
Pathophysiology of Cerebral Malaria: Implications of MSCs as A Regenerative Medicinal Tool


1
Parasite-Host Biology, National Institute of Malaria Research, New Delhi 110077, India
2
AcSIR, Ghaziabad 201002, India
*
Author to whom correspondence should be addressed.
Bioengineering 2022, 9(6), 263; https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9060263
Submission received: 10 April 2022
/
Revised: 2 May 2022
/
Accepted: 3 May 2022
/
Published: 20 June 2022
(This article belongs to the Special Issue New Sight of Biomaterials and Tissue Regeneration in Medicine Research: Updates and Future Direction)
{kind=link}
{kind=link}
Abstract
:The severe form of malaria, i.e., cerebral malaria caused by Plasmodium falciparum, is a complex neurological syndrome. Surviving persons have a risk of behavioral difficulties, cognitive disorders, and epilepsy. Cerebral malaria is associated with multiple organ dysfunctions. The adhesion and accumulation of infected RBCs, platelets, and leucocytes (macrophages, CD4+ and CD8+ T cells, and monocytes) in the brain microvessels play an essential role in disease progression. Micro-vascular hindrance by coagulation and endothelial dysfunction contributes to neurological damage and the severity of the disease. Recent studies in human cerebral malaria and the murine model of cerebral malaria indicate that different pathogens as well as host-derived factors are involved in brain microvessel adhesion and coagulation that induces changes in vascular permeability and impairment of the blood-brain barrier. Efforts to alleviate blood-brain barrier dysfunction and de-sequestering of RBCs could serve as adjunct therapies. In this review, we briefly summarize the current understanding of the pathogenesis of cerebral malaria, the role of some factors (NK cells, platelet, ANG-2/ANG-1 ratio, and PfEMP1) in disease progression and various functions of Mesenchymal stem cells. This review also highlighted the implications of MSCs as a regenerative medicine.
1. Introduction
Plasmodium falciparum causes over 90 percent of all malaria infections. Children under the age of 5 years and pregnant women were the most susceptible groups affected by malaria. The World Health Organization (WHO) has characterized malaria as severe and uncomplicated. Delays in the detection and treatment of an uncomplicated infection of P. falciparum malaria lead to complications of severe cerebral malaria (CM). CM is usually caused by P. falciparum, but Plasmodium vivax is rarely responsible for CM complications. CM is a severe neurological complication caused by Plasmodium falciparum infection, resulting in high mortality rates. CM is characterized by brain tissue hemorrhage, the accumulation of infected red blood cells and mononuclear cells in brain microvessels, and blood-brain barrier (BBB) disruption. The precise mechanism of BBB disruption and brain injury is still unclear. Understanding the mechanism of brain injury and loss of the BBB is vital for further developments of therapeutic approaches [1]. CM is the leading cause of neural disability in children in the African region. Over 25% of children treated for cerebral malaria suffer from long-term neurocognitive impairment [2]. Main symptoms and risks include intracranial hypertension, hyperglycemia, repeated seizures, and deep coma. Altogether, these factors result in cerebral nervous system dysfunction and death [1]. The severity of cerebral malaria including mortality rate, depth of coma, and neurological symptoms in children differ from the adults. Moreover, intravascular hemolysis, hyperglycemia, and acidosis are common during cerebral malaria in children. On the other hand, cases of vital organ failure in children are less common than adults. Clinical reports suggest the involvement of acute renal failure in mortality; however, persisting renal failure is less reported in children. There have been 15–20% deaths reported in children even after the suggested treatment with anti-malarial drugs, i.e., cinchonoids or artemisinin derivatives. However, treatment with intravenous artesunate results in a lower mortality rate in adults [3].
The BBB separating brain parenchyma from peripheral blood is a semipermeable membrane. The binding of infected RBCs to the brain endothelium cells causes sequestering of infected RBCs in brain microvessels, leading to inflammation and cerebral swelling. Experiments confirm the presence of parasites within brain endothelial cells. The loss of the BBB facilitates Plasmodium falciparum plasma protein and fluid leakage into parenchymal and perivascular space, leading to cerebral vasogenic edema [4]. Electron microscopy reveals that BBB disruption results in parenchymal fluid increase and brain swelling [5].
Moreover, adhesion of infected RBCs to endothelial cells obstructs blood vessels, leading to sudden swelling in the brain and may result in death due to respiratory failure [4]. Although the sequestering of infected RBCs in the luminal site of brain microvessels is a keynote characteristic of human cerebral malaria (HCM) [6], only a few or no sequestrating of infected RBCs were observed in brain microvessels during experimental cerebral malaria (ECM) [7]. Moreover, no intracerebral accumulation of CD8+ T cells was observed during HCM [8]. There was a prominent pro-inflammatory cytokine response in the brain during ECM. Intracerebral accumulation of both CD8+ T cells was found essential for the development of ECM [9]. Moreover, infected RBCs were observed to be perivascular relative to several organs, followed by a morphological change in brain endothelial surface [10]. Targeting BBB disruption may be an optional therapeutic approach to reduce brain swelling and edema [5]. Moreover, the preventions of sequestration and de-sequestration of the infected RBCs are attractive therapeutic approaches.
2. Pathophysiology during Cerebral Malaria Progression
Several hypotheses have been reported to explain the mechanism of cerebral malaria development. One such hypothesis affirms that infected RBCs adherence to brain endothelium leads to blockage in microvessels, causing nutrient deprivation and hypoxia in nearby brain tissue. As a result, brain tissues cannot maintain membrane potential, which causes water inflow from extracellular to intracellular compartments, ultimately leading to cell death and tissue damage [11]. According to other groups, the sequestration of infected RBCs and inflammation enhance binding of infected RBCs with non-infected RBCs to form rosette [11] and the adherence of infected RBCs and leukocytes to brain endothelium (Figure 1) [12]. Moreover, platelet-mediated clumping of infected RBCs during infected RBCs sequestration [13] causes excess activation of the endothelial wall with pro-inflammatory mediators secretion [14] and an imbalanced release of endothelin-1 and angiopoietin-2 vasoactive mediators [15]. Finally, the brain endothelial cell barrier becomes impaired with leakage in brain parenchyma, resulting in breakage of the BBB.
In addition to acting as a transport system, endothelial cells also facilitate the binding of infected RBCs expressing Plasmodium falciparum erythrocyte membrane protein-1 (PfEMP1) linked with CM and act as phagocytic cells. Intercellular adhesion molecule (ICAM) dependent internalization of infected RBCs by endothelial cells results in brain injury and impairment of BBB [16]. After phagocytosis, infected RBCs undergo degradation and exposes host cells to cellular content and toxins [17]. ICAM-1 facilitates the entry of leukocytes in the endothelium by forming ring-like projections [18].
Transfer of Pf antigens from infected RBCs to brain endothelial cells by direct contact (i) or by production of antigen-carrying extracellular vesicles (ii); Pf antigen picked up and expressed by MHC I (iii); interaction of cytotoxic T lymphocytes (CTL) with endothelial cells expressing antigens, through their CD8 and TCR receptors (iv); granzyme B secreted by CTL (v); infected RBCs interact with uninfected RBCs leading to rosette formation (vi); platelet-mediated clumping (vii) disruption of the blood-brain barrier, leading to vasogenic edema (viii) (this figure is adopted and modified from Renia et al. (2020) [9].
In the case of infected RBCs, it is suggested that the mechanism is similar; however, infected RBCs show slower transit than leukocytes, as infected RBCs are deficient in machinery required for motility [18]. Preclinical studies showed that activated protein C (aPC), an anticoagulant serine protease, binds to endothelial protein C receptor (EPCR) and exerts its anti-inflammatory and anti-apoptotic activities, thereby protecting endothelial barrier functions [19,20]. EPCR, expressed at low levels by the brain endothelium, interact with infected RBCs and caused a loss of aPC, leading to a loss of cytoprotection [21].
Consequently, infected RBCs binding with EPCR turn off cytoprotection via the CIDRα1 domain, whereas the ICAM-1 binding DBLβ domain incites binding and phagocytic response by the endothelium and assists cerebral malaria pathology through cerebral swelling and BBB disruption [16]. The link between ICAM-1 and EPCR to cerebral malaria symptoms has been established [16], but the mechanism of pathogenesis is still ambiguous. The activation of endothelial cells is regulated by cytokines such as Tumour Necrosis Factor alpha (TNF-α), Tumour Necrosis Factor beta (TNF-β), and interferon-γ [22]. The upregulation of TNF cytokine was observed in the blood and the brain cells of human and murine models of cerebral malaria [23]. In vitro TNF exposure induces the upregulation of infected RBCs adhesion factors and endothelial C activation markers, leading to increases in adherence of infected RBCs on human [24] or murine [25] brain microvascular cells (Figure 2). Furthermore, the upregulation of endothelial cells activation markers such as E-Selectin, von Willebrand Factor (vWF), ICAM-1, and CD36 all can act as adhesion targets for infected RBCs and contribute to endothelial activation.
Malaria parasite during infection suppresses erythropoiesis in the bone marrow, and hemozoin deposition in the tissues are mostly responsible for malarial anemia. Dyserythropoiesis results in the removal of infected and uninfected erythrocytes from the system and suppressed the production of mature erythrocytes in the bone marrow, which are primary contributors to the severity of malaria infection [26,27], followed by the development of severe malarial anemia leading to increased mortality and morbidity. The proliferation of erythroid progenitor cells is challenged during malaria infection due to decreased expression of erythroid-specific transcription factors such as GATA-1 and GATA-2 [28,29]. It has been reported that erythroblasts, BFU-E, and CFU-E numbers decrease after infection with P. berghei [30]. GATA-1 facilitates the survival and late-stage differentiation of BFU-E to CFU-E, which is facilitated by the mediator subunit MED1/TRAP220 [28,29], whereas GATA-2 is critical for the maintenance and proliferation of hematopoietic progenitors.
3. Platelets: A New Player in Infected RBCs Cytoadherence
Platelets are considered as critical contributors to CM by providing an alternate, indirect mechanism for infected RBCs adhesion [32]. Platelet sequestering observed only in CM brain and not in non CM brain [22]. Platelets accumulation were found colocalized with malarial pigments on the endothelial surface in Malawian patients died from CM, suggesting that the platelet closely interact with infected RBCs and endothelial cells during CM pathology [33]. Platelet-induced in vitro clumping experiments revealed the occurrence of infected RBCs clumping after incubation with Platelet-rich plasma. In the case of cerebral malaria, a high level of clumping was observed compared to uncomplicated malaria. However, in the presence of Platelet poor plasma, no clumping of infected RBC was observed in any of the patient groups after co-culture for 120 min [34].
Three platelet receptors CD36, globular C1q (gC1qR/HABP1/p32), and P-selectin have been identified for involvement in clumping [34,35,36]. CD36, a major receptor for infected RBCs, is constitutively express on platelets [37], suggesting that platelets could affect the adherence of infected RBCs on the brain endothelium, promoting vascular obstruction [38]. Both CD36 and P-selectin are known to be receptors for PfEMP-1 receptor of infected RBCs [39,40,41]. Infected RBCs are able to activate platelets [42] that lead to the transfer of the internal P-selectin to its surface [43]. Its expression on platelet membranes increased dramatically during clumping, confirming that infected RBCs activate platelets. Intercepting the clumping phenomenon using antibody-mediated blocking of CD36 and P-selectin encourages the idea of synergism between both receptors during clumping [34]. P-selectin increases the adherence of infected RBCs to CD36 on the endothelium [44,45] and may act as a trigger for the amplification of the clumping phenomenon. Platelets can form bridges between endothelial cells and infected RBCs [32,46,47] after bonding to the TNF activated endothelial cells with the help of specific molecules such as endothelial CD40 and platelet CD41. In addition to direct cytoadherence (infected RBCs to the endothelial wall), platelet-associated clumping and bridging may also contribute to the obstruction of the brain microvessels in CM patients.
Moreover, platelet activation leads to massive releases of transforming growth factor TGF-β1 from their a-granules [48]. Recent reports revealed that after amplified release and accumulation in brain microvessels, TGF-β1 might act together with TNF to induce endothelial apoptosis [14]. In addition, platelets are characterized as an important factor in assisting inflammatory reactions and immune responses [49]. The synchronized expression of adhesive and immune receptors on the platelet’s surface and release of some cytokines and inflammatory mediators can activate the interaction with leukocytes and increase their recruitment in brain microvessels. [50,51]. With the help of fibrinogen and ICAM-2, the IL-1β mediator can initiate the adhesion of leukocyte relative to endothelial cells [52,53]. Furthermore, IL-1β [54] can initiate leukocyte adhesion to endothelial cells that contribute to the obstruction of brain microvessels.
4. The Emerging Role of NK Cells in CM Pathogenesis
Natural killer (NK) cells are one of the earliest cells observed in the brain of Plasmodium berghei infected mice brain. In initial reports, no role of NK cells was found in ECM [55], but recent reports using anti-Asialo GM1 antibody confirms the active role of NK cells in ECM [56,57]. The antibody-mediated depletion of NK cells results in the inhibition of T cell recruitment in the infected brain of mice and confirms the role of NK cells in the regulation of adaptive immune response by modulation of the ability of T lymphocytes to migrate to the site of inflammation. [56]. T cells accumulation in brain microvessels plays a vital role in cerebral malaria pathogenesis [58]. The roles of CD4+ [55] and CD8+ [58,59] cells in disease assistance have been determined experimentally. Studies suggested that the sequestration of leukocytes and parasitized RBC in brain microvessels helps in disease induction [60,61]. Factors associated with the innate immune system are also involved in cerebral malaria induction. Based on the host’s genetic background, CD1d-restricted natural killer T (NKT) cells arbitrate resistance or susceptibility relative to cerebral malaria in mice [62].
Other than its pathological role, NK cells induce the production of IL-10 that inhibits pathological CD8+ cells response and protect against cerebral malaria in mice [63]. The disease model confirms the production of IL-10 by NK cells during infections [64,65,66]. A large quantity of interferon gamma (IFN-γ) is secreted by NK cells that play a critical role in immune-mediated brain damage in experimental cerebral malaria [67,68]. The treatment of brain endothelial cells of mice with IFN-γ upregulates the expression of intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which increases the adhesion of T lymphocytes by two- to three-folds [69]. Additionally, infected RBCs pretreated with IFN-γ showed significantly higher binding than that of untreated infected RBCs and normal RBCs.
5. Effect of Ang-2/Ang-1 Ratio in CM
Vascular endothelium activation and dysfunction are linked with the production of several biomarkers such as angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) in infectious diseases. The study on the role of Ang-1 and Ang-2 in endothelial cell serenity has become an essential and preferential topic lately [70]. Smooth muscle cells and pericytes continuously produce Ang-1, while endothelial cells produce Ang-2 that is stored in Weibel-Palade bodies. At the time of infection, Ang-2 is resealed rapidly from Weibel- Palade bodies [71]. Both Ang-1 and Ang-2 bind to the Tie-2 receptor, which is a member of the vascular tyrosine kinase receptor family. Under normal conditions, Ang-1 is present in high concentrations that allow it to bind with the Tie-2 receptor, leading to the activation of pro-survival pathways and inhibition of pro-inflammatory pathways. However, after infection, the rapid release of Ang-2 allows it to bind preferentially to tie-2 and promotes pro-inflammatory pathways [72]. A high concentration of plasma Ang-2 was observed in patients with severe malaria [72]. The plasma Ang-2/Ang-1 ratio is critical for endothelial activation, and it also act as a biomarker to differentiate uncomplicated malaria to CM. Elevated levels of Ang-2 are linked with mortality in patients with CM, while elevated levels of Ang-1 is related with uncomplicated malaria [73]. Since endothelial cell activation is a primary event during cerebral malaria, the biomarkers of the activated endothelium can be detected earliest before traditional disease markers and can be valuable biomarkers of disease severity.
6. Role of PfEMP1 in Infected RBCs Cytoadherence
The PfEMP1 is expressed on the membrane of infected RBCs and encourage the adherence of the infected RBCs on brain endothelial cells [74,75,76]. Unforeseen features of infected RBCs expressing PfEMP1 protein were discovered by a 3D spheroid model of the blood-brain barrier. The single haploid genome of Plasmodium falciparum encodes nearly 60 different PfEMP1variants, facilitating Plasmodium falciparum to undergo antigenic variation and dodges the immune system. PfEMP1 binds to a variety of receptors present on brain endothelial cells, including VCAM-1, cytokine-activated EPCR, and ICAM-1 [13,77,78], thereby fixing the infected RBCs on brain endothelial cells. Infected RBCs expressing PfEMP1 interact with ICAM-1 with their DBLβ motif [77]. Parasite-expressing PfEMP1 shows expansions in patient with cerebral malaria [77,79]. Moreover, the in vitro deletion of the DBLβ motif of PfEMP-1 results in a reduction in ICAM-1-specific adhesion of erythrocytes on the endothelium cells. Patients with high anti-PfEMP1 antibodies had a significantly reduced risk of developing symptomatic malaria [80]. After attachment to endothelium wall receptors, PfEMP1 may produce several signaling pathways that lead to restructuring tight junction complexes. Ultimately all these events result in impairment and breakage of the blood-brain barrier. Studies on leukocyte movement in brain endothelium, using murine and human models, suggest that the interaction of PfEMP1 and ICAM-1 promotes cytoskeleton-associated proteins phosphorylation, such as cortactin, paxillin, FAK, and p130Cas, that results in redesigning the endothelial cytoskeleton, which eventually assists in opening the blood-brain barrier [81].
7. Therapeutic Approaches
The sequestration of infected RBCs can also be prevented by using drugs targeting cytoadherence on the endothelium. In experimental cerebral malaria reports, the use of rapamycin restricts the cytoadherence of infected RBCs on endothelial cells by VCAM-1 and ICAM-1 reduction [82]. Moreover, the fungal ethanolic extract of Trichoderma stromaticum helps in reducing inflammation in experimental cerebral malaria by reducing the expression of VCAM-1 and ICAM-1, thus protecting the blood-brain barrier’s integrity [83].
The activation of endothelial cells by cytoadherence of infected RBCs and the pro-inflammatory effects of released cytokines play important roles in cerebral malaria pathophysiology [47]. Anti-TNF treatments can be helpful in managing the severity of disease and mortality rate. Endothelium activation triggers a TNF dependent pro-apoptotic pathway [84]. The difference in response of endothelial cells to TNF can affect the severity of the disease [85]. Anti-TNF therapy showed a positive result in vitro. However, it showed no reduction in mortality in the patients. Moreover, the use of antibodies against endothelial protein C receptor inhibited PfEMP1′s ability to bind to human endothelial cells [78]. Further studies on endothelial cells confirm the activation of Rac1 signaling via cross binding of VCAM-1 that results in Rho-dependent inductions of stress fibers, leading to the weakening of tight junctions [81].
Moreover, the interaction of EPCR with activated protein C is prevented by binding PfEMP1 to EPCR. This binding shows two impacts: (i) promoting the activation of tissue factors Va and VIIIa, which results in the disablement of the aPC-mediated anti-coagulative pathway. These factors are responsible for thrombin generation on activation, which eventually results in fibrin deposition. (ii) The initiation of NF-kB and Rho A pathways through thrombin-mediated scissions of PAR1 is also observed. The activation of these pathways generates a pro-inflammatory response, which leads to impairment and breakage of the blood-brain barrier [20,79,86]. The administration of aPC may prevent blood-brain barrier dysfunction by inhibiting thrombin activity [87]. Other approaches embrace the utilization of endothelial cells isolated from patients suffering with cerebral malaria. Moreover, measuring the consequences of Ang-1 on endothelial cells pre-treated with TNF will be crucial for the development of new additional therapies and improving disease outcomes in CM. Since Ang-1 acts as a serenity agent for endothelial cells in the exquisite model of sepsis [88], maintaining a higher Ang-1concetration in Ang-2/Ang-1 ratio during infection can potentially block and reverse ongoing inflammatory processes in CM patients.
8. MSCs as A Regenerative Therapy
8.1. MSCs Mediated Cellular Mechanism of Protection
Mesenchymal stem cells (MSCs) first observed in the bone marrow are multipotent cells [89], which can differentiate into various cell lines lineages such as adipocytes, chondrocytes, and osteoblasts and display strong tissue protective and restorative properties. During malaria infection, MSCs become accumulated in primary and secondary lymphoid organs. Different surface antigens are expressed by MSCs in different tissues, which facilitate tissue repair and regeneration through the secretion of various soluble factors. Souza MC et al. demonstrated that, in P. berghei-infected mice, neurons were damaged, with an increased number of astrocytes and oligodendrocytes. However, in P. berghei-infected mice treated with BM-MSCs, brain damage was repaired, which leads to a reduction in parasitemia and mortality [90]. There was a substantial increase in phagocytic neutrophils in the brain [91]; hepatocytes and Kupffer cell regeneration in MSC-infused mice indicate regenerative ability of MSCs. MSCs promote hematopoiesis by releasing different critical molecules involved in the self-renewal, proliferation, and differentiation of hematopoietic stem and progenitor cells (HSPCs). Other than that, MSCs encourage the formation of colony-forming units-erythroid (CFU-E) cells in the bone marrow [92], which ultimately helps in preventing malarial anemia. These observations suggest that cell-based therapeutics for intervention in malaria may be useful in achieving sterile clearance and preventing disease reactivation.
8.2. MSCs Mediated Molecular Aspects of Protection
Studies on anti -CD3−, -CD19−, -Sca-1+, and -CD34+ antibodies revealed a significant elevation in Sca- 1+ and CD34+ cells in the lymph node and spleen of MSC-treated mice as compared to untreated mice [91]. A drop in GATA-1 and GATA-2 expression was observed in plasmodium-infected mice. Since GATA-1 is a key erythroid cell differentiation factor, the low level of GATA-1 expression may be responsible for reduced RBC formation in malaria-infected animals. However, after MSCs treatment, an increase in the expression of GATA-1 and GATA-2 has been reported in animals infused with MSCs. Increased CFU-E formation and reduced hemozoin content in MSC-infused mice support the conclusion that MSCs elicits signals to support erythropoiesis [93].
8.3. Application of MSCs in Animal Model of Cerebral Malaria
Malaria infection causes a reduction in CD4+ T cells in mice models of cerebral malaria, which are crucial for immunity development against malaria, leading to impaired T cell-mediated immunity [94]. However, an increase in the number of CD4+ and CD8+ T cells was reported in MSC-infused mice, and MSCs are able to rescue the proliferation of CD4+ T cells [92]. Ongoing research studies are focused on signaling molecules engaged in restoring immune responses by MSCs. MSCs also inhibited the induction of the negative co-stimulatory receptor programmed death-1 by T cells in recipient animals. Taken together, MSCs help in the protection against malaria infection by reprogramming hematopoiesis, by enhancing the differentiation of CD34+ cells, restoring CD4+ and CD8+ T cell proliferation, and by suppressing the expression of negative co-stimulators on T cells. The increased production of interleukin IL-12, which is crucial for self-renewal and differentiation of multipotent progenitor cells and suppressed production of IL-10, was reported in MSC infused animals. The transfer of MSCs isolated from secondary lymphoid organs of P. berghei-infected mice conferred host resistance against malaria through the enhanced production of pro-inflammatory cytokines IL-6, IL-12, and TNF-α, and the suppression of IL-10 [93,95].
Preclinical studies on several metabolic disorders, tissue regeneration, cancer, heart disorders, and other disorders suggest the robust use of MSC-based therapy [96]. Despite multiple preclinical studies on MSC-based therapy, no promising clinical studies are available for cerebral malaria. The multipotent nature and the potential of MSCs of modifying the tissue microenvironment makes them appropriate candidates for the development of stem cell-based regenerative medicinal therapy.
9. Conclusions
Current therapeutic approaches have been unsuccessful to prevent neuro-cognitive complications and deaths in cerebral malaria cases. Stem cell therapy is a newly emerging treatment for a wide range of disorders. A number of studies have pointed out the importance of MSC-based treatments in many disease conditions such as cancer, Parkinson’s disease, Huntington’s disease, transplantation, etc. Reports of experimental cerebral malaria suggest that bone marrow-derived MSC treatment reduces mortality and parasitemia; hemozoin accumulation deposition in the spleen, liver, kidney, and lung; and increases survival [90]. An appropriate evaluation of both preclinical clinical and clinical efficacy and the analysis of mechanisms by which MSCs protect the host against malaria should be performed. A further understanding of the immune-modulatory properties and impact of MSCs on erythropoiesis would make it possible to establish MSCs as a novel regenerative medicine in malaria research.
Author Contributions
J.D. and A.C. designed the original idea and developed it in detail, reviewed the literature, and wrote the manuscript. A.C. generated all figures and arranged references. P.K. and N.S. helped in reviewing the literature. All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding.
Acknowledgments
We are thankful to the ICMR National Institute of Malaria Research ICMR-NIMR) for infrastructural support. Amrendra Chaudhary is a recipient of a junior research fellowship from the Dept of biotechnology (DBT), Govt. of India. Poonam Kataria is a recipient of a junior research fellowship from the university grant commission (UGC) Govt. of India. Neha Surela is a recipient of a junior research fellowship from the university grant commission (UGC) Govt. of India. We also thank to all lab members for their generous support during this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R. Cerebral malaria: Mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatric Res. 2010, 68, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.; Mungala-Odera, V.; Neville, B.G.; Murira, G.; Mturi, N.; Musumba, C. Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J. Neurol. Neurosurg. Psychiatry Apr. 2005, 76, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dondorp, A.; Nosten, F.; Stepniewska, K.; Day, N.; White, N. Artesunate versus quinine for treatment of severe falciparum malaria: A randomised trial. Lancet 2005, 366, 717–725. [Google Scholar] [PubMed] [Green Version]
- Seydel, K.B.; Kampondeni, S.D.; Valim, C.; Potchen, M.J.; Milner, D.A.; Muwalo, F.W.; Birbeck, G.L.; Bradley, W.G.; Fox, L.L.; Glover, S.J. Brain swelling and death in children with cerebral malaria. N. Engl. J. Med. 2015, 372, 1126–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Jin, J.; Aida, M.; Wai, C.H.; Mohanty, S.; Sahu, P.K.; Pattnaik, R.; Pirpamer, L.; Fischer, M.; Heiland, S. Reversible brain edema in experimental cerebral malaria is associated with transcellular blood-brain barrier disruption and delayed microhemorrhages. bioRxiv 2021. [Google Scholar] [CrossRef]
- Storm, J.; Craig, A.G. Pathogenesis of cerebral malaria--inflammation and cytoadherence. Front. Cell Infect. Microbiol. 2014, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Strangward, P.; Haley, M.J.; Shaw, T.N.; Schwartz, J.-M.; Greig, R.; Mironov, A.; de Souza, J.B.; Cruickshank, S.M.; Craig, A.G.; Milner Jr, D.A. A quantitative brain map of experimental cerebral malaria pathology. PLoS Pathog. 2017, 13, e1006267. [Google Scholar] [CrossRef]
- White, N.J.; Turner, G.D.; Medana, I.M.; Dondorp, A.M.; Day, N.P. The murine cerebral malaria phenomenon. Trends Parasitol. 2010, 26, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Rénia, L.; Grau, G.E.; Wassmer, S.C. CD8+ T cells and human cerebral malaria: A shifting episteme. J. Clin. Investig. 2020, 130, 1109–1111. [Google Scholar] [CrossRef]
- Mota, M.M.; Jarra, W.; Hirst, E.; Patnaik, P.K.; Holder, A.A. Plasmodium chabaudi-infected erythrocytes adhere to CD36 and bind to microvascular endothelial cells in an organ-specific way. Infect. Immun. 2000, 68, 4135–4144. [Google Scholar] [CrossRef] [Green Version]
- Wassmer, S.C.; Grau, G.E.R. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, D.; Roth, E.J.; Nagel, R.; Howard, R.; Handunnetti, S. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood 1991, 78, 812–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, J.A.; Claessens, A.; Corrigan, R.A.; Arman, M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 2009, 11, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassmer, S.C.; de Souza, J.B.; Frère, C.; Candal, F.J.; Juhan-Vague, I.; Grau, G.E. TGF-β1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis: A new mechanism for microvascular lesion during cerebral malaria. J. Immunol. 2006, 176, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prapansilp, P.; Medana, I.; Mai, N.T.H.; Day, N.P.; Phu, N.H.; Yeo, T.W.; Hien, T.T.; White, N.J.; Anstey, N.M.; Turner, G.D. A clinicopathological correlation of the expression of the angiopoietin-Tie-2 receptor pathway in the brain of adults with Plasmodium falciparum malaria. Malar. J. 2013, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Adams, Y.; Olsen, R.W.; Bengtsson, A.; Dalgaard, N.; Zdioruk, M.; Satpathi, S.; Behera, P.K.; Sahu, P.K.; Lawler, S.E.; Qvortrup, K. Plasmodium falciparum erythrocyte membrane protein 1 variants induce cell swelling and disrupt the blood–brain barrier in cerebral malaria. J. Exp. Med. 2021, 218, e20201266. [Google Scholar] [CrossRef]
- Gallego-Delgado, J.; Rodriguez, A. Rupture and Release: A Role for Soluble Erythrocyte Content in the Pathology of Cerebral Malaria. Trends Parasitol. 2017, 33, 832–835. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sun, T.; McIntosh, M.T.; Bucala, R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 2009, 183, 5208–5220. [Google Scholar] [CrossRef]
- Carman, C.V.; Jun, C.-D.; Salas, A.; Springer, T.A. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J. Immunol. 2003, 171, 6135–6144. [Google Scholar] [CrossRef] [Green Version]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef]
- Bernabeu, M.; Smith, J.D. EPCR and malaria severity: The center of a perfect storm. Trends Parasitol. 2017, 33, 295–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moxon, C.A.; Wassmer, S.C.; Milner Jr, D.A.; Chisala, N.V.; Taylor, T.E.; Seydel, K.B.; Molyneux, M.E.; Faragher, B.; Esmon, C.T.; Downey, C. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood J. Am. Soc. Hematol. 2013, 122, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.H.; Grau, G.E. Cytokines: Accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003, 24, 491–499. [Google Scholar] [CrossRef]
- Grau, G.E.; Fajardo, L.F.; Piguet, P.-F.; Allet, B.; Lambert, P.-H.; Vassalli, P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987, 237, 1210–1212. [Google Scholar] [CrossRef]
- Jambou, R.; Combes, V.; Jambou, M.-J.; Weksler, B.B.; Couraud, P.-O.; Grau, G.E. Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog. 2010, 6, e1001021. [Google Scholar] [CrossRef] [Green Version]
- El-Assaad, F.; Wheway, J.; Mitchell, A.J.; Lou, J.; Hunt, N.H.; Combes, V.; Grau, G.E.R. Cytoadherence of Plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect. Immun. 2013, 81, 3984–3991. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-H.; Stevenson, M.M. Malarial anaemia: Mechanisms and implications of insufficient erythropoiesis during blood-stage malaria. Int. J. Parasitol. 2004, 34, 1501–1516. [Google Scholar] [CrossRef]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood J. Am. Soc. Hematol. 2008, 112, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Borggrefe, T.; Waskow, C.; Roeder, R.G.; Stumpf, M. Severely Impaired Erythropoiesis in Mice Lacking Mediator Subunit Med1/TRAP220. Am. Soc.Hematol. 2004, 104, 1611. [Google Scholar] [CrossRef]
- Tang, Y.; Joyner, C.J.; Cabrera-Mora, M.; Saney, C.L.; Lapp, S.A.; Nural, M.V.; Pakala, S.B.; DeBarry, J.D.; Soderberg, S.; Kissinger, J.C. Integrative analysis associates monocytes with insufficient erythropoiesis during acute Plasmodium cynomolgi malaria in rhesus macaques. Malar. J. 2017, 16, 384. [Google Scholar] [CrossRef] [Green Version]
- Wassmer, S.C.; Taylor, T.E.; Rathod, P.K.; Mishra, S.K.; Mohanty, S.; Arevalo-Herrera, M.; Duraisingh, M.T.; Smith, J.D. Investigating the Pathogenesis of Severe Malaria: A Multidisciplinary and Cross-Geographical Approach. Am. J. Trop. Med. Hyg. 2015, 93 (Suppl. S3), 42–56. [Google Scholar] [CrossRef] [PubMed]
- Maggio-Price, L.; Brookoff, D.; Weiss, L. Changes in hematopoietic stem cells in bone marrow of mice with Plasmodium berghei malaria. Blood 1985, 66, 1080–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heyde, H.C.; Nolan, J.; Combes, V.; Gramaglia, I.; Grau, G.E. A unified hypothesis for the genesis of cerebral malaria: Sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006, 22, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Grau, G.E.; Mackenzie, C.D.; Carr, R.A.; Redard, M.; Pizzolato, G.; Allasia, C.; Cataldo, C.; Taylor, T.E.; Molyneux, M.E. Platelet Accumulation in Brain Microvessels in Fatal Pediatric Cerebral Malaria. J. Infect. Dis. 2003, 187, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Taylor, T.; Maclennan, C.A.; Kanjala, M.; Mukaka, M.; Molyneux, M.E.; Grau, G.E. Platelet-induced clumping of Plasmodium falciparum-infected erythrocytes from Malawian patients with cerebral malaria-possible modulation in vivo by thrombocytopenia. J. Infect. Dis. 2008, 197, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Pain, A.; Ferguson, D.J.; Kai, O.; Urban, B.C.; Lowe, B.; Marsh, K.; Roberts, D.J. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc. Natl Acad Sci USA 2001, 98, 1805–1810. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.K.; Hafiz, A.; Banerjee, B.; Kim, K.S.; Datta, K.; Chitnis, C.E. Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathog. 2007, 3, 1271–1280. [Google Scholar] [CrossRef] [Green Version]
- Barnwell, J.W.; Asch, A.S.; Nachman, R.L.; Yamaya, M.; Aikawa, M.; Ingravallo, P. A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. J. Clin. Investig. 1989, 84, 765–772. [Google Scholar] [CrossRef]
- Cooke, B.M.; Coppel, R.L. Cytoadhesion and Falciparum Malaria: Going with the flow. Parasitol. Today 1995, 11, 282–287. [Google Scholar] [CrossRef]
- Ockenhouse, C.F.; Magowan, C.; Chulay, J.D. Activation of monocytes and platelets by monoclonal antibodies or malaria-infected erythrocytes binding to the CD36 surface receptor in vitro. J. Clin. Investig. 1989, 84, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S. Platelet membrane glycoproteins in haemostasis. Clin. Lab. 2002, 48, 247–262. [Google Scholar] [PubMed]
- Udomsangpetch, R.; Reinhardt, P.H.; Schollaardt, T.; Elliott, J.F.; Kubes, P.; Ho, M. Promiscuity of clinical Plasmodium falciparum isolates for multiple adhesion molecules under flow conditions. J. Immunol. 1997, 158, 4358–4364. [Google Scholar]
- Polack, B.; Peyron, F.; Zadiuddin, S.; Kolodie, L.; Ambroise-Thomas, P. Erythrocytes infected by Plasmodium falciparum activate human platelets. Comptes Rendus L’academie Sci. Serie III Sci. Vie 1990, 310, 577–582. [Google Scholar]
- Israels, S.J.; Gerrard, J.M.; Jacques, Y.V.; McNicol, A.; Cham, B.; Nishibori, M.; Bainton, D.F. Platelet dense granule membranes contain both granulophysin and P-selectin (GMP-140). Blood 1992, 80, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Anand, S.; Schollaardt, T.; Patel, K.D.; Looareesuwan, S.; Ho, M. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum–infected erythrocytes to microvascular endothelial cells under flow. Blood 2000, 96, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Lépolard, C.; Traoré, B.; Pouvelle, B.; Gysin, J.; Grau, G.E. Platelets ReorientPlasmodium falciparum–Infected Erythrocyte Cytoadhesion to Activated Endothelial Cells. J. Infect. Dis. 2004, 189, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Moxon, C.A.; Heyderman, R.S.; Wassmer, S.C. Dysregulation of coagulation in cerebral malaria. Mol. Biochem. Parasitol. 2009, 166, 99–108. [Google Scholar] [CrossRef]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [CrossRef]
- Männel, D.N.; Grau, G.E. Role of platelet adhesion in homeostasis and immunopathology. Mol. Pathol 1997, 50, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [Green Version]
- Hundelshausen, P.; Petersen, F.; Brandt, E. Platelet-derived chemokines in vascular biology. Thromb. Haemost. 2007, 97, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijper, P.H.M.; Torres, H.I.G.; Lammers, J.W.J.; Sixma, J.J.; Koenderman, L.; Zwaginga, J.J. Platelet Associated Fibrinogen and ICAM-2 Induce Firm Adhesion of Neutrophils under Flow Conditions. Thromb. Haemost. 1998, 80, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Hawrylowicz, C.M.; Howells, G.L.; Feldmann, M. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J. Exp. Med. 1991, 174, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Yañez, D.M.; Manning, D.D.; Cooley, A.J.; Weidanz, W.P.; Van Der Heyde, H. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 1996, 157, 1620–1624. [Google Scholar]
- Hansen, D.S.; Bernard, N.J.; Nie, C.Q.; Schofield, L. NK Cells Stimulate Recruitment of CXCR3+T Cells to the Brain duringPlasmodium berghei-Mediated Cerebral Malaria. J. Immunol. 2007, 178, 5779–5788. [Google Scholar] [CrossRef] [Green Version]
- Ryg-Cornejo, V.; Nie, C.Q.; Bernard, N.J.; Lundie, R.J.; Evans, K.J.; Crabb, B.S.; Schofield, L.; Hansen, D.S. NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology 2013, 218, 263–271. [Google Scholar] [CrossRef]
- Belnoue, E.; Kayibanda, M.; Vigario, A.M.; Deschemin, J.-C.; Rooijen, N.v.; Viguier, M.; Snounou, G.; Rénia, L. On the Pathogenic Role of Brain-Sequestered αβ CD8+T Cells in Experimental Cerebral Malaria. J. Immunol. 2002, 169, 6369–6375. [Google Scholar] [CrossRef] [Green Version]
- Nitcheu, J.; Bonduelle, O.; Combadiere, C.; Tefit, M.; Seilhean, D.; Mazier, D.; Combadiere, B. Perforin-Dependent Brain-Infiltrating Cytotoxic CD8+ T Lymphocytes Mediate Experimental Cerebral Malaria Pathogenesis. J. Immunol. 2003, 170, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Grau, G.E.; Bieler, G.; Pointaire, P.; De Kossodo, S.; Tacchini-Cotier, F.; Vassalli, P.; Piguet, P.F.; Lambert, P.H. Significance of cytokine production and adhesion molecules in malarial immunopathology. Immunol. Lett. 1990, 25, 189–194. [Google Scholar] [CrossRef]
- Baptista, F.G.; Pamplona, A.; Pena, A.C.; Mota, M.M.; Pied, S.; Vigário, A.M. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect. Immun. 2010, 78, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.S.; Siomos, M.-A.; Buckingham, L.; Scalzo, A.A.; Schofield, L. Regulation of Murine Cerebral Malaria Pathogenesis by CD1d-Restricted NKT Cells and the Natural Killer Complex. Immunity 2003, 18, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Burrack, K.S.; Huggins, M.A.; Taras, E.; Dougherty, P.; Henzler, C.M.; Yang, R.; Alter, S.; Jeng, E.K.; Wong, H.C.; Felices, M.; et al. Interleukin-15 Complex Treatment Protects Mice from Cerebral Malaria by Inducing Interleukin-10-Producing Natural Killer Cells. Immunity 2018, 48, 760–772.e764. [Google Scholar] [CrossRef] [Green Version]
- Perona-Wright, G.; Mohrs, K.; Szaba, F.M.; Kummer, L.W.; Madan, R.; Karp, C.L.; Johnson, L.L.; Smiley, S.T.; Mohrs, M. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe 2009, 6, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.-S.; Sherratt, S.; Riley, E.M. NK Cells: Uncertain Allies against Malaria. Front. Immunol. 2017, 8, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkal, M.; Chauhan, R.; Thakur, R.S.; Tiwari, M.; Pande, V.; Das, J. IL-10 Producing Regulatory B Cells Mediated Protection against Murine Malaria Pathogenesis. Biology 2022, 11, 669. [Google Scholar] [CrossRef]
- Grau, G.E.; Heremans, H.; Piguet, P.F.; Pointaire, P.; Lambert, P.H.; Billiau, A.; Vassalli, P. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5572–5574. [Google Scholar] [CrossRef] [Green Version]
- Villegas-Mendez, A.; Greig, R.; Shaw, T.N.; de Souza, J.B.; Gwyer Findlay, E.; Stumhofer, J.S.; Hafalla, J.C.R.; Blount, D.G.; Hunter, C.A.; Riley, E.M.; et al. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol 2012, 189, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Faveeuw, C.; Di Mauro, M.E.; Price, A.A.; Ager, A. Roles of α4 integrins/VCAM-1 and LFA-1/ICAM-1 in the binding and transendothelial migration of T lymphocytes and T lymphoblasts across high endothelial venules. Int. Immunol. 2000, 12, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, A.; Fulton, D.; Mahboubi, K.; Kalb, R.G.; O’Connor, D.S.; Li, F.; Altieri, D.C.; Sessa, W.C. Angiopoietin-1 Inhibits Endothelial Cell Apoptosis via the Akt/Survivin Pathway. J. Biol. Chem. 2000, 275, 9102–9105. [Google Scholar] [CrossRef] [Green Version]
- van Meurs, M.; Kümpers, P.; Ligtenberg, J.J.M.; Meertens, J.H.J.M.; Molema, G.; Zijlstra, J.G. Bench-to-bedside review: Angiopoietin signalling in critical illness—A future target? Crit. Care 2009, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, T.W.; Lampah, D.A.; Gitawati, R.; Tjitra, E.; Kenangalem, E.; Piera, K.; Price, R.N.; Duffull, S.B.; Celermajer, D.S.; Anstey, N.M. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc. Natl. Acad. Sci. USA 2008, 105, 17097–17102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.V.; Liles, W.C. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence 2013, 4, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rug, M.; Prescott, S.W.; Fernandez, K.M.; Cooke, B.M.; Cowman, A.F. The role of KAHRP domains in knob formation and cytoadherence of P falciparum-infected human erythrocytes. Blood 2006, 108, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Kataria, P.; Surela, N.; Chaudhary, A.; Das, J. MiRNA: Biological Regulator in Host-Parasite Interaction during Malaria Infection. Int. J. Environ. Res. Public Health 2022, 19, 2395. [Google Scholar] [CrossRef]
- Kalkal, M.; Kalkal, A.; Dhanda, S.K.; Das, E.; Pande, V.; Das, J. A comprehensive study of epitopes and immune reactivity among Plasmodium species. BMC Microbiol. 2022, 22, 74. [Google Scholar] [CrossRef]
- Lennartz, F.; Adams, Y.; Bengtsson, A.; Olsen, R.W.; Turner, L.; Ndam, N.T.; Ecklu-Mensah, G.; Moussiliou, A.; Ofori, M.F.; Gamain, B.; et al. Structure-Guided Identification of a Family of Dual Receptor-Binding PfEMP1 that Is Associated with Cerebral Malaria. Cell Host Microbe 2017, 21, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Avril, M.; Bernabeu, M.; Benjamin, M.; Brazier, A.J.; Smith, J.D. Interaction between Endothelial Protein C Receptor and Intercellular Adhesion Molecule 1 to Mediate Binding of Plasmodium falciparum-Infected Erythrocytes to Endothelial Cells. mBio 2016, 7, e00615-16. [Google Scholar] [CrossRef] [Green Version]
- Kessler, A.; Dankwa, S.; Bernabeu, M.; Harawa, V.; Danziger, S.A.; Duffy, F.; Kampondeni, S.D.; Potchen, M.J.; Dambrauskas, N.; Vigdorovich, V.; et al. Linking EPCR-Binding PfEMP1 to Brain Swelling in Pediatric Cerebral Malaria. Cell Host Microbe 2017, 22, 601–614.e605. [Google Scholar] [CrossRef]
- Chan, J.-A.; Howell, K.B.; Reiling, L.; Ataide, R.; Mackintosh, C.L.; Fowkes, F.J.I.; Petter, M.; Chesson, J.M.; Langer, C.; Warimwe, G.M.; et al. Targets of antibodies against Plasmodium falciparum-infected erythrocytes in malaria immunity. J. Clin. Investig. 2012, 122, 3227–3238. [Google Scholar] [CrossRef] [Green Version]
- Wittchen, E.S. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front. Biosci. 2009, 14, 2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejia, P.; Treviño-Villarreal, J.H.; Reynolds, J.S.; De Niz, M.; Thompson, A.; Marti, M.; Mitchell, J.R. A single rapamycin dose protects against late-stage experimental cerebral malaria via modulation of host immunity, endothelial activation and parasite sequestration. Malar. J. 2017, 16, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariaco, Y.; Lima, W.R.; Sousa, R.; Nascimento, L.A.C.; Briceño, M.P.; Fotoran, W.L.; Wunderlich, G.; Dos Santos, J.L.; Silva, N.M. Ethanolic extract of the fungus Trichoderma stromaticum decreases inflammation and ameliorates experimental cerebral malaria in C57BL/6 mice. Sci Rep. 2018, 8, 1547. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Combes, V.; Grau, G.E. Pathophysiology of Cerebral Malaria. Ann. N. Y. Acad. Sci. 2003, 992, 30–38. [Google Scholar] [CrossRef]
- Combes, V.; Coltel, N.; Faille, D.; Wassmer, S.C.; Grau, G.E. Cerebral malaria: Role of microparticles and platelets in alterations of the blood–brain barrier. Int. J. Parasitol. 2006, 36, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.V.; Ardeshirylajimi, A.; Dinarvand, P.; Yang, L.; Rezaie, A.R. Occupancy of human EPCR by protein C induces β-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood 2016, 128, 1884–1893. [Google Scholar] [CrossRef] [Green Version]
- Glennon, E.K.K.; Dankwa, S.; Smith, J.D.; Kaushansky, A. Opportunities for Host-targeted Therapies for Malaria. Trends Parasitol. 2018, 34, 843–860. [Google Scholar] [CrossRef]
- Mei, S.H.J.; McCarter, S.D.; Deng, Y.; Parker, C.H.; Liles, W.C.; Stewart, D.J. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007, 4, e269. [Google Scholar] [CrossRef]
- Kalkal, M.; Tiwari, M.; Thakur, R.S.; Awasthi, V.; Pande, V.; Chattopadhyay, D.; Das, J. Mesenchymal Stem Cells: A Novel Therapeutic Approach to Enhance Protective Immunomodulation and Erythropoietic Recovery in Malaria. Stem Cell Rev. Rep. 2021, 17, 1993–2002. [Google Scholar] [CrossRef]
- Souza, M.C.; Silva, J.D.; Pádua, T.A.; Torres, N.D.; Antunes, M.A.; Xisto, D.G.; Abreu, T.P.; Capelozzi, V.L.; Morales, M.M.; Sa Pinheiro, A.A. Mesenchymal stromal cell therapy attenuated lung and kidney injury but not brain damage in experimental cerebral malaria. Stem Cell Res. Ther. 2015, 6, 102. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Z.; Sendo, F. Neutrophils play a critical role in the pathogenesis of experimental cerebral malaria. Clin. Exp. Immunol. 2000, 120, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.S.; Awasthi, V.; Sanyal, A.; Chatterjee, S.; Rani, S.; Chauhan, R.; Kalkal, M.; Tiwari, M.; Pande, V.; Das, J. Mesenchymal stem cells protect against malaria pathogenesis by reprogramming erythropoiesis in the bone marrow. Cell Death Discov. 2020, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.S.; Tousif, S.; Awasthi, V.; Sanyal, A.; Atul, P.; Punia, P.; Das, J. Mesenchymal stem cells play an important role in host protective immune responses against malaria by modulating regulatory T cells. Eur. J. Immunol. 2013, 43, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wipasa, J.; Yan, H.; Zeng, M.; Makobongo, M.O.; Finkelman, F.D.; Kelso, A.; Good, M.F. The mechanism and significance of deletion of parasite-specific CD4+ T cells in malaria infection. J. Exp. Med. 2002, 195, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.S.; Schofield, L. Natural regulatory T cells in malaria: Host or parasite allies? PLoS Pathog. 2010, 6, e1000771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, R.; Spohn, G.; Baer, P.C. Mesenchymal stem/stromal cells in regenerative medicine: Can preconditioning strategies improve therapeutic efficacy. Transfus. Med. Hemotherapy 2016, 43, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Factors involved in blood-brain barrier breakdown during cerebral malaria.

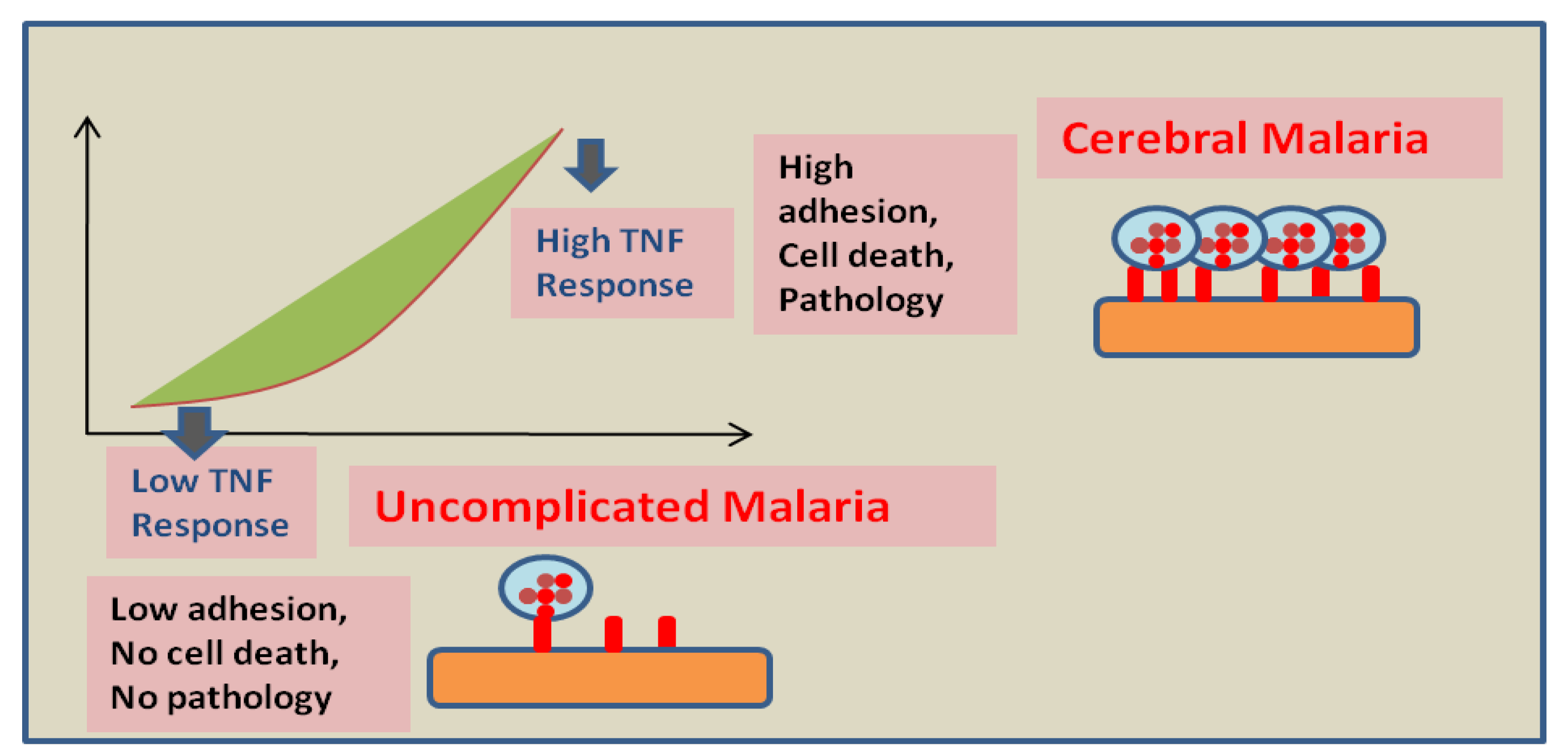
Figure 2.
Proposed effect of the host endothelial response to TNF on disease severity: Low endothelium response to TNF leads to a minimal adhesion of infected RBCs and host cells and might be responsible for the absence of pathology. However, high endothelium response to TNF leads to elevated adhesion of infected RBCs. Moreover, a strong pro-apoptotic signal is generated in high responders that possibly results in BBB breakage and vasogenic edema (this figure is adopted and modified from Wassmer SC et al. [31]).
Figure 2.
Proposed effect of the host endothelial response to TNF on disease severity: Low endothelium response to TNF leads to a minimal adhesion of infected RBCs and host cells and might be responsible for the absence of pathology. However, high endothelium response to TNF leads to elevated adhesion of infected RBCs. Moreover, a strong pro-apoptotic signal is generated in high responders that possibly results in BBB breakage and vasogenic edema (this figure is adopted and modified from Wassmer SC et al. [31]).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaudhary, A.; Kataria, P.; Surela, N.; Das, J. Pathophysiology of Cerebral Malaria: Implications of MSCs as A Regenerative Medicinal Tool. Bioengineering 2022, 9, 263. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9060263
AMA Style
Chaudhary A, Kataria P, Surela N, Das J. Pathophysiology of Cerebral Malaria: Implications of MSCs as A Regenerative Medicinal Tool. Bioengineering. 2022; 9(6):263. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9060263
Chicago/Turabian StyleChaudhary, Amrendra, Poonam Kataria, Neha Surela, and Jyoti Das. 2022. "Pathophysiology of Cerebral Malaria: Implications of MSCs as A Regenerative Medicinal Tool" Bioengineering 9, no. 6: 263. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9060263
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.