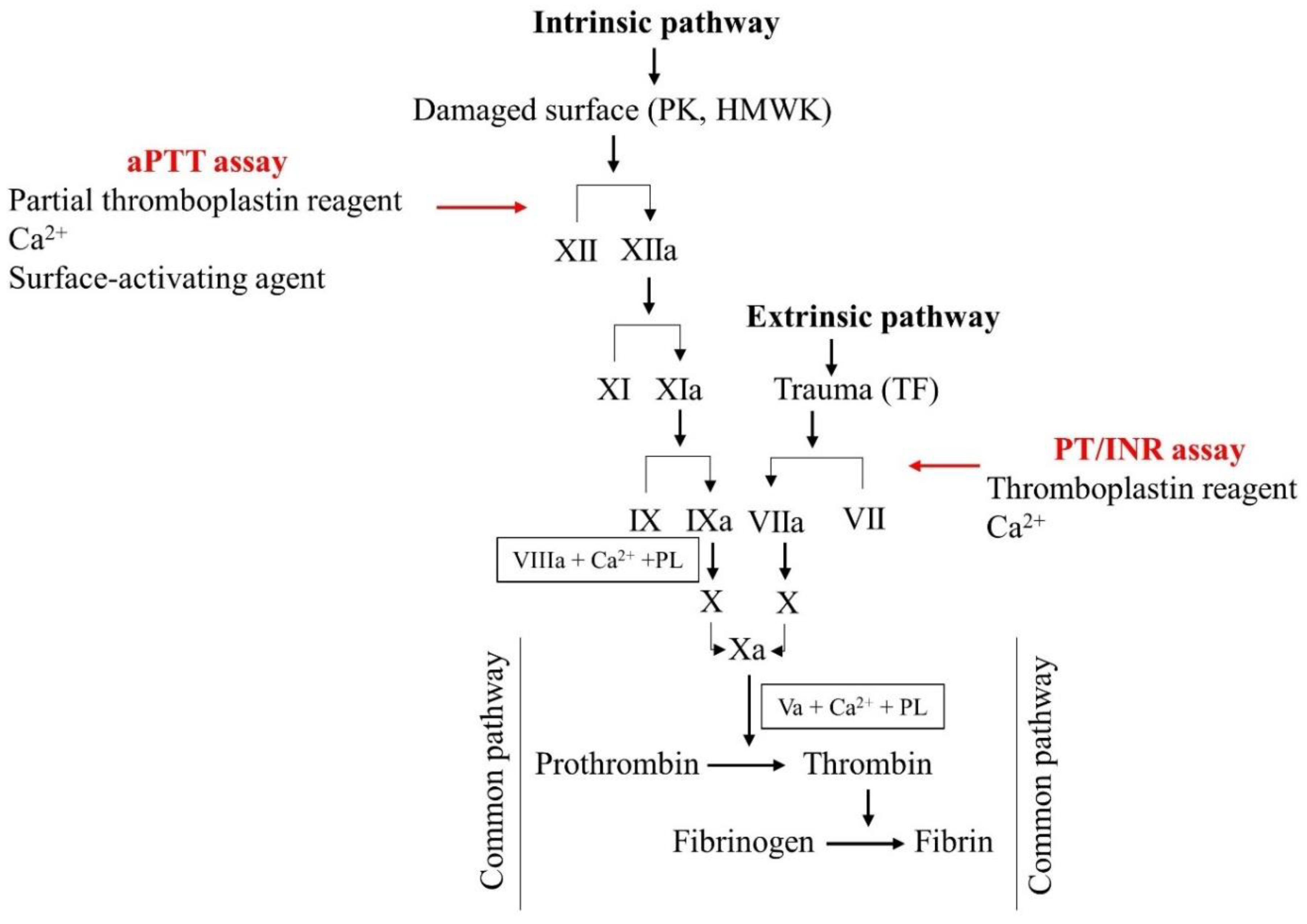
3. Laboratory Assessment of Anticoagulant Therapy
3.1. Monitoring VKAs and UFH
3.2. Monitoring LMWH Therapy
3.3. Monitoring DOAC Therapy
4. Coagulation Test Performance of Drug Delivery Systems
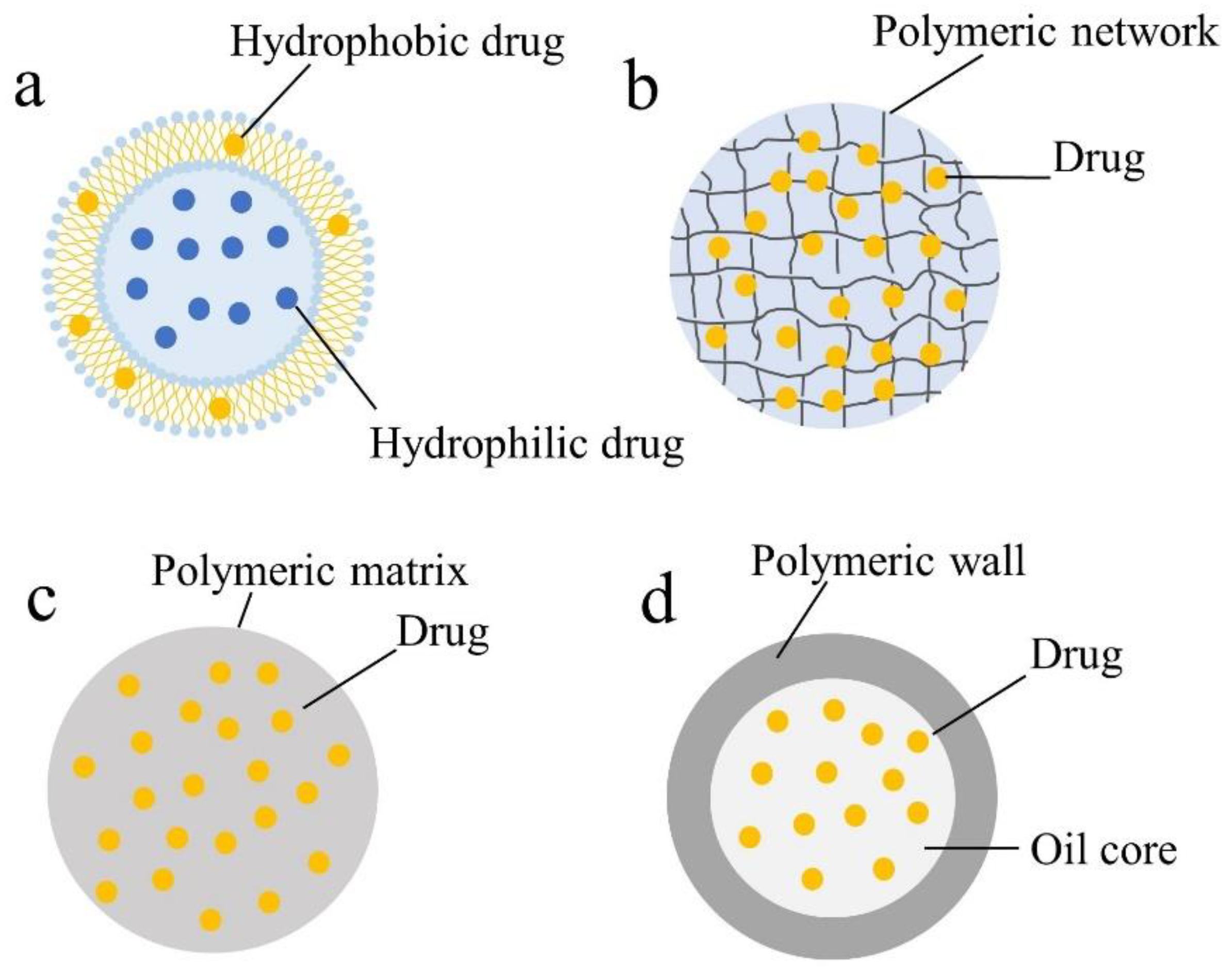
4.1. Liposomes
4.2. Hydrogels
4.3. Polymeric Nanoparticles (PNPs)
4.4. Other Drug Delivery Systems
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS | chitosan |
| Chol | cholesterol |
| CTAB | hexadecyltrimethyl ammonium bromide |
| DSPE | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine |
| DSPE–PEG | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine polyethyleneglycol |
| E80 | egg yolk lecithin |
| Egg PC | phosphatidylcholine from egg yolk |
| GMO | glyceryl monooleate |
| HPMC | hydroxypropylmethylcellulose |
| IPM | isopropyl myristate |
| MBA | N,N-methylenebis (acrylamide) |
| NA | not available |
| PEI | polyethylenimine |
| PC | phosphatidylcholine |
| Plys | ε-polylysine |
| PNP | polymeric nanoparticle |
| SA | stearyl amine |
| SEDDS | self-emulsifying drug delivery system |
| SLN | solid lipid nanoparticles |
| SNEDDS | self-nanoemulsifying drug delivery system |
| TEA | triethylamine |
References
- Patel, V.F.; Liu, F.; Brown, M.B. Advances in oral transmucosal drug delivery. J. Control. Release 2011, 153, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aláez-Versón, C.R.; Lantero, E.; Fernàndez-Busquets, X. Heparin: New life for an old drug. Nanomedicine 2017, 12, 1727–1744. [Google Scholar] [CrossRef] [PubMed]
- Motlekar, N.A.; Youan, B.-B.C. The quest for non-invasive delivery of bioactive macromolecules: A focus on heparins. J. Control. Release 2006, 113, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das Kurmi, B.; Tekchandani, P.; Paliwal, R.; Rai Paliwal, S. Nanocarriers in Improved Heparin Delivery: Recent Updates. Curr. Pharm. Des. 2015, 21, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yeo, Y. Controlled drug release from pharmaceutical nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Wang, X.; Liu, C.; Zhang, X.; Zhang, X.; Chen, X.; Kou, Y.; Mao, S. Chitosan based polymer-lipid hybrid nanoparticles for oral delivery of enoxaparin. Int. J. Pharm. 2018, 547, 499–505. [Google Scholar] [CrossRef]
- Charoongchit, P.; Suksiriworapong, J.; Mao, S.; Sapin-Minet, A.; Maincent, P.; Junyaprasert, V.B. Investigation of cationized triblock and diblock poly(ε-caprolactone)-co-poly(ethylene glycol) copolymers for oral delivery of enoxaparin: In vitro approach. Acta Biomater. 2017, 61, 180–192. [Google Scholar] [CrossRef]
- Rawat, A.; Majumder, Q.H.; Ahsan, F. Inhalable large porous microspheres of low molecular weight heparin: In vitro and in vivo evaluation. J. Control. Release 2008, 128, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Gupta, V.; Ahsan, F. PEG–PLGA based large porous particles for pulmonary delivery of a highly soluble drug, low molecular weight heparin. J. Control. Release 2012, 162, 310–320. [Google Scholar] [CrossRef]
- Loira-Pastoriza, C.; Sapin-Minet, A.; Diab, R.; Grossiord, J.L.; Maincent, P. Low molecular weight heparin gels, based on nanoparticles, for topical delivery. Int. J. Pharm. 2012, 426, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Walenga, J.; Hoppensteadt, D. Monitoring the new antithrombotic drugs. Semin. Thromb. Hemost. 2005, 30, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Samama Meyer, M.; Guinet, C. Laboratory assessment of new anticoagulants. Clin. Chem. Lab. Med. 2011, 49, 761–772. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Torri, G.; Naggi, A. Heparin centenary—An ever-young life-saving drug. Int. J. Cardiol. 2016, 212, S1–S4. [Google Scholar] [CrossRef] [Green Version]
- Onishi, A.; St Ange, K.; Dordick, J.S.; Linhardt, R.J. Heparin and anticoagulation. Front. Biosci. (Landmark Ed). 2016, 21, 1372–1392. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Mulloy, B.; Barrowcliffe, T.W. Heparin and low-molecular-weight heparin. Thromb. Haemost. 2008, 99, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harter, K.; Levine, M.; Henderson, S.O. Anticoagulation drug therapy: A review. West. J. Emerg. Med. 2015, 16, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Vene, N.; Mavri, A. An overview of the anticoagulant drugs used in routine clinical practice. In Anticoagulant Drugs; Božič-Mijovski, M., Ed.; IntechOpen: London, UK, 2018; p. 10. [Google Scholar] [CrossRef]
- Casu, B.; Oreste, P.; Torri, G.; Zoppetti, G.; Choay, J.; Lormeau, J.C.; Petitou, M.; Sinaɕ, P. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Biochem. J. 1981, 197, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Bäckström, G.; Höök, M.; Thunberg, L.; Fransson, L.-A.; Linker, A. Structure of the antithrombin-binding site in heparin. Proc. Natl. Acad. Sci. USA 1979, 76, 3198–3202. [Google Scholar] [CrossRef] [Green Version]
- Cosmi, B.; Palareti, G. Old and new heparins. Thromb. Res. 2012, 129, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K. Low- and ultra-low-molecular-weight heparins. Best Pract. Res. Clin. Haematol. 2004, 17, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.S.; Osman, R.; Awad, G.A.S.; Mortada, N.D.; Geneidy, A.-S. Low molecular weight heparins for current and future uses: Approaches for micro- and nano-particulate delivery. Drug. Deliv. 2016, 23, 2661–2667. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Byun, Y. Recent advances in anticoagulant drug delivery. Expert Opin. Drug Deliv. 2016, 13, 421–434. [Google Scholar] [CrossRef]
- Merli, G.J.; Vanscoy, G.J.; Rihn, T.L.; Groce Iii, J.B.; McCormick, W. Applying scientific citeria to therapeutic interchange: A balanced analysis of low-molecular-weight heparins. J. Thromb. Thrombolysis 2001, 11, 247–259. [Google Scholar] [CrossRef]
- Hirsh, J.; O’Donnell, M.; Eikelboom, J.W. Beyond unfractionated heparin and warfarin. Circulation 2007, 116, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Walenga, J.M.; Lyman, G.H. Evolution of heparin anticoagulants to ultra-low-molecular-weight heparins: A review of pharmacologic and clinical differences and applications in patients with cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 1–18. [Google Scholar] [CrossRef]
- Masuko, S.; Linhardt, R.J. Chemoenzymatic synthesis of the next generation of ultralow MW heparin therapeutics. Future Med. Chem. 2012, 4, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.A. Fondaparinux sodium: A selective inhibitor of factor Xa. Am. J. Health Syst. Pharm. 2001, 58, S14–S17. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Lecumberri, R.; Rocha, E.; Pozo-Hernández, C.; Vargas-Castrillón, E. New parenteral anticoagulants in development. Ther. Adv. Cardiovasc. Dis. 2011, 5, 33–59. [Google Scholar] [CrossRef]
- Savi, P.; Herault, J.P.; Duchaussoy, P.; Millet, L.; Schaeffer, P.; Petitou, M.; Bono, F.; Herbert, J.M. Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy. J. Thromb. Haemost. 2008, 6, 1697–1706. [Google Scholar] [CrossRef]
- Paty, I.; Trellu, M.; Destors, J.-M.; Cortez, P.; Boëlle, E.; Sanderink, G. Reversibility of the anti-FXa activity of idrabiotaparinux (biotinylated idraparinux) by intravenous avidin infusion. J. Thromb. Haemost. 2010, 8, 722–729. [Google Scholar] [CrossRef]
- Falkon, L.; Garí, M.; Barbanoj, M.; Amiral, J.; Fontcuberta, J. Tissue factor pathway inhibitor and anti-FXa kinetic profiles of a new low-molecular-mass heparin, bemiparin, at therapeutic subcutaneous doses. Blood Coagul. Fibrinolysis 1998, 9, 137–141. [Google Scholar] [CrossRef]
- Ciccone, M.M.; Cortese, F.; Corbo, F.; Corrales, N.E.; Al-Momen, A.K.; Silva, A.; Zito, A.; Pinto, M.; Gesualdo, M.; Scicchitano, P. Bemiparin, an effective and safe low molecular weight heparin: A review. Vasc. Pharmacol. 2014, 62, 32–37. [Google Scholar] [CrossRef]
- Akhtar, F.; Wan, X.; Wu, G.; Kesse, S.; Wang, S.; He, S. Low-Molecular-Weight Heparins: Reduced Size Particulate Systems for Improved Therapeutic Outcomes. Molecules 2018, 23, 1757. [Google Scholar] [CrossRef] [Green Version]
- Schluter, A.; Lamprecht, A. Current Developments for the Oral Delivery of Heparin. Curr. Pharm. Biotechnol. 2014, 15, 640–649. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Kamali, F.; Daly, A.K.; Wadelius, M. Oral anticoagulation: A critique of recent advances and controversies. Trends Pharm. Sci. 2015, 36, 153–163. [Google Scholar] [CrossRef]
- Ferreira, J.L.; Wipf, J.E. Pharmacologic therapies in anticoagulation. Med. Clin. N. Am. 2016, 100, 695–718. [Google Scholar] [CrossRef]
- Ibrahim, T.F.; Maxwell, S.; Iqbal, O. Current anticoagulation drugs and mechanisms of action. In Anticoagulation and Hemostasis in Neurosurgery; Loftus, C.M., Ed.; Springer: Cham, Switzerland, 2016; pp. 33–46. [Google Scholar] [CrossRef]
- Hirsh, J. Oral anticoagulant drugs. N. Engl. J. Med. 1991, 324, 1865–1875. [Google Scholar] [CrossRef]
- Ageno, W.; Gallus, A.S.; Wittkowsky, A.; Crowther, M.; Hylek, E.M.; Palareti, G. Oral anticoagulant therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 2012, 141, e44S–e88S. [Google Scholar] [CrossRef] [Green Version]
- Wadelius, M.; Pirmohamed, M. Pharmacogenetics of warfarin: Current status and future challenges. Pharmacogenomics J. 2006, 7, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Ufer, M. Comparative pharmacokinetics of vitamin K antagonists. Clin. Pharmacokinet. 2005, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Simon, T. Pharmacology of antithrombotic drugs: An assessment of oral antiplatelet and anticoagulant treatments. Lancet 2015, 386, 281–291. [Google Scholar] [CrossRef]
- Wysowski, D.K.; Nourjah, P.; Swartz, L. Bleeding complications with warfarin use: A prevalent adverse effect resulting in regulatory action. Arch. Intern. Med. 2007, 167, 1414–1419. [Google Scholar] [CrossRef]
- Franchini, M.; Liumbruno, G.M.; Bonfanti, C.; Lippi, G. The evolution of anticoagulant therapy. Blood. Transfus. 2016, 14, 175–184. [Google Scholar] [CrossRef]
- Riva, N.; Ageno, W. Pros and cons of vitamin K antagonists and non–vitamin K antagonist oral anticoagulants. Semin. Thromb. Hemost. 2015, 41, 178–187. [Google Scholar] [CrossRef]
- Schulman, S. Advantages and limitations of the new anticoagulants. J. Intern. Med. 2014, 275, 1–11. [Google Scholar] [CrossRef]
- Bauer, K.A. Pros and cons of new oral anticoagulants. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Stangier, J. Clinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilate. Clin. Pharmacokinet. 2008, 47, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zacconi, F.C. FXa direct synthetic inhibitors. In Anticoagulant Drugs; Božič-Mijovski, M., Ed.; IntechOpen: London, UK, 2018; pp. 11–37. [Google Scholar] [CrossRef]
- Hinojar, R.; Jiménez-Natcher, J.J.; Fernández-Golfín, C.; Zamorano, J.L. New oral anticoagulants: A practical guide for physicians. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 1, 134–145. [Google Scholar] [CrossRef]
- Laux, V.; Perzborn, E.; Kubitza, D.; Misselwitz, F. Preclinical and clinical characteristics of rivaroxaban: A novel, oral, direct factor Xa inhibitor. Semin. Thromb. Hemost. 2007, 33, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; He, B.; Grace, J.E.; Xin, B.; Zhang, D.; Pinto, D.J.; Luettgen, J.M.; Knabb, R.M.; Lam, P.Y.S.; Wexler, R.R.; et al. Preclinical pharmacokinetic and metabolism of apixaban, a potent and selective factor Xa inhibitor. Blood 2006, 108, 910. [Google Scholar] [CrossRef]
- Minguet, J.; Sims, H.M.; Smith, K.H.; Bramlage, P. The factor Xa inhibitor edoxaban for the prevention of stroke and systemic embolism in patients with atrial fibrillation. Expert Rev. Clin. Pharmacol. 2017, 10, 5–15. [Google Scholar] [CrossRef]
- Poulakos, M.; Walker, J.N.; Baig, U.; David, T. Edoxaban: A direct oral anticoagulant. Am. J. Health Syst. Pharm. 2017, 74, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Parasrampuria, D.A.; Truitt, K.E. Pharmacokinetics and pharmacodynamics of edoxaban, a non-vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin. Pharmacokinet. 2016, 55, 641–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garland, S.G.; DeRemer, C.E.; Smith, S.M.; Gums, J.G. Betrixaban: A new oral factor Xa inhibitor for extended venous thromboembolism prophylaxis in high-risk hospitalized patients. Ann. Pharmacother. 2018, 52, 554–561. [Google Scholar] [CrossRef]
- Huisman, M.V.; Klok, F.A. Pharmacological properties of betrixaban. Eur. Heart J. Suppl. 2018, 20, E12–E15. [Google Scholar] [CrossRef]
- Thoenes, M.; Minguet, J.; Bramlage, K.; Bramlage, P.; Ferrero, C. Betrixaban—the next direct factor Xa inhibitor? Expert Rev. Hematol. 2016, 9, 1111–1117. [Google Scholar] [CrossRef]
- Cosmi, B. An update on the pharmaceutical management of thrombosis. Expert Opin. Pharmacother. 2016, 17, 2149–2164. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Lecumberri, R.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Direct-acting oral anticoagulants: Pharmacology, indications, management, and future perspectives. Eur. J. Haematol. 2015, 95, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, E.J.; Lippi, G.; Koutts, J. Laboratory testing of anticoagulants: The present and the future. Pathology 2011, 43, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Czuprynska, J.; Patel, J.P.; Arya, R. Current challenges and future prospects in oral anticoagulant therapy. Br. J. Haematol. 2017, 178, 838–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, K.; Lewis, C. Laboratory monitoring of oral vitamin K anticoagulation. Semin. Thromb. Hemost. 2017, 43. [Google Scholar] [CrossRef]
- Langdell, R.D.; Wagner, R.H.; Brinkhous, K.M. Effect of antihemophilic factor on one-stage clotting tests. A presumptive test for hemophilia and a simple one-stage antihemophilic factor assay procedure. J. Lab. Clin. Med. 1953, 41, 637–647. [Google Scholar]
- Kitchens, C.S. To bleed or not to bleed? Is that the question for the PTT? J. Thromb. Haemost. 2005, 3, 2607–2611. [Google Scholar] [CrossRef]
- Chee, Y.L.; Greaves, M. Role of coagulation testing in predicting bleeding risk. Hematol. J. 2003, 4, 373–378. [Google Scholar] [CrossRef]
- Bonhomme, F.; Fontana, P. Laboratory testing of hemostasis. In Perioperative hemostasis; Marcucci, C.E., Schoettker, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 13–24. [Google Scholar] [CrossRef]
- Avecilla, S.T.; Roshal, M.; Reyes Gil, M.; Erdman, P.A. Laboratory monitoring for heparins, fondaparinux, direct thrombin inhibitors, and oral anti-Xa medications. In Transfusion Medicine and Hemostasis, 3rd ed.; Shaz, B.H., Hillyer, C.D., Reyes Gil, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 933–938. [Google Scholar] [CrossRef]
- Cuker, A. Unfractionated heparin for the treatment of venous thromboembolism: Best practices and areas of uncertainty. Semin. Thromb. Hemost. 2012, 38, 593–599. [Google Scholar] [CrossRef]
- Marlar, R.A.; Clement, B.; Gausman, J. Activated partial thromboplastin time monitoring of unfractionated heparin therapy: Issues and recommendations. Semin. Thromb. Hemost. 2017, 43, 253–260. [Google Scholar] [CrossRef]
- Shapiro, G.A.; Huntzinger, S.W.; Wilson, J.E. Variation among commercial activated partial thromboplastin time reagents in response to heparin. Am. J. Clin. Pathol. 1977, 67, 477–480. [Google Scholar] [CrossRef]
- Watson, H.G.; Greaves, M. Can we predict bleeding? Semin. Thromb. Hemost. 2008, 34, 97–103. [Google Scholar] [CrossRef]
- Cuker, A.; Raby, A.; Moffat, K.; Flynn, G.; Crowther, M. Interlaboratory variation in heparin monitoring: Lessons from the quality management program of Ontario coagulation surveys. Thromb. Haemost. 2010, 104, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Babin, J.L.; Traylor, K.; Witt, D. Laboratory monitoring of low-molecular-weight heparin and fondaparinux. Semin. Thromb. Hemost. 2017, 43, 261–269. [Google Scholar] [CrossRef] [PubMed]
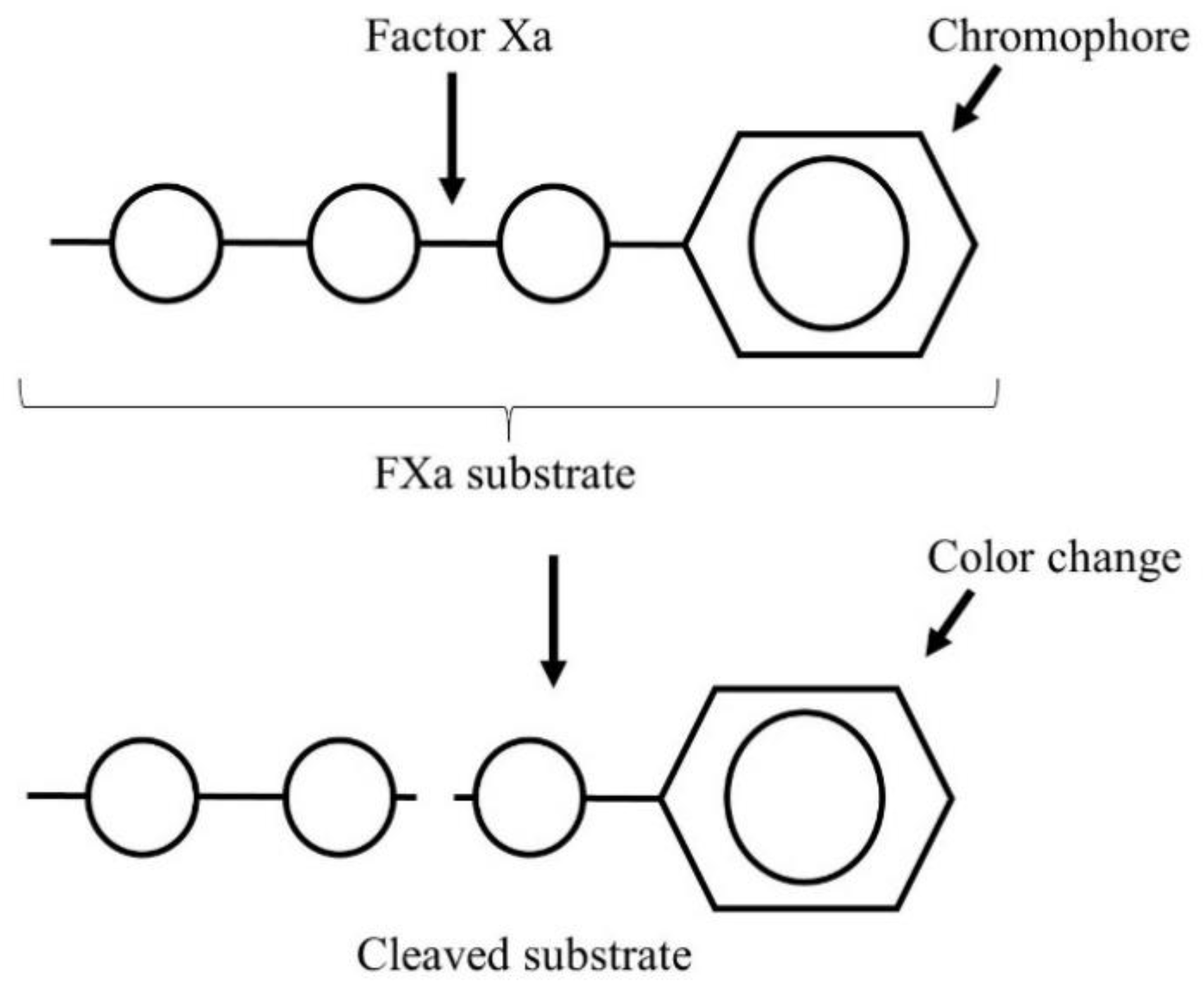
- Gehrie, E.; Laposata, M. Test of the month: The chromogenic antifactor Xa assay. Am. J. Hematol. 2012, 87, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.M.; Weitz, J.I. Coagulation assays. Circulation 2005, 112, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newall, F. Anti-factor Xa (anti-Xa) assay. In Haemostasis. Methods in Molecular Biology (Methods and Protocols); Monagle, P., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 992, pp. 265–272. [Google Scholar]
- Funk, D.M. Coagulation assays and anticoagulant monitoring. Hematol. Am. Soc. Hematol. Educ. Program. 2012, 2012, 460–465. [Google Scholar] [CrossRef]
- Samuelson, B.T.; Cuker, A. Measurement and reversal of the direct oral anticoagulants. Blood Rev. 2017, 31, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Samuelson, B.T.; Cuker, A.; Siegal, D.M.; Crowther, M.; Garcia, D.A. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: A systematic review. Chest 2017, 151, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Eby, C. Novel anticoagulants and laboratory testing. Int. J. Lab. Hematol. 2013, 35, 262–268. [Google Scholar] [CrossRef]
- Adcock, D.M.; Gosselin, R. Direct oral anticoagulants (DOACs) in the laboratory: 2015 review. Thromb. Res. 2015, 136, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, E.M.H.; Boonen, K.; van den Heuvel, D.J.A.; van Dongen, J.L.J.; Schellings, M.W.M.; Emmen, J.M.A.; van der Graaf, F.; Brunsveld, L.; van de Kerkhof, D. Determination of dabigatran, rivaroxaban and apixaban by ultra-performance liquid chromatography—tandem mass spectrometry (UPLC-MS/MS) and coagulation assays for therapy monitoring of novel direct oral anticoagulants. J. Thromb. Haemost. 2014, 12, 1636–1646. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E. Recent guidelines and recommendations for laboratory assessment of the direct oral anticoagulants (DOACs): Is there consensus? Clin. Chem. Lab. Med. 2015, 53, 185–197. [Google Scholar] [CrossRef]
- Dale, B.J.; Chan, N.C.; Eikelboom, J.W. Laboratory measurement of the direct oral anticoagulants. Br. J. Haematol. 2016, 172, 315–336. [Google Scholar] [CrossRef] [Green Version]
- van Ryn, J.; Stangier, J.; Härtter, S.; Liesenfeld, K.-H.; Wienen, W.; Feuring, M.; Clemens, A. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: Interpretation of coagulation assays and reversal of anticoagulant activity. Thromb. Haemost. 2010, 103, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Pollack, C.V. Coagulation assessment with the new generation of oral anticoagulants. Emerg. Med. J. 2016, 33, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douxfils, J.; Mullier, F.; Robert, S.; Chatelain, C.; Chatelain, B.; Dogne, J.M. Impact of dabigatran on a large panel of routine or specific coagulation assays. Laboratory recommendations for monitoring of dabigatran etexilate. Thromb. Haemost. 2012, 107, 985–997. [Google Scholar] [CrossRef]
- Nowak, G. The ecarin clotting time, a universal method to quantify direct thrombin inhibitors. Pathophysiol. Haemost. Thromb. 2003, 33, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, R.C.; Dwyre, D.M.; Dager, W.E. Measuring dabigatran concentrations using a chromogenic ecarin clotting time assay. Ann. Pharmacother. 2013, 47, 1635–1640. [Google Scholar] [CrossRef]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.H.; Korma, S.A.; Lipeng, Q.; Jinghua, C. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2019, 27, 742–761. [Google Scholar] [CrossRef]
- Liu, G.; Hou, S.; Tong, P.; Li, J. Liposomes: Preparation, Characteristics, and Application Strategies in Analytical Chemistry. Crit. Rev. Anal. Chem. 2020, 1–21. [Google Scholar] [CrossRef]
- Song, Y.-K.; Hyun, S.Y.; Kim, H.-T.; Kim, C.-K.; Oh, J.-M. Transdermal delivery of low molecular weight heparin loaded in flexible liposomes with bioavailability enhancement: Comparison with ethosomes. J. Microencapsul. 2011, 28, 151–158. [Google Scholar] [CrossRef]
- Bai, S.; Ahsan, F. Inhalable liposomes of low molecular weight heparin for the treatment of venous thromboembolism. J. Pharm. Sci. 2010, 99, 4554–4564. [Google Scholar] [CrossRef] [PubMed]
- Croisfelt, F.M.; Tundisi, L.L.; Ataide, J.A.; Silveira, E.; Tambourgi, E.B.; Jozala, A.F.; Souto, E.M.B.; Mazzola, P.G. Modified-release topical hydrogels: A ten-year review. J. Mater. Sci. 2019, 54, 10963–10983. [Google Scholar] [CrossRef]
- Narayanaswamy, R.; Torchilin, V.P. Hydrogels and Their Applications in Targeted Drug Delivery. Molecules (Basel, Switzerland) 2019, 24, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, S.; Nair, A.B.; Shah, J.; Sreeharsha, N.; Gupta, S.; Shinu, P. Emerging Role of Hydrogels in Drug Delivery Systems, Tissue Engineering and Wound Management. Pharmaceutics 2021, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Matanović, M.R.; Grabnar, P.A.; Voinovich, D.; Golob, S.; Mijovski, M.B.; Grabnar, I. Development and preclinical pharmacokinetics of a novel subcutaneous thermoresponsive system for prolonged delivery of heparin. Int. J. Pharm. 2015, 496, 583–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, S.; Rathore, A.; Dave, V.; Reddy, K.R.; Chouhan, R.S.; Sadhu, V. Recent developments in functionalized polymer nanoparticles for efficient drug delivery system. Nano-Struct. Nano-Objects 2019, 20, 100397. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharma. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Ramadan, A.; Lagarce, F.; Tessier-Marteau, A.; Thomas, O.; Legras, P.; Macchi, L.; Saulnier, P.; Benoit, J.-P. Oral fondaparinux: Use of lipid nanocapsules as nanocarriers and in vivo pharmacokinetic study. Int. J. Nanomed. 2011, 6, 2941–2951. [Google Scholar] [CrossRef] [Green Version]
- Souto, E.B.; Baldim, I.; Oliveira, W.P.; Rao, R.; Yadav, N.; Gama, F.M.; Mahant, S. SLN and NLC for topical, dermal and transdermal drug delivery. Expert Opin. Drug Deliv. 2020, 17, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Mahant, S.; Rao, R.; Souto, E.B.; Nanda, S. Analytical tools and evaluation strategies for nanostructured lipid carrier based topical delivery systems. Expert Opin. Drug Deliv. 2020. [Google Scholar] [CrossRef]
- Paliwal, R.; Paliwal, S.R.; Agrawal, G.P.; Vyas, S.P. Biomimetic solid lipid nanoparticles for oral bioavailability enhancement of low molecular weight heparin and its lipid conjugates: In vitro and in vivo evaluation. Mol. Pharm. 2011, 8, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Rehman, F.U.; Shah, K.U.; Shah, S.U.; Khan, I.U.; Khan, G.M.; Khan, A. From nanoemulsions to self-nanoemulsions, with recent advances in self-nanoemulsifying drug delivery systems (SNEDDS). Expert Opin. Drug Deliv. 2017, 14, 1325–1340. [Google Scholar] [CrossRef] [PubMed]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef]
- Zupančič, O.; Grieβinger, J.A.; Rohrer, J.; Pereira de Sousa, I.; Danninger, L.; Partenhauser, A.; Sündermann, N.E.; Laffleur, F.; Bernkop-Schnürch, A. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur. J. Pharm. Biopharm. 2016, 109, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, N.; Muzib, Y.I.; Aukunuru, J.; Balekari, U. Preparation and evaluation of a novel oral delivery system for low molecular weight heparin. Int. J. Pharm. Investig. 2016, 6, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, G.; Zhou, J.; Qian, Y.; Gou, J.; Yang, X.; Tang, B. Development and evaluation of thermo-sensitive hydrogel system with nanocomplexes for prolonged subcutaneous delivery of enoxaparin. J. Drug Deliv. Sci. Technol. 2018, 48, 118–124. [Google Scholar] [CrossRef]
- Gritsch, L.; Motta, F.L.; Contessi Negrini, N.; Yahia, L.H.; Farè, S. Crosslinked gelatin hydrogels as carriers for controlled heparin release. Mater. Lett. 2018, 228, 375–378. [Google Scholar] [CrossRef]
- Eleraky, N.E.; Swarnakar, N.K.; Mohamed, D.F.; Attia, M.A.; Pauletti, G.M. Permeation-Enhancing Nanoparticle Formulation to Enable Oral Absorption of Enoxaparin. AAPS PharmSciTech 2020, 21, 88. [Google Scholar] [CrossRef]
- Tang, B.; Qian, Y.; Fang, G. Development of Lipid-Polymer Hybrid Nanoparticles for Improving Oral Absorption of Enoxaparin. Pharmaceutics 2020, 12, 607. [Google Scholar] [CrossRef] [PubMed]
- Soltani, Y.; Goodarzi, N.; Mahjub, R. Preparation and characterization of self nano-emulsifying drug delivery system (SNEDDS) for oral delivery of heparin using hydrophobic complexation by cationic polymer of β-cyclodextrin. Drug Dev. Ind. Pharm. 2017, 43, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Cao, M.; Ren, L.; Qian, Y.; Chen, G. Preparation and Optimization of Rivaroxaban by Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Enhanced Oral Bioavailability and No Food Effect. AAPS PharmSciTech 2018, 19, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Abouhussein, D.M.N.; Bahaa El Din Mahmoud, D.; Mohammad, F.E. Design of a liquid nano-sized drug delivery system with enhanced solubility of rivaroxaban for venous thromboembolism management in paediatric patients and emergency cases. J. Liposome Res. 2019, 29, 399–412. [Google Scholar] [CrossRef]



| Features | Factor IIa Inhibitor | Factor Xa Inhibitor | References | |||
|---|---|---|---|---|---|---|
| Dabigatran | Apixaban | Rivaroxaban | Edoxaban | Betrixaban | ||
| Mw a (Da b) | 628 | 460 | 436 | 548 | 452 | [62] |
| Bioavailability (%) | 6 | 50 | 80–100 | 62 | 34 | [62,63] |
| Protein binding (%) | 35 | 87 | 92–95 | 55 | 60 | [61,63] |
| Tmax c (h) | 1–3 | 1–4 | 2–4 | 1–2 | 3–4 | [61] |
| Half-life (h) | 12–17 | 12 | 5–13 | 9–11 | 19–27 | [53,60] |
| Reversal agents | Yes | No | No | No | No | [62] |
| Renal clearance (%) | >80 | 25 | 66 | 35–50 | <7 | [61] |
| Drug Delivery System | Active (Class) | Composition | Size Range | Major Outcomes | Ref |
|---|---|---|---|---|---|
| Liposomes | (LMWH) | Egg PC Tween® 20 Ethanol | 80–90 nm | LMWH-loaded flexosomes showed higher antifactor Xa (Anti-Xa) max than LMWH-loaded ethosome. | [96] |
| Ardeparin (LMWH) | PC Chol DSPE DSPE-PEG-2000 and DSPE–PEG-5000 | 100–150 nm | Liposomal formulations showed sustained release and longer half-life compared to the plain solution or subcutaneous route. | [97] | |
| Enoxaparin (LMWH) | Soybean PC Chol SA Eudragit® S 100 | 100–200 nm | Eudragit-coated liposomes showed higher permeation and oral bioavailability when compared to uncoated liposomes. | [113] | |
| Hydrogels | Bemiparin (LMWH) Nadroparin (LMWH) Tinzaparin (LMWH) | Eudragit® RS 30D | 130 nm | Gel formulations were able to deliver LMWHs across the skin barrier, and after 24 h, the drug was not detected in plasma. | [11] |
| Heparin (UFH) | CS hydrochloride Lutrol® F127 Lutrol® F68 HPMC | 150–400 nm | The dual system enabled the lowest absorption rate of heparin into systemic circulation and provided heparin concentrations above the prophylaxis threshold for 5 days. | [101] | |
| Enoxaparin (LMWH) | CS Pluronic® F127 PEI Plys | 100–1000 nm | Thermo-sensitive hydrogels were able to prolong the enoxaparin release. | [114] | |
| Heparin (UFH) | Gelatin MBA TEA | NA | Heparin-loaded hydrogels showed sustained release for 60 h and platelet adhesion was significantly reduced. | [115] | |
| PNP | Enoxaparin (LMWH) | CS GMO Pluronic® F127 | 290–320 nm | The optimized formulation showed higher oral bioavailability compared with the drug solution. | [7] |
| Fondaparinux (Synthetic analogues) | Labrafac® WL 1349 Lipoïd® S75-3 Lipoïd® GMBH Solutol® HS 15 CTAB SA | 40–65 nm | Cationic lipid nanocapsules showed increased oral bioavailability and longer half-life when compared to fondaparinux control solutions (oral and intravenous). | [106] | |
| Enoxaparin (LMWH) | Pluronic® F-68 CTAB Dextran sulfate PLGA Precirol ATO 5 E80 Tween 80 Poloxamer 407 | 180–195 nm | Enoxaparin/CTAB nanoparticles showed three-fold improved gastrointestinal permeation when compared with the drug solution. | [116] | |
| Enoxaparin (LMWH) | 145–160 nm | LPHNs improved the drug’s intestinal permeation, enhanced the oral bioavailability, and showed therapeutic efficacy. | [117] | ||
| SLN | (LMWH) | Compritol 888 ATO Stearic, palmitic and myristic acid PC | 280–380 nm | The SLNs were able to improve the LMWH bioavailability in comparison to the free drug solution. | [109] |
| SNEDDDS | Enoxaparin (LMWH) | Capmul MCM EP, Capmul PG-8 EP/NF Captex 8000, Peceol Labrafil M 1944 CS, Labrasol Maisine 35-1, Transcutol HP Myglyol 840, Cremophor EL PEG, triacetin, olive and sesame oil | 30–245 nm | SEDDS formulations showed sustained enoxaparin release and two-fold bioavailability. | [118] |
| Rivaroxaban (Factor Xa inhibitor) | IPM Ethyl oleate, Tween20, and Tween80 Cremophor, Cremophor HEL, and Transutol | 50–105 nm | SNEDDS showed higher dissolution than the commercial formulation. The SNEDDS technology used in rivaroxaban successfully enhanced drug bioavailability in fasting conditions, and no food effects were observed in the rivaroxaban–SNEDDS formulation. | [119] | |
| Rivaroxaban (Factor Xa inhibitor) | Transcutol HP Capryol TM 90 Maisine TM 35-1 Castor oil, oleic acid, triacetin, IPM Cremophore EL PEG 300 and PEG 400 Tween 20, Tween 80, and Span 80 | 10–115 nm | Safe SNEDDS formulations enhanced oral and intravenous bioavailability in comparison to the drug suspension. Moreover, SNEDDS exhibited anticoagulant efficacy in a rat thrombosis model. | [120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patriota, Y.B.G.; Chaves, L.L.; Gocke, E.H.; Severino, P.; Soares, M.F.R.; Soares-Sobrinho, J.L.; Souto, E.B. Applied Nanotechnologies in Anticoagulant Therapy: From Anticoagulants to Coagulation Test Performance of Drug Delivery Systems. Appl. Nano 2021, 2, 98-117. https://0-doi-org.brum.beds.ac.uk/10.3390/applnano2020009
Patriota YBG, Chaves LL, Gocke EH, Severino P, Soares MFR, Soares-Sobrinho JL, Souto EB. Applied Nanotechnologies in Anticoagulant Therapy: From Anticoagulants to Coagulation Test Performance of Drug Delivery Systems. Applied Nano. 2021; 2(2):98-117. https://0-doi-org.brum.beds.ac.uk/10.3390/applnano2020009
Chicago/Turabian StylePatriota, Yuri B. G., Luíse L. Chaves, Evren H. Gocke, Patricia Severino, Mônica F. R. Soares, José L. Soares-Sobrinho, and Eliana B. Souto. 2021. "Applied Nanotechnologies in Anticoagulant Therapy: From Anticoagulants to Coagulation Test Performance of Drug Delivery Systems" Applied Nano 2, no. 2: 98-117. https://0-doi-org.brum.beds.ac.uk/10.3390/applnano2020009