
Seasonal Aerosol Acidity, Liquid Water Content and Their Impact on Fine Urban Aerosol in SE Canada


Abstract
:1. Introduction
2. Methods
2.1. Study Location
2.2. PM2.5 Sample Collection and Analysis
2.3. Estimation of Aerosol pH
3. Results and Discussion
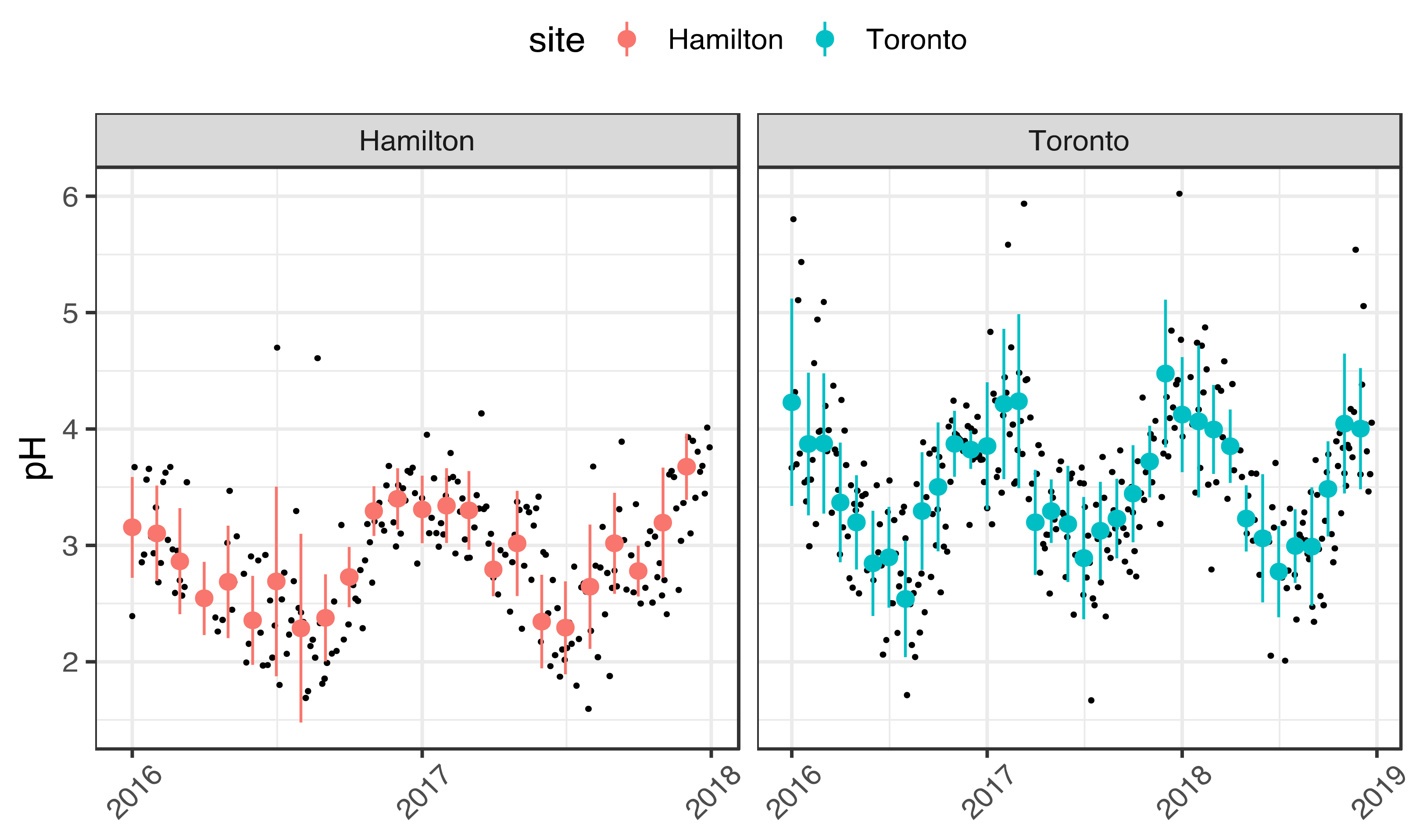
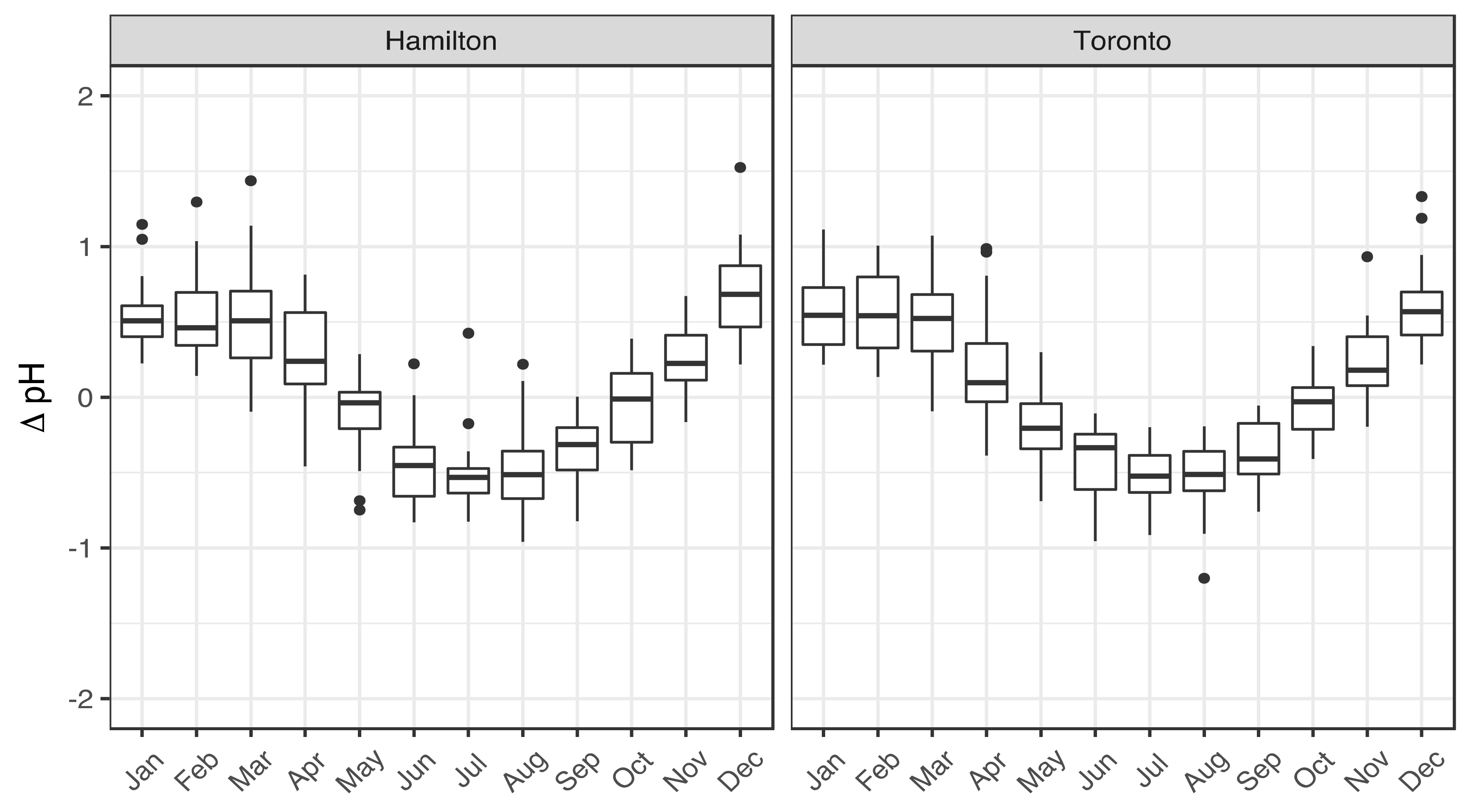
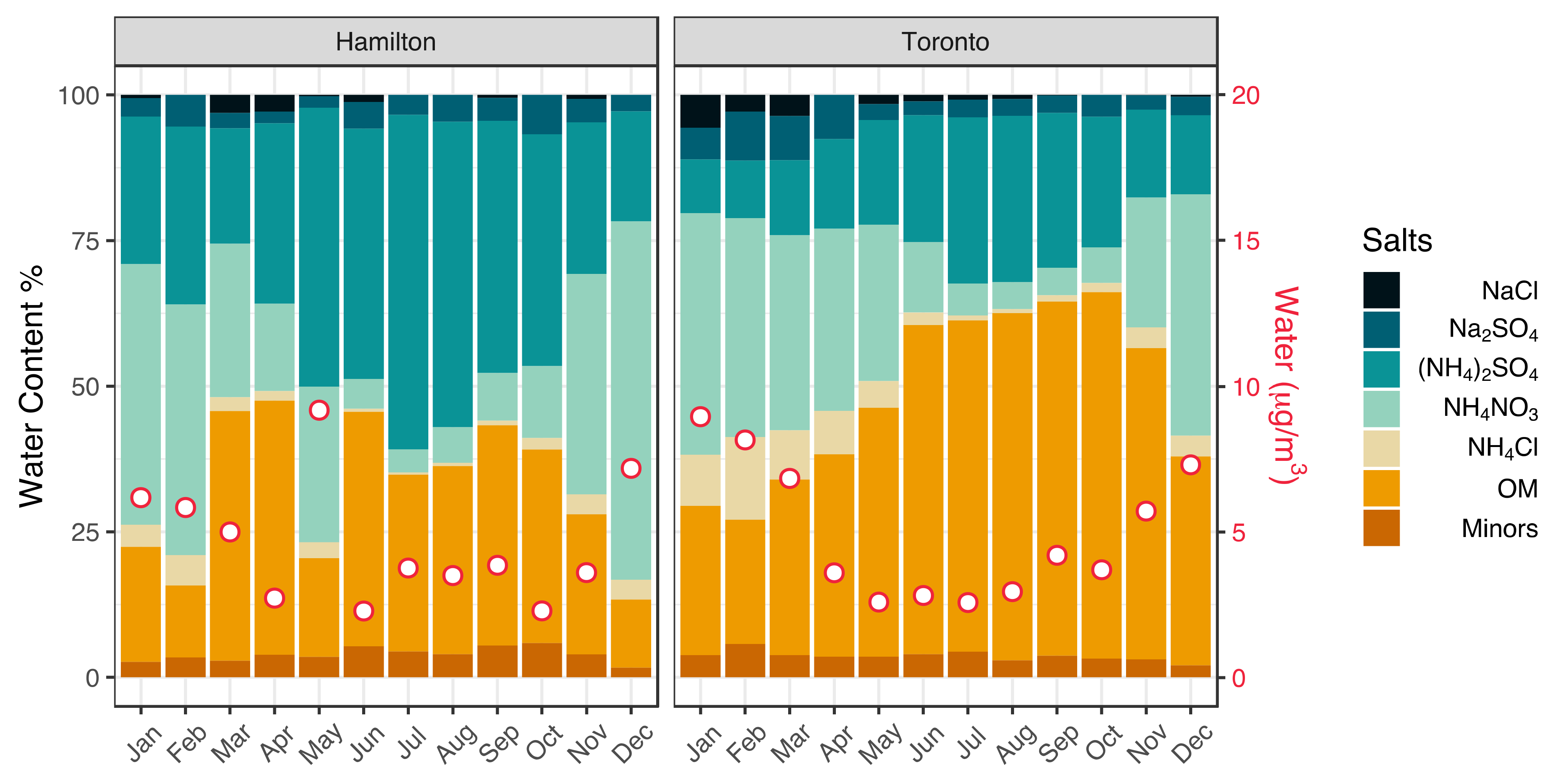
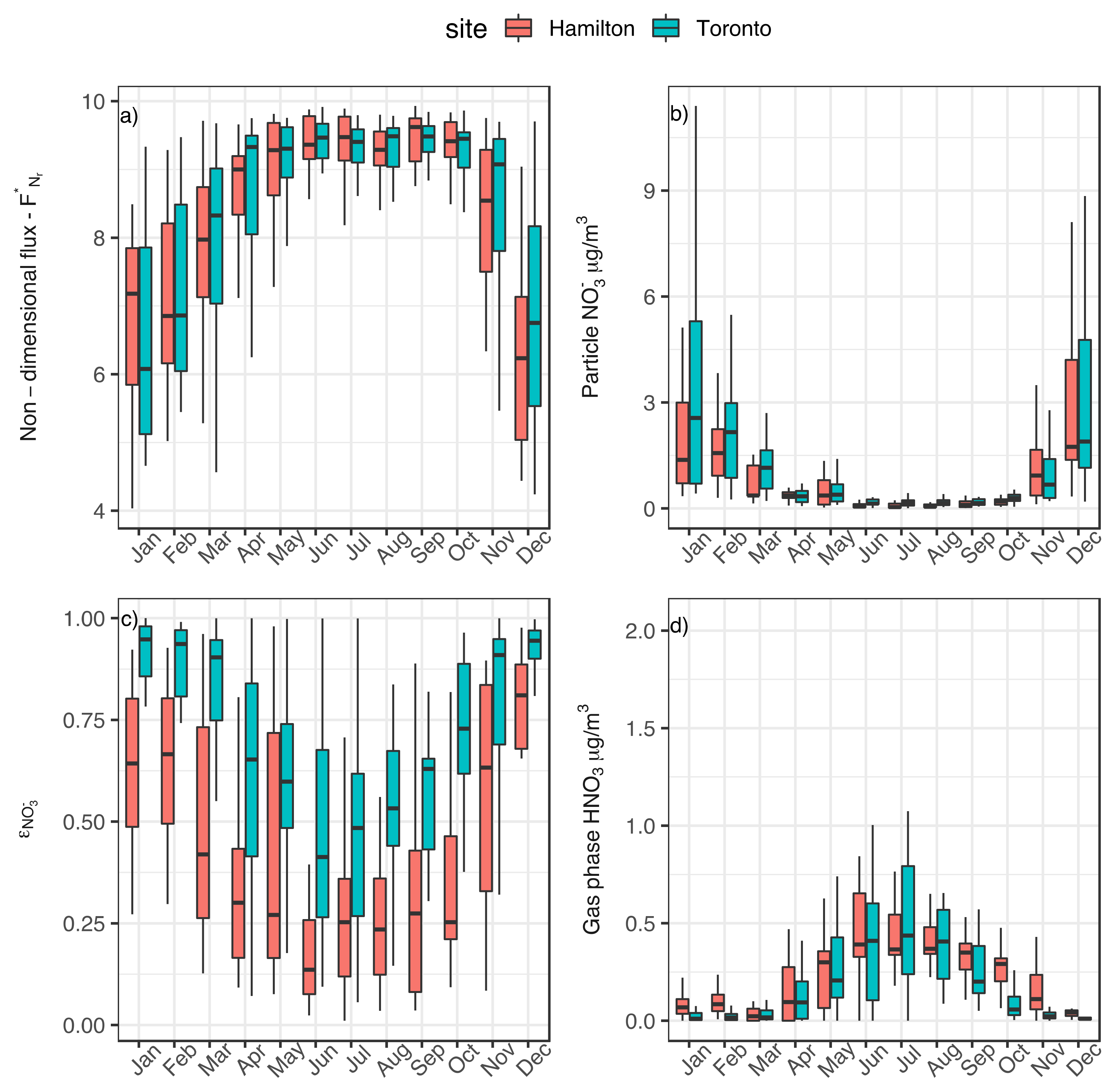
3.1. Seasonality of Aerosol Acidity
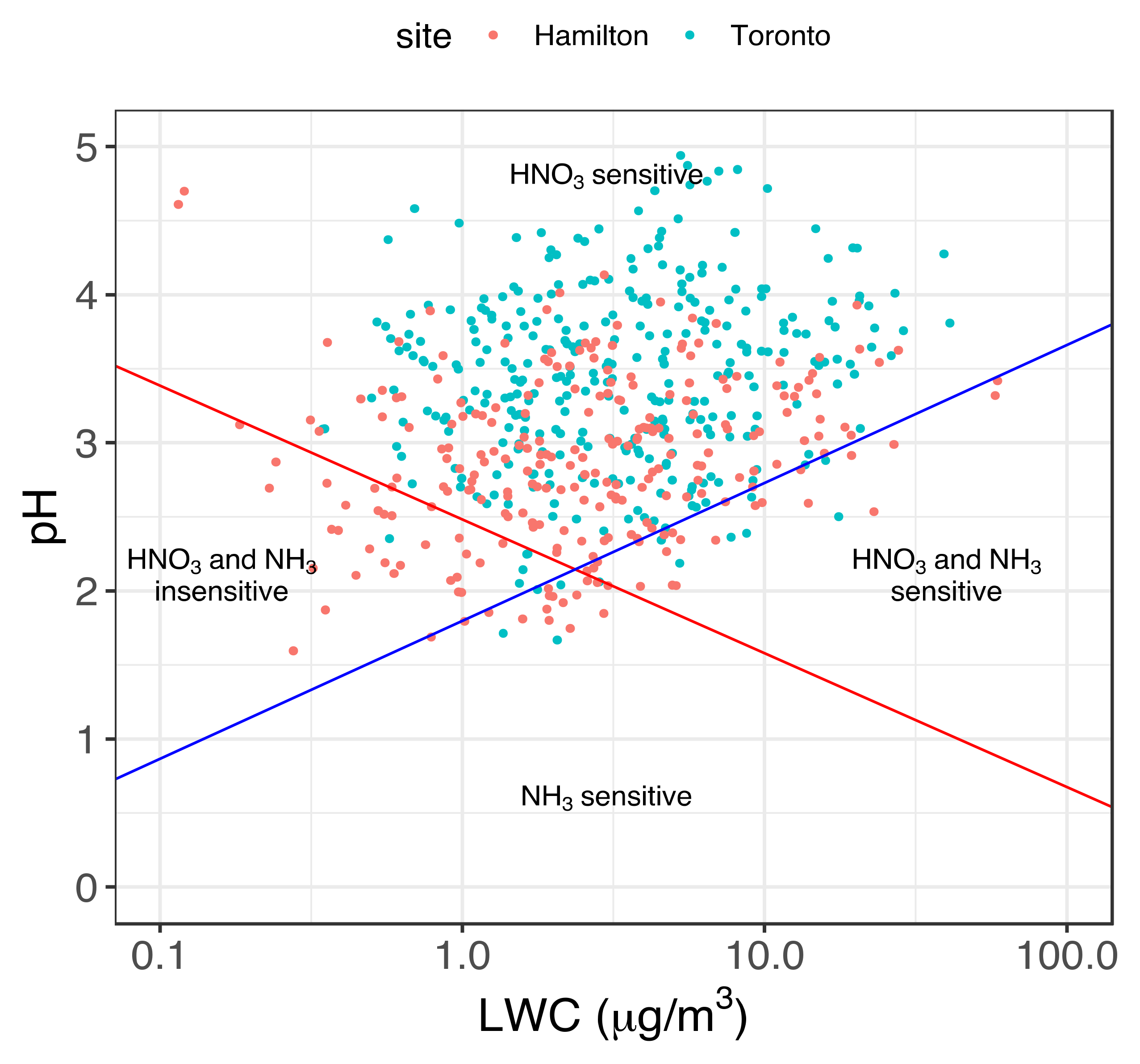
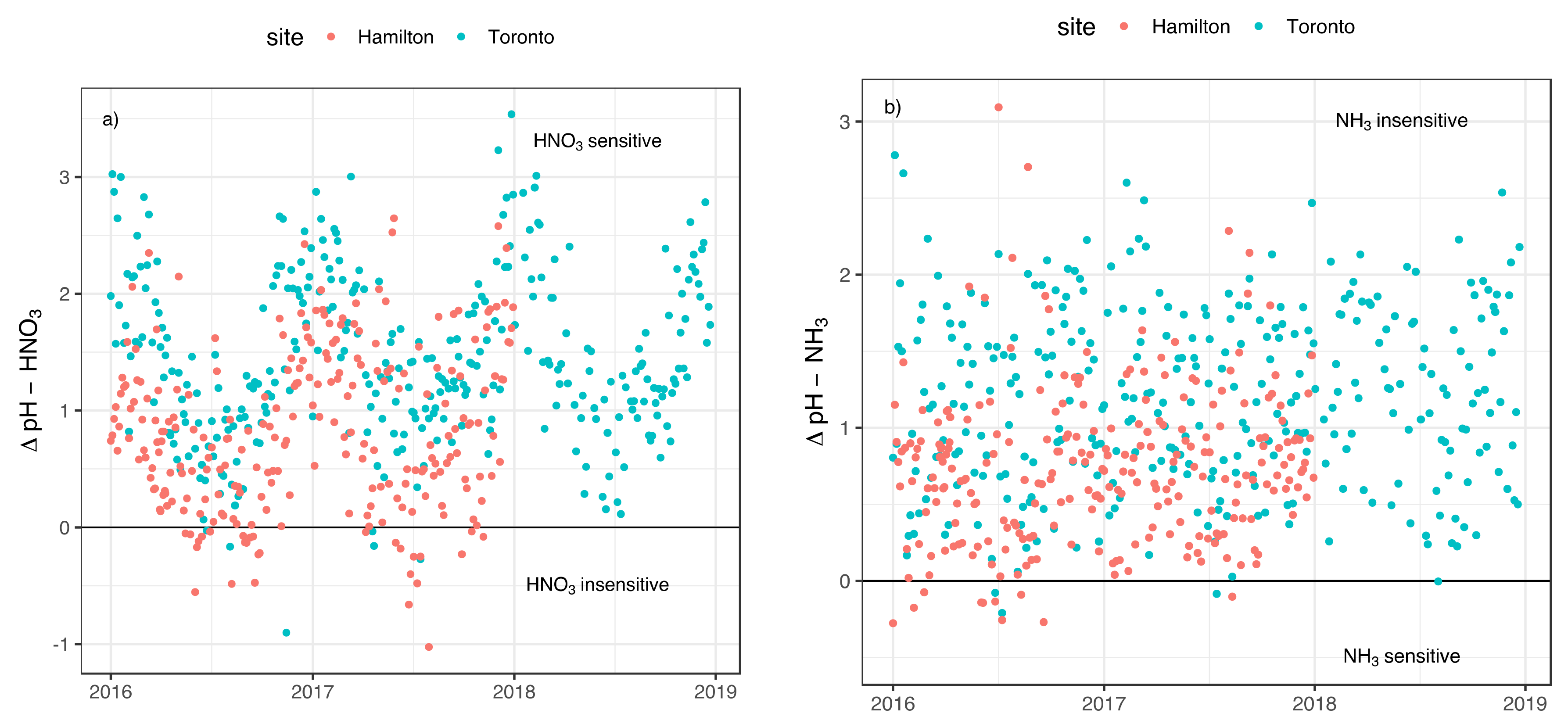
3.2. Implication of Aerosol Acidity Variability on Nitrogen Dry Deposition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, T.; Guo, H.; Zeng, L.; Verma, V.; Nenes, A.; Weber, R.J. Highly Acidic Ambient Particles, Soluble Metals, and Oxidative Potential: A Link between Sulfate and Aerosol Toxicity. Environ. Sci. Technol. 2017, 51, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Shahpoury, P.; Zhang, Z.W.; Arangio, A.; Celo, V.; Dabek-Zlotorzynska, E.; Harner, T.; Nenes, A. The Influence of Chemical Composition, Aerosol Acidity, and Metal Dissolution on the Oxidative Potential of Fine Particulate Matter and Redox Potential of the Lung Lining Fluid. Environ. Int. 2021, 148, 106343. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Murphy, J.G. The Sensitivity of PM2.5 Acidity to Meteorological Parameters and Chemical Composition Changes: 10-Year Records from Six Canadian Monitoring Sites. Atmos. Chem. Phys. 2019, 19, 9309–9320. [Google Scholar] [CrossRef] [Green Version]
- Lawal, A.S.; Guan, X.; Liu, C.; Henneman, L.R.F.; Vasilakos, P.; Bhogineni, V.; Weber, R.J.; Nenes, A.; Russell, A.G. Linked Response of Aerosol Acidity and Ammonia to SO2 and NOx Emissions Reductions in the United States. Environ. Sci. Technol. 2018, 52, 9861–9873. [Google Scholar] [CrossRef]
- Pye, H.O.T.; Nenes, A.; Alexander, B.; Ault, A.P.; Barth, M.C.; Clegg, S.L.; Collett, J.L., Jr.; Fahey, K.M.; Hennigan, C.J.; Herrmann, H.; et al. The Acidity of Atmospheric Particles and Clouds. Atmos. Chem. Phys. 2020, 20, 4809–4888. [Google Scholar] [CrossRef] [Green Version]
- Meskhidze, N. Iron Mobilization in Mineral Dust: Can Anthropogenic SO2 Emissions Affect Ocean Productivity? Geophys. Res. Lett. 2003, 30, 2085. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Xu, L.; Bougiatioti, A.; Cerully, K.M.; Capps, S.L.; Hite, J.R.; Carlton, A.G.; Lee, S.-H.; Bergin, M.H.; Ng, N.L.; et al. Fine-Particle Water and PH in the Southeastern United States. Atmos. Chem. Phys. 2015, 15, 5211–5228. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Weber, R.J.; Nenes, A. High Levels of Ammonia Do Not Raise Fine Particle PH Sufficiently to Yield Nitrogen Oxide-Dominated Sulfate Production. Sci. Rep. 2017, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Nenes, A.; Pandis, S.N.; Weber, R.J.; Russell, A. Aerosol PH and Liquid Water Content Determine When Particulate Matter Is Sensitive to Ammonia and Nitrate Availability. Atmos. Chem. Phys. 2020, 20, 3249–3258. [Google Scholar] [CrossRef] [Green Version]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd ed.; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic Aerosol and Global Climate Modelling: A Review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef] [Green Version]
- Sardar, S.B. Seasonal and Spatial Variability of the Size-Resolved Chemical Composition of Particulate Matter (PM) in the Los Angeles Basin. J. Geophys. Res. 2005, 110, D07S08. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes: Ubiquity and Dominance of Oxigenated OA. Geophys. Res. Lett. 2007, 34, L13801. [Google Scholar] [CrossRef] [Green Version]
- Pinder, R.W.; Adams, P.J.; Pandis, S.N. Ammonia Emission Controls as a Cost-Effective Strategy for Reducing Atmospheric Particulate Matter in the Eastern United States. Environ. Sci. Technol. 2007, 41, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinder, R.W.; Dennis, R.L.; Bhave, P.V. Observable Indicators of the Sensitivity of PM2.5 Nitrate to Emission Reductions—Part I: Derivation of the Adjusted Gas Ratio and Applicability at Regulatory-Relevant Time Scales. Atmos. Environ. 2008, 42, 1275–1286. [Google Scholar] [CrossRef]
- Nenes, A.; Pandis, S.N.; Kanakidou, M.; Russell, A.G.; Song, S.; Vasilakos, P.; Weber, R.J. Aerosol Acidity and Liquid Water Content Regulate the Dry Deposition of Inorganic Reactive Nitrogen. Atmos. Chem. Phys. 2021, 21, 6023–6033. [Google Scholar] [CrossRef]
- Canadian Council of Ministers of the Environment: Ambient Air Monitoring and Quality Assurance/Quality Control Guidelines, National Air Pollution Surveillance Program; Canadian Council of Ministers of the Environment: Ottawa, ON, Canada, 2019.
- Dabek-Zlotorzynska, E.; Dann, T.F.; Kalyani Martinelango, P.; Celo, V.; Brook, J.R.; Mathieu, D.; Ding, L.; Austin, C.C. Canadian National Air Pollution Surveillance (NAPS) PM2.5 Speciation Program: Methodology and PM2.5 Chemical Composition for the Years 2003–2008. Atmos. Environ. 2011, 45, 673–686. [Google Scholar] [CrossRef]
- Yu, X.; Lee, T.; Ayres, B.; Kreidenweis, S.; Malm, W.; Collettjr, J. Loss of Fine Particle Ammonium from Denuded Nylon Filters. Atmos. Environ. 2006, 40, 4797–4807. [Google Scholar] [CrossRef]
- Babich, P.; Davey, M.; Allen, G.; Koutrakis, P. Method Comparisons for Particulate Nitrate, Elemental Carbon, and PM 2.5 Mass in Seven U.S. Cities. J. Air Waste Manag. Assoc. 2000, 50, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Hennigan, C.J.; Izumi, J.; Sullivan, A.P.; Weber, R.J.; Nenes, A. A Critical Evaluation of Proxy Methods Used to Estimate the Acidity of Atmospheric Particles. Atmos. Chem. Phys. 2015, 15, 2775–2790. [Google Scholar] [CrossRef] [Green Version]
- Bougiatioti, A.; Nikolaou, P.; Stavroulas, I.; Kouvarakis, G.; Weber, R.; Nenes, A.; Kanakidou, M.; Mihalopoulos, N. Particle Water and PH in the Eastern Mediterranean: Source Variability and Implications for Nutrient Availability. Atmos. Chem. Phys. 2016, 16, 4579–4591. [Google Scholar] [CrossRef] [Green Version]
- Kakavas, S.; Pandis, S.N.; Nenes, A. ISORROPIA-Lite: A Comprehensive Atmospheric Aerosol Thermodynamics Module for Earth System Models. Tellus B Chem. Phys. Meteorol. 2022, 74, 1–23. [Google Scholar] [CrossRef]
- Fountoukis, C.; Nenes, A. ISORROPIA II: A Computationally Efficient Thermodynamic Equilibrium Model for K+–Ca2+–Mg2+–NH4+–Na+–SO42−–NO3−–Cl−–H2O aerosols. Atmos. Chem. Phys. 2007, 7, 4639–4659. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Gao, M.; Xu, W.; Shao, J.; Shi, G.; Wang, S.; Wang, Y.; Sun, Y.; McElroy, M.B. Fine-Particle PH for Beijing Winter Haze as Inferred from Different Thermodynamic Equilibrium Models. Atmos. Chem. Phys. 2018, 18, 7423–7438. [Google Scholar] [CrossRef] [Green Version]
- Vasilakos, P.; Russell, A.; Weber, R.; Nenes, A. Understanding Nitrate Formation in a World with Less Sulfate. Atmos. Chem. Phys. 2018, 18, 12765–12775. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.A., Jr.; Weber, R.J.; Nenes, A.; Hennigan, C.J. Effects of Water-Soluble Organic Carbon on Aerosol PH. Atmos. Chem. Phys. 2019, 19, 14607–14620. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.A.; Douglas, S.; Hennigan, C.J. Effect of the Urban Heat Island on Aerosol PH. Environ. Sci. Technol. 2017, 51, 13095–13103. [Google Scholar] [CrossRef]
- Farren, N.J.; Davison, J.; Rose, R.A.; Wagner, R.L.; Carslaw, D.C. Underestimated Ammonia Emissions from Road Vehicles. Environ. Sci. Technol. 2020, 54, 15689–15697. [Google Scholar] [CrossRef]
- Arndt, R.L.; Carmichael, G.R.; Streets, D.G.; Bhatti, N. Sulfur Dioxide Emissions and Sectorial Contributions to Sulfur Deposition in Asia. Atmos. Environ. 1997, 31, 1553–1572. [Google Scholar] [CrossRef]
- Garg, A.; Shukla, P.R.; Bhattacharya, S.; Dadhwal, V.K. Sub-Region (District) and Sector Level SO2 and NOx Emissions for India: Assessment of Inventories and Mitigation Flexibility. Atmos. Environ. 2001, 35, 703–713. [Google Scholar] [CrossRef]
- Serbula, S.; Ţivkovic, D.; Radojevic, A.; Kalinovic, T.; Kalinovic, J. Emission of SO2 and SO42− from Copper Smelter and Its Influence on the Level of Total s in Soil and Moss in Bor and the Surroundings. Hem. Ind. 2015, 69, 51–58. [Google Scholar] [CrossRef]
- Duyzer, J. Dry Deposition of Ammonia and Ammonium Aerosols over Heathland. J. Geophys. Res. 1994, 99, 18757. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Season | Hamilton | Toronto | Significance (p-Value) |
|---|---|---|---|---|
| Gas–NH3 (μg m−3) | Winter | 1.17 ± 0.02 | 3.17 ± 0.02 | <0.05 |
| Spring | 2.66 ± 0.05 | 3.93 ± 0.02 | <0.05 | |
| Summer | 3.27 ± 0.02 | 4.66 ± 0.02 | <0.05 | |
| Fall | 3.53 ± 0.09 | 4.68 ± 0.03 | <0.05 | |
| Particle–NH4+ (μg m−3) | Winter | 0.94 ± 0.01 | 1.02 ± 0.01 | 0.50 |
| Spring | 0.71 ± 0.01 | 0.47 ± 0.01 | <0.05 | |
| Summer | 0.42 ± 0.01 | 0.32 ± 0.01 | 0.14 | |
| Fall | 0.42 ± 0.01 | 0.37 ± 0.01 | 0.38 | |
| Total–NH4 (μg m−3) | Winter | 2.10 ± 0.02 | 4.19 ± 0.03 | <0.05 |
| Spring | 3.37 ± 0.06 | 4.40 ± 0.03 | <0.05 | |
| Summer | 3.69 ± 0.02 | 4.99 ± 0.02 | <0.05 | |
| Fall | 3.96 ± 0.09 | 5.05 ± 0.03 | <0.05 | |
| Gas–HNO3 (μg m−3) | Winter | 0.10 ± 0.01 | 0.03 ± 0.001 | <0.05 |
| Spring | 0.24 ± 0.005 | 0.18 ± 0.01 | 0.14 | |
| Summer | 0.46 ± 0.003 | 0.49 ± 0.01 | 0.49 | |
| Fall | 0.26 ± 0.003 | 0.14 ± 0.002 | <0.05 | |
| Particle–NO3− (μg m−3) | Winter | 2.29 ± 0.03 | 2.85 ± 0.03 | 0.15 |
| Spring | 0.92 ± 0.02 | 0.96 ± 0.02 | 0.14 | |
| Summer | 0.09 ± 0.02 | 0.20 ± 0.02 | <0.05 | |
| Fall | 0.54 ± 0.02 | 0.64 ± 0.01 | 0.51 | |
| Total–NO3 (μg m−3) | Winter | 2.39 ± 0.03 | 2.87 ± 0.03 | 0.21 |
| Spring | 1.17 ± 0.02 | 1.14 ± 0.02 | 0.89 | |
| Summer | 0.55 ± 0.003 | 0.70 ± 0.004 | <0.05 | |
| Fall | 0.81 ± 0.01 | 0.78 ± 0.01 | 0.85 | |
| SO42− (μg m−3) | Winter | 1.18 ± 0.01 | 1.09 ± 0.01 | 0.33 |
| Spring | 1.71± 0.03 | 0.98 ± 0.01 | <0.05 | |
| Summer | 1.51 ± 0.02 | 1.12 ± 0.02 | 0.05 | |
| Fall | 1.15 ± 0.02 | 0.90 ± 0.01 | 0.12 | |
| OM (μg m−3) | Winter | 1.68 ± 1.88 | 3.92 ± 0.67 | <0.05 |
| Spring | 1.68 ± 1.97 | 4.56 ± 0.74 | <0.05 | |
| Summer | 2.48 ± 2.37 | 6.91 ± 0.51 | <0.05 | |
| Fall | 1.72 ± 2.62 | 5.43 ± 0.61 | <0.05 | |
| pH | Winter | 3.34 ± 0.37 | 4.08 ± 0.62 | <0.05 |
| Spring | 2.92 ± 0.45 | 3.57 ± 0.59 | <0.05 | |
| Summer | 2.44 ± 0.60 | 2.91 ± 0.47 | <0.05 | |
| Fall | 2.90 ± 0.45 | 3.50 ± 0.53 | <0.05 |
| FNO3T (μmol m−2 s−1) | FNH3T (μmol m−2 s−1) | |||||
|---|---|---|---|---|---|---|
| Toronto | Hamilton | p-Value | Toronto | Hamilton | p-Value | |
| Winter | 30.1 | 40.3 | <0.05 | 50.0 | 23.9 | <0.05 |
| Spring | 22.6 | 24.5 | 0.357 | 54 | 25.8 | <0.05 |
| Summer | 26.2 | 26.5 | 0.889 | 60.6 | 42.4 | <0.05 |
| Fall | 14.0 | 25.4 | <0.05 | 57.5 | 38.1 | <0.05 |
| Average | 23.2 | 29.2 | 55.5 | 32.5 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arangio, A.M.; Shahpoury, P.; Dabek-Zlotorzynska, E.; Nenes, A. Seasonal Aerosol Acidity, Liquid Water Content and Their Impact on Fine Urban Aerosol in SE Canada. Atmosphere 2022, 13, 1012. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos13071012
Arangio AM, Shahpoury P, Dabek-Zlotorzynska E, Nenes A. Seasonal Aerosol Acidity, Liquid Water Content and Their Impact on Fine Urban Aerosol in SE Canada. Atmosphere. 2022; 13(7):1012. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos13071012
Chicago/Turabian StyleArangio, Andrea M., Pourya Shahpoury, Ewa Dabek-Zlotorzynska, and Athanasios Nenes. 2022. "Seasonal Aerosol Acidity, Liquid Water Content and Their Impact on Fine Urban Aerosol in SE Canada" Atmosphere 13, no. 7: 1012. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos13071012