
Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review




Abstract
:Simple Summary
Abstract
1. Introduction
2. Sirtuins and Metabolism
3. Metabolism and Its (De)regulation
3.1. Metabolic Control
3.2. Metabolites Acting as Intracellular Signals
3.3. Metabolic Diseases
3.4. Metabolic Syndrome
3.5. Linking Metabolism to the Cell Cycle
4. The Cell Cycle and Its (De)regulation
4.1. Cell Cycle Control
4.2. Linking the Cell Cycle to Metabolism
5. Sirtuins Substrates Involved in Regulating Metabolism
5.1. AMPK—The Metabolic Swich
5.2. The PI3K/AKT/mTOR Pathway
5.3. PPAR Transcription Factors
5.4. Forkhead Box Proteins
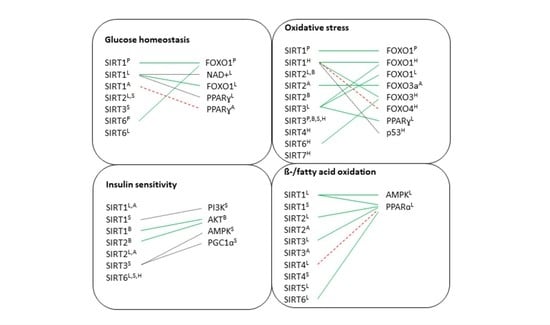
6. The Role of Sirtuins in Metabolic Tissues
6.1. Pancreas
6.2. Liver
6.3. Brain
6.4. Adipose Tissue
6.5. Skeletal Muscle
6.6. Heart
7. Discussion
7.1. Pancreas
7.2. Liver
7.3. Brain
7.4. Adipose Tissue
7.5. Skeletal Muscle
7.6. Heart
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A
Appendix A.1. Systematic Search Strategy
Appendix A.2. Study Eligibility
Appendix A.3. Study Selection Procedure

References
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 Family of Protein Deactylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiss, S.; Gartner, A. Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol. Cells 2009, 28, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cencioni, C.; Spallotta, F.; Mai, A.; Martelli, F.; Farsetti, A.; Zeiher, A.M.; Gaetano, C. Sirtuin function in aging heart and vessels. J. Mol. Cell. Cardiol. 2015, 83, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Redondo, P.; Vaquero, A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer 2013, 4, 148–163. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Simone, C.; Peserico, A. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar]
- Chen, D.; Vollmar, M.; Rossi, M.N.; Phillips, C.; Kraehenbuehl, R.; Slade, D.; Mehrotra, P.V.; von Delft, F.; Crosthwaite, S.K.; Gileadi, O.; et al. Identification of macrodomain proteins as novel O-acetyl-ADP-ribose deacetylases. J. Biol. Chem. 2011, 286, 13261–13271. [Google Scholar] [CrossRef] [Green Version]
- Bishop, N.A.; Guarente, L. Genetic links between diet and lifespan: Shared mechanisms from yeast to humans. Nat. Rev. Genet. 2007, 8, 835–844. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó-Mirabet, P.; Bermejo-Nogales, A.; Calduch-Giner, J.A.; Pérez-Sánchez, J. Tissue-specific gene expression and fasting regulation of sirtuin family in gilthead sea bream (Sparus aurata). J. Comp. Physiol. B 2017, 187, 153–163. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of Sirtuin Inhibition by Nicotinamide: Altering the NAD+ Cosubstrate Specificity of a Sir2 Enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic Shuttling of the NAD+-dependent Histone Deacetylase SIRT1. J. Biol. Chem. 2006, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Auwerx, J. Targeting Sirtuin 1 to Improve Metabolism: All You Need Is NAD+? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Verdin, E. Interphase Nucleo-Cytoplasmic Shuttling and Localization of SIRT2 during Mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Beirowski, B.; Gustin, J.; Armour, S.M.; Yamamoto, H.; Viader, A.; North, B.J.; Michan, S.; Baloh, R.H.; Golden, J.P.; Schmidt, R.E.; et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, E952–E961. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, W.C.; Albaugh, B.N.; Denu, J.M. Where in the cell is SIRT3?—Functional localization of an NAD+-dependent protein deacetylase. Biochem. J. 2008, 411, e11–e13. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Ogura, M.; Tanaka, D.; Inagaki, N. Localization of mouse mitochondrial SIRT proteins: Shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem. Biophys. Res. Commun. 2008, 366, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.J.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 Coordinates the Balance between Lipid Synthesis and Catabolism by Repressing Malonyl CoA Decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Baré, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.M.; Skinner, M.E.; et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 Homolog SIRT6 Is a Nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.A.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.F. A Role for SIRT6 in Secretion. Sci. Signal. 2013, 6, ec81. [Google Scholar] [CrossRef]
- Ardestani, P.M.; Liang, F. Sub-cellular localization, expression and functions of Sirt6 during the cell cycle in HeLa cells. Nucleus 2012, 3, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Kiran, S.; Chatterjee, N.; Singh, S.; Kaul, S.C.; Wadhwa, R.; Ramakrishna, G. Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. FEBS J. 2013, 280, 3451–3466. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Jo, Y.S.; Lo Sasso, G.; Stein, S.; Zhang, H.; Perino, A.; Lee, J.U.; Zeviani, M.; Romand, R.; Hottiger, M.O.; et al. A SIRT7-Dependent Acetylation Switch of GABPβ1 Controls Mitochondrial Function. Cell Metab. 2014, 20, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. SIRT7 and hepatic lipid metabolism. Front. Cell Dev. Biol. 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liang, Y.; Zhang, Q.; Wang, D.; Lei, M.; Qu, J.; He, X.; Lei, Q.; Wang, Y. Arginine methylation of SIRT 7 couples glucose sensing with mitochondria biogenesis. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, J.R. Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem. 1980, 49, 877–919. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins-emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Glycolysis and Gluconeogenesis. In Biochemistry, 5th ed.; Freeman, W.H., Ed.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Banerjee, R.; Becker, D.F.; Dickman, M.B.; Gladyshev, V.N.; Ragsdale, S.W. (Eds.) Redox Biochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; ISBN 9780471786245. [Google Scholar]
- Hsia, C.C.W.; Schmitz, A.; Lambertz, M.; Perry, S.F.; Maina, J.N. Evolution of air breathing: Oxygen homeostasis and the transitions from water to land and sky. Compr. Physiol. 2013, 3, 849–915. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, C.G. Physiology and genetics of sulfur-oxidizing bacteria. Adv. Microb. Physiol. 1998, 39, 235–289. [Google Scholar]
- Pace, N.R. The universal nature of biochemistry. Proc. Natl. Acad. Sci. USA 2001, 98, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Ebenhöh, O.; Heinrich, R. Evolutionary optimization of metabolic pathways. Theoretical reconstruction of the stoichiometry of ATP and NADH producing systems. Bull. Math. Biol. 2001, 63, 21–55. [Google Scholar] [CrossRef]
- Henze, K.; Martin, W. Evolutionary biology: Essence of mitochondria. Nature 2003, 426, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; Freeman, W.H., Ed.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Brand, M.D. Regulation analysis of energy metabolism. J. Exp. Biol. 1997, 200 Pt 2, 193–202. [Google Scholar]
- Soyer, O.S.; Salathé, M.; Bonhoeffer, S. Signal transduction networks: Topology, response and biochemical processes. J. Theor. Biol. 2006, 238, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Groen, A.K.; Wanders, R.J.A. Modern theories of metabolic control and their applications. Biosci. Rep. 1984, 4, 1–22. [Google Scholar] [CrossRef]
- Shaw-Dunn, J. Essentials of Human Anatomy and Physiology. J. Anat. 1991, 179, 206. [Google Scholar]
- Fell, D.A.; Thomas, S. Physiological control of metabolic flux: The requirement for multisite modulation. Biochem. J. 1995, 311 Pt 1, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Samoilov, M.; Plyasunov, S.; Arkin, A.P. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc. Natl. Acad. Sci. USA 2005, 102, 2310–2315. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, W.A. Transduction of biochemical signals across cell membranes. Q. Rev. Biophys. 2005, 38, 321–330. [Google Scholar] [CrossRef]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef]
- Kennedy, H.J.; Pouli, A.E.; Ainscow, E.K.; Jouaville, L.S.; Rizzuto, R.; Rutter, G.A. Glucose generates sub-plasma membrane ATP microdomains in single islet β-cells. Potential role for strategically located mitochondria. J. Biol. Chem. 1999, 274, 13281–13291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, S.; Dinh, T.T.; Zhang, Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res. 2000, 856, 37–47. [Google Scholar] [CrossRef]
- Zhang, M.; Galdieri, L.; Vancura, A. The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol. Cell. Biol. 2013, 33, 4701–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, M.; Sachs, K.; Heckelmann, M.; Maretzki, D.; Hofmann, K.P.; Schmidt, M.F.G. Palmitoylation of rhodopsin with S-protein acyltransferase: Enzyme catalyzed reaction versus autocatalytic acylation. Biochim. Biophys. Acta Lipids Lipid Metab. 1998, 1394, 90–98. [Google Scholar] [CrossRef]
- Blatnik, M.; Frizzell, N.; Thorpe, S.R.; Baynes, J.W. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: Formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes 2008, 57, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Trepanowski, J.F.; Canale, R.E.; Marshall, K.E.; Kabir, M.M.; Bloomer, R.J. Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: A summary of available findings. Nutr. J. 2011, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.K.; Allard, J.S.; Younts, C.M.; Ward, T.M.; de Cabo, R. Dietary interventions to extend life span and health span based on calorie restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A. Ketogenic diet for obesity: Friend or foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef] [Green Version]
- Madeo, F.; Pietrocola, F.; Eisenberg, T.; Kroemer, G. Caloric restriction mimetics: Towards a molecular definition. Nat. Rev. Drug Discov. 2014, 13, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Peregrín-Alvarez, J.M.; Sanford, C.; Parkinson, J. The conservation and evolutionary modularity of metabolism. Genome Biol. 2009, 10, R63. [Google Scholar] [CrossRef] [Green Version]
- Vernon, H.J. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 2015, 169, 778–782. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Handisurya, A. Metabolic diseases and associated complications: Sex and gender matter! Eur. J. Clin. Investig. 2009, 39, 631–648. [Google Scholar] [CrossRef]
- Weiss, K.M.; Ferrell, R.F.; Hanis, C.L. A new world syndrome of metabolic diseases with a genetic and evolutionary basis. Am. J. Phys. Anthropol. 1984, 27, 153–178. [Google Scholar] [CrossRef]
- Vilbergsson, S.; Sigurdsson, G.; Sigvaldason, H.; Hreidarsson, A.B.; Sigfusson, N. Prevalence and incidence of NIDDM in Iceland: Evidence for stable incidence among males and females 1967–1991—The Reykjavik Study. Diabet. Med. 1997, 14, 491–498. [Google Scholar] [CrossRef]
- Hales, C.N. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozanne, S.E.; Hales, C.N. Early programming of glucose-insulin metabolism. Trends Endocrinol. Metab. 2002, 13, 368–373. [Google Scholar] [CrossRef]
- Stöger, R. The thrifty epigenotype: An acquired and heritable predisposition for obesity and diabetes? Bioessays 2008, 30, 156–166. [Google Scholar] [CrossRef]
- Watve, M.G.; Yajnik, C.S. Evolutionary origins of insulin resistance: A behavioral switch hypothesis. BMC Evol. Biol. 2007, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, K.B.; Storey, J.M. Freeze tolerance and intolerance as strategies of winter survival in terrestrially-hibernating amphibians. Comp. Biochem. Physiol. Part A Physiol. 1986, 83, 613–617. [Google Scholar] [CrossRef]
- Moalem, S.; Storey, K.B.; Percy, M.E.; Peros, M.C.; Perl, D.P. The sweet thing about Type 1 diabetes: A cryoprotective evolutionary adaptation. Med. Hypotheses 2005, 65, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Jung, C.-H.; Park, J.-S.; Rhee, E.-J.; Kim, S.-W. Effects of smoking, alcohol, exercise, education, and family history on the metabolic syndrome as defined by the ATP III. Diabetes Res. Clin. Pract. 2005, 67, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The critical role of metabolic pathways in aging. Diabetes 2012, 61, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, K.; Ciardiello, F.; Giugliano, D. Unhealthy diets: A common soil for the association of metabolic syndrome and cancer. Endocrine 2014, 46, 39–42. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Halberg, N.; Wernstedt-Asterholm, I.; Scherer, P.E. The adipocyte as an endocrine cell. Endocrinol. Metab. Clin. N. Am. 2008, 37, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch. Eur. J. Physiol. 2007, 455, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Sethi, J.K. TNF-alpha and adipocyte biology. FEBS Lett. 2008, 582, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Gao, Z.; He, Q.; Zhou, D.; Guo, Z.; Ye, J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E333–E342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2003, 111, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Berg, H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 3, pp. 1–58. ISBN 2040-4603. [Google Scholar]
- Kim, S.H.; Reaven, G.M. Insulin resistance and hyperinsulinemia. Diabetes Care 2008, 31, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Stumvoll, M.; Jacob, S.; Wahl, H.G.; Hauer, B.; Löblein, K.; Grauer, P.; Becker, R.; Nielsen, M.; Renn, W.; Häring, H. Suppression of systemic, intramuscular, and subcutaneous adipose tissue lipolysis by insulin in humans. J. Clin. Endocrinol. Metab. 2000, 85, 3740–3745. [Google Scholar] [CrossRef]
- Steil, G.M.; Trivedi, N.; Jonas, J.C.; Hasenkamp, W.M.; Sharma, A.; Bonner-Weir, S.; Weir, G.C. Adaptation of beta-cell mass to substrate oversupply: Enhanced function with normal gene expression. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E788–E796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Q.; Jetton, T.L.; Leahy, J.L. β-cell adaptation to insulin resistance. Increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic zucker fatty rats. J. Biol. Chem. 2002, 277, 39163–39168. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Hribal, M.L.; Lin, H.V.; Bennett, W.R.; Ward, A.; Accili, D. Role of the forkhead protein FoxO1 in β cell compensation to insulin resistance. J. Clin. Investig. 2006, 116, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Mizutani, M.; Oikawa, S.; Hiraku, Y.; Kawanishi, S. Oxidative DNA damage by hyperglycemia-related aldehydes and its marked enhancement by hydrogen peroxide. FEBS Lett. 2003, 554, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Shen, N.; Zhang, M.-L.; Pan, F.-Y.; Wang, C.; Jia, W.-P.; Liu, C.; Gao, Q.; Gao, X.; Xue, B.; et al. Egr-1 decreases adipocyte insulin sensitivity by tilting PI3K/Akt and MAPK signal balance in mice. EMBO J. 2011, 30, 3754–3765. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, M.; Yamaguchi, H.; Hirata, A.; Daimon, M.; Tominaga, M.; Kato, T. Insulin activates p38 mitogen-activated protein (MAP) kinase via a MAP kinase kinase (MKK) 3/MKK 6 pathway in vascular smooth muscle cells. Eur. J. Clin. Investig. 2000, 30, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Cramer, H.; Schmenger, K.; Heinrich, K.; Horstmeyer, A.; Böning, H.; Breit, A.; Piiper, A.; Lundstrom, K.; Müller-Esterl, W.; Schroeder, C. Coupling of endothelin receptors to the ERK/MAP kinase pathway roles of palmitoylation and Gαq. Eur. J. Biochem. 2001, 268, 5449–5459. [Google Scholar] [CrossRef]
- Ramachandran, V.; Saravanan, R. Glucose uptake through translocation and activation of GLUT4 in PI3K/Akt signaling pathway by asiatic acid in diabetic rats. Hum. Exp. Toxicol. 2015, 34, 884–893. [Google Scholar] [CrossRef]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temelkova-Kurktschiev, T.; Hanefeld, M. The Lipid Triad in Type 2 Diabetes—Prevalence and Relevance of Hypertriglyceridaemia/Low High-Density Lipoprotein Syndrome in Type 2 Diabetes. Exp. Clin. Endocrinol. Diabetes 2004, 112, 75–79. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.-K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.-J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.F.; Uffelman, K.D.; Szeto, L.W.; Steiner, G. Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL Apo B production in normal weight and obese individuals. Diabetes 1993, 42, 833–842. [Google Scholar] [CrossRef]
- Sparks, J.D.; O’Dell, C.; Chamberlain, J.M.; Sparks, C.E. Insulin-dependent apolipoprotein B degradation is mediated by autophagy and involves class I and class III phosphatidylinositide 3-kinases. Biochem. Biophys. Res. Commun. 2013, 435, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.K.; Russell, C.D.; Grauso, N.L.; Brolin, R.E. Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J. Clin. Investig. 1993, 92, 2191–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panarotto, D.; Rémillard, P.; Bouffard, L.; Maheux, P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur. J. Clin. Investig. 2002, 32, 84–92. [Google Scholar] [CrossRef]
- Murdoch, S.J.; Breckenridge, W.C. Influence of lipoprotein lipase and hepatic lipase on the transformation of VLDL and HDL during lipolysis of VLDL. Atherosclerosis 1995, 118, 193–212. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. Lipoprotein retention- and clues for atheroma regression. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1536–1540. [Google Scholar] [CrossRef] [Green Version]
- Packard, C.J.; Shepherd, J. Lipoprotein heterogeneity and apolipoprotein B metabolism. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3542–3556. [Google Scholar] [CrossRef] [PubMed]
- Rocchini, A.P.; Moorehead, C.; DeRemer, S.; Goodfriend, T.L.; Ball, D.L. Hyperinsulinemia and the aldosterone and pressor responses to angiotensin II. Hypertension 1990, 15, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Tack, G.J.J.; Smits, P.; Willemsen, J.J.; Lenders, J.W.M.; Thien, T.; Lutterman, J.A. Effects of insulin on vascular tone and sympathetic nervous system in NIDDM. Diabetes 1996, 45, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeli, S.; Schling, P.; Gorzelniak, K.; Boschmann, M.; Janke, J.; Ailhaud, G.; Teboul, M.; Massiéra, F.; Sharma, A.M. The adipose-tissue renin-angiotensin-aldosterone system: Role in the metabolic syndrome? Int. J. Biochem. Cell Biol. 2003, 35, 807–825. [Google Scholar] [CrossRef]
- Teng, M.H.; Bartholomew, J.C.; Bissell, M.J. Insulin effect on the cell cycle: Analysis of the kinetics of growth parameters in confluent chick cells. Proc. Natl. Acad. Sci. USA 1976, 73, 3173–3177. [Google Scholar] [CrossRef] [Green Version]
- Cho, R.J.; Campbell, M.J.; Winzeler, E.A.; Steinmetz, L.; Conway, A.; Wodicka, L.; Wolfsberg, T.G.; Gabrielian, A.E.; Landsman, D.; Lockhart, D.J.; et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 1998, 2, 65–73. [Google Scholar] [CrossRef]
- Spellman, P.T.; Sherlock, G.; Zhang, M.Q.; Iyer, V.R.; Anders, K.; Eisen, M.B.; Brown, P.O.; Botstein, D.; Futcher, B. Comprehensive Identification of Cell Cycle-regulated Genes of the Yeast Saccharomyces cerevisiae by Microarray Hybridization. Mol. Biol. Cell 1998, 9, 3273–3297. [Google Scholar] [CrossRef]
- Tu, B.P.; Kudlicki, A.; Rowicka, M.; McKnight, S.L. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.P.; Mohler, R.E.; Liu, J.C.; Dombek, K.M.; Young, E.T.; Synovec, R.E.; McKnight, S.L. Cyclic changes in metabolic state during the life of a yeast cell. Proc. Natl. Acad. Sci. USA 2007, 104, 16886–16891. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic control of the cell division cycle in yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A family of human cdc2-related protein kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [Green Version]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Larochelle, S.; Pandur, J.; Fisher, R.P.; Salz, H.K.; Suter, B. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998, 12, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Queralt, E.; Uhlmann, F. Cdk-counteracting phosphatases unlock mitotic exit. Curr. Opin. Cell Biol. 2008, 20, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kornbluth, S. All aboard the cyclin train: Subcellular trafficking of cyclins and their CDK partners. Trends Cell Biol. 1999, 9, 207–210. [Google Scholar] [CrossRef]
- Errico, A.; Deshmukh, K.; Tanaka, Y.; Pozniakovsky, A.; Hunt, T. Identification of substrates for cyclin dependent kinases. Adv. Enzyme Regul. 2010, 50, 375–399. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, H.; Zhou, T.; Zhou, J.; Herrup, K. Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J. Biol. Chem. 2012, 287, 25985–25994. [Google Scholar] [CrossRef] [Green Version]
- Barberis, M.; Linke, C.; Adrover, M.À.; González-Novo, A.; Lehrach, H.; Krobitsch, S.; Posas, F.; Klipp, E. Sic1 plays a role in timing and oscillatory behaviour of B-type cyclins. Biotechnol. Adv. 2012, 30, 108–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, A.W.; Kirschner, M.W. Cyclin synthesis drives the early embryonic cell cycle. Nature 1989, 339, 275–280. [Google Scholar] [CrossRef]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008, 9, R149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyczkowski, J.; Vingron, M. Comparative analysis of cell cycle regulated genes in eukaryotes. Genome Inform. 2005, 16, 125–131. [Google Scholar] [PubMed]
- Mill, P.; Mo, R.; Hu, M.C.; Dagnino, L.; Rosenblum, N.D.; Hui, C.C. Shh controls epithelial proliferation via independent pathways that converge on N-Myc. Dev. Cell 2005, 9, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrs, S.; Kutzner, N.; Vlad, A.; Grunwald, T.; Ziegler, S.; Müller, O. Chronological expression of Wnt target genes Ccnd1, Myc, Cdkn1a, Tfrc, Plf1 and Ramp3. Cell Biol. Int. 2009, 33, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, D.; Jones, A.B.; Diwan, B.A.; Perantoni, A.O.; Hochadel, J.F.; Anderson, L.M. Epidermal growth factor and transforming growth factor-α-associated overexpression of cyclin D1, Cdk4, and c-Myc during hepatocarcinogenesis in Helicobacter hepaticus-infected A/JCr mice. Cancer Res. 1998, 58, 3590–3597. [Google Scholar] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, C.; Dittrich, O.; Kiermaier, A.; Dohmann, K.; Menkel, A.; Eilers, M.; Lüscher, B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001, 15, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartel, A.L.; Radhakrishnan, S.K. Lost in transcription: P21 repression, mechanisms, and consequences. Cancer Res. 2005, 65, 3980–3985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Weber, J.D. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem. J. 2007, 407, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014, 3, e02872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Dean, D.C. Rb-mediated chromatin structure regulation and transcriptional repression. Oncogene 2001, 20, 3134–3138. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Roger, I.; Solomon, D.L.; Sewing, A.; Land, H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene 1997, 14, 2373–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandramohan, V.; Mineva, N.D.; Burke, B.; Jeay, S.; Wu, M.; Shen, J.; Yang, W.; Hann, S.R.; Sonenshein, G.E. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27Kip1 cyclin dependent kinase inhibitor. J. Cell. Biochem. 2008, 104, 2091–2106. [Google Scholar] [CrossRef]
- Ohtani, K.; DeGregori, J.; Nevins, J.R. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef] [Green Version]
- Resnitzky, D.; Reed, S.I. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol. Cell. Biol. 1995, 15, 3463–3469. [Google Scholar] [CrossRef] [Green Version]
- Coverley, D.; Laman, H.; Laskey, R. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 2002, 4, 523–528. [Google Scholar] [CrossRef]
- Thangue, L.N. Regulation of E2F transcription by cyclin ECdk2 kinase mediated through p300/CBP coactivators. Nat. Cell Biol. 2000, 2, 232–239. [Google Scholar] [CrossRef]
- Zhu, H.; Nie, L.; Maki, C.G. Cdk2-dependent inhibition of p21 stability via a C-terminal cyclin-binding motif. J. Biol. Chem. 2005, 280, 29282–29288. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999, 9, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef]
- Yam, C.H.; Fung, T.K.; Poon, R.Y.C. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Sørensen, C.S.; Kramer, E.; Santoni-Rugiu, E.; Lindeneg, C.; Peters, J.M.; Bartek, J.; Lukas, J. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 1999, 401, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Alvarez, M.; Meijer, L.T.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.R.; Medema, R.H. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Indjeian, V.B.; McManus, M.; Wang, L.; Dynlacht, B.D. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev. Cell 2002, 3, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Suzuki, A.; Sugimura, K.; Okumura, K.; Zineldeen, D.H.; Shimada, M.; Niida, H.; Mizuno, T.; Hanaoka, F.; Nakanishi, M. Cyclin A-Cdk1 regulates the origin firing program in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3184–3189. [Google Scholar] [CrossRef] [Green Version]
- Gong, D.; Pomerening, J.R.; Myers, J.W.; Gustavsson, C.; Jones, J.T.; Hahn, A.T.; Meyer, T.; Ferrell, J.E. Cyclin A2 Regulates Nuclear-Envelope Breakdown and the Nuclear Accumulation of Cyclin B1. Curr. Biol. 2007, 17, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; Desouza, C.; Gabrielli, B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 2774, 4261–4268. [Google Scholar] [CrossRef] [Green Version]
- Gong, D.; Ferrell, J.E. The Roles of Cyclin A2, B1, and B2 in Early and Late Mitotic Events. Mol. Biol. Cell 2010, 21, 3149–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, F.; Vidal-Laliena, M.; Canela, N.; Busino, L.; Martinez-Balbas, M.A.; Pagano, M.; Agell, N.; Bachs, O. Degradation of cyclin A is regulated by acetylation. Oncogene 2009, 28, 2654–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciortino, S.; Gurtner, A.; Manni, I.; Fontemaggi, G.; Dey, A.; Sacchi, A.; Ozato, K.; Piaggio, G. The cyclin B1 gene is actively transcribed during mitosis in HeLa cells. EMBO Rep. 2001, 2, 1018–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H. Cdc20: A WD40 Activator for a Cell Cycle Degradation Machine. Mol. Cell 2007, 27, 3–16. [Google Scholar] [CrossRef]
- Cross, F.R.; Buchler, N.E.; Skotheim, J.M. Evolution of networks and sequences in eukaryotic cell cycle control. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3532–3544. [Google Scholar] [CrossRef] [Green Version]
- Nevins, J.R. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar] [CrossRef]
- Blanchet, E.; Annicotte, J.-S.; Lagarrigue, S.; Aguilar, V.; Clapé, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef] [Green Version]
- Raimundo, N.; Song, L.; Shutt, T.E.; McKay, S.E.; Cotney, J.; Guan, M.-X.; Gilliland, T.C.; Hohuan, D.; Santos-Sacchi, J.; Shadel, G.S. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 2012, 148, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Annicotte, J.-S.; Blanchet, E.; Chavey, C.; Iankova, I.; Costes, S.; Assou, S.; Teyssier, J.; Dalle, S.; Sardet, C.; Fajas, L. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat. Cell Biol. 2009, 11, 1017–1023. [Google Scholar] [CrossRef]
- Naaz, A.; Holsberger, D.R.; Iwamoto, G.A.; Nelson, A.; Kiyokawa, H.; Cooke, P.S. Loss of cyclin-dependent kinase inhibitors produces adipocyte hyperplasia and obesity. FASEB J. 2004, 18, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, A.; Wada, J.; Makino, H. Cell cycle abnormality in metabolic syndrome and nuclear receptors as an emerging therapeutic target. Acta Med. Okayama 2013, 67, 129–134. [Google Scholar]
- Koopman, R.; Ly, C.H.; Ryall, J.G. A metabolic link to skeletal muscle wasting and regeneration. Front. Physiol. 2014, 5, 32. [Google Scholar] [CrossRef]
- Dungan, C.M.; Williamson, D.L. High-Fat Diet Regulation of Cell Cycle. Int. J. Exerc. Sci. Conf. Proc. 2014, 9, 15. [Google Scholar]
- Tahergorabi, Z.; Khazaei, M. Imbalance of angiogenesis in diabetic complications: The mechanisms. Int. J. Prev. Med. 2012, 3, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Cell cycle regulation in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, S59–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Watt, M.J.; Febbraio, M.A. Cytokine Regulation of AMPK signalling. Front. Biosci. 2009, 14, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Marcinko, K.; Steinberg, G.R. The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp. Physiol. Exp. Physiol. 2014, 9912, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Winder, W.W.; Hardie, D.G. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol. Leg. Content 1999, 277, E1–E10. [Google Scholar] [CrossRef]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 5741, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Banko, M.R.; Brunet, A. AMP-activated protein kinase and FoxO transcription factors in dietary restriction-induced longevity. Ann. N. Y. Acad. Sci. 2009, 1170, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banko, M.R.; Allen, J.J.; Schaffer, B.E.; Wilker, E.W.; Tsou, P.P.; White, J.L.; Villén, J.; Wang, B.; Kim, S.R.; Sakamoto, K.; et al. Chemical Genetic Screen for AMPKα2 Substrates Uncovers a Network of Proteins Involved in Mitosis. Mol. Cell 2011, 44, 878–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-C.; Day, R.M. Angiotensin II regulates activation of Bim via Rb/E2F1 during apoptosis: Involvement of interaction between AMPKβ1/2 and Cdk4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L228–L238. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Leevers, S.J.; Panayotou, G.; Waterfield, M.D.; Waterfield, M.D. Phosphoinositide 3-kinases: A conserved family of signal transducers. Trends Biochem. Sci. 1997, 22, 267–272. [Google Scholar] [CrossRef]
- Foster, F.M.; Traer, C.J.; Abraham, S.M.; Fry, M.J. The phosphoinositide (PI) 3-kinase family. J. Cell Sci. 2003, 116, 3037–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rameh, L.E.; Cantley, L.C. The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 1999, 274, 8347–8350. [Google Scholar] [CrossRef] [Green Version]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.-H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.-M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, C.; Yoshino, K.I.; Yonezawa, K. mTOR integrates amino acid- and energy-sensing pathways. Biochem. Biophys. Res. Commun. 2004, 313, 443–446. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Lemberger, T.; Saladin, R.; Vazquez, M.; Assimacopoulos, F.; Staels, B.; Desvergne, B.; Wahli, W.; Auwerx, J. Expression of the peroxisome proliferator-activated receptor alpha gene is stimulated by stress and follows a diurnal rhythm. J. Biol. Chem. 1996, 271, 1764–1769. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Okamoto, S.; Lee, S.; Saito, K.; Shiuchi, T.; Minokoshi, Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor a gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the a2 form of AMP-activated protein kinase. Mol. Cell. Biol. 2007, 27, 4317–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalev, A.; Siegrist-Kaiser, C.A.; Yen, P.M.; Wahli, W.; Burger, A.G.; Chin, W.W.; Meier, C.A. The peroxisome proliferator-activated receptor alpha is a phosphoprotein: Regulation by insulin. Endocrinology 1996, 137, 4499–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yessoufou, A.; Atègbo, J.-M.; Attakpa, E.; Hichami, A.; Moutairou, K.; Dramane, K.L.; Khan, N. a Peroxisome proliferator-activated receptor-alpha modulates insulin gene transcription factors and inflammation in adipose tissues in mice. Mol. Cell. Biochem. 2009, 323, 101–111. [Google Scholar] [CrossRef]
- Rosenson, R.S. Fenofibrate: Treatment of hyperlipidemia and beyond. Expert Rev. Cardiovasc. Ther. 2008, 6, 1319–1330. [Google Scholar] [CrossRef]
- Seo, Y.S.; Kim, J.H.; Jo, N.Y.; Choi, K.M.; Baik, S.H.; Park, J.-J.; Kim, J.S.; Byun, K.S.; Bak, Y.-T.; Lee, C.H.; et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J. Gastroenterol. Hepatol. 2008, 23, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Andreelli, F.; Auboeuf, D.; Roques, M.; Vallier, P.; Riou, J.P.; Auwerx, J.; Laville, M.; Vidal, H. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes 1999, 48, 699–705. [Google Scholar] [CrossRef]
- Medina-Gomez, G.; Gray, S.; Vidal-Puig, A. Adipogenesis and lipotoxicity: Role of peroxisome proliferator-activated receptor gamma (PPARgamma) and PPARgammacoactivator-1 (PGC1). Public Health Nutr. 2007, 10, 1132–1137. [Google Scholar] [CrossRef]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; O’Reilly, C.P.; Chapes, S.K.; Mora, S. Adiponectin and leptin are secreted through distinct trafficking pathways in adipocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abella, A.; Dubus, P.; Malumbres, M.; Rane, S.G.; Kiyokawa, H.; Sicard, A.; Vignon, F.; Langin, D.; Barbacid, M.; Fajas, L. Cdk4 promotes adipogenesis through PPARγ activation. Cell Metab. 2005, 2, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iankova, I.; Petersen, R.K.; Annicotte, J.-S.; Chavey, C.; Hansen, J.B.; Kratchmarova, I.; Sarruf, D.; Benkirane, M.; Kristiansen, K.; Fajas, L. Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol. 2006, 20, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, V.; Annicotte, J.S.; Escote, X.; Vendrell, J.; Langin, D.; Fajas, L. Cyclin G2 regulates adipogenesis through PPARγ coactivation. Endocrinology 2010, 151, 5247–5254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Christopher Corton, J.; Lynis Dohm, G.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) α knock-out mice. Evidence for compensatory regulation by PPARδ. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hon, M.; Evans, R.M. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- Luquet, S.; Gaudel, C.; Holst, D.; Lopez-Soriano, J.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of PPAR delta in lipid absorption and metabolism: A new target for the treatment of type 2 diabetes. Biochim. Biophys. Acta 2005, 1740, 313–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef]
- Kim, J.E.; Chen, J. Regulation of Peroxisome Proliferator–Activated Receptor-γ Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Sozio, M.S.; Lu, C.; Zeng, Y.; Liangpunsakul, S.; Crabb, D.W. Activated AMPK inhibits PPAR- and PPAR- transcriptional activity in hepatoma cells. AJP Gastrointest. Liver Physiol. 2011, 301, G739–G747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado de Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Davis, K.E.; Moldes, M.; Farmer, S.R. The forkhead transcription factor FoxC2 inhibits white adipocyte differentiation. J. Biol. Chem. 2004, 279, 42453–42461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigel, D.; Jäckle, H. The fork head domain: A novel DNA binding motif of eukaryotic transcription factors? Cell 1990, 63, 455–456. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Knöchel, W.; Martínez, D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000, 14, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuteja, G.; Kaestner, K.H. SnapShot: Forkhead Transcription Factors I. Cell 2007, 130, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuteja, G.; Kaestner, K.H. SnapShot: Forkhead Transcription Factors II. Cell 2007, 131, 192. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.E.; Kundaje, A.; Brunet, A. Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 2016, 15, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Coffer, P.J. The extending network of FOXO transcriptional target genes. Antioxid. Redox Signal. 2011, 14, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs, W.H.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 2003, 4, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; DePinho, R.A.; Accili, D. Impaired Regulation of Hepatic Glucose Production in Mice Lacking the Forkhead Transcription Factor Foxo1 in Liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, J.; Oki, M.; Cao, Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008, 582, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [Green Version]
- Linke, C.; Klipp, E.; Lehrach, H.; Barberis, M.; Krobitsch, S. Fkh1 and Fkh2 associate with Sir2 to control CLB2 transcription under normal and oxidative stress conditions. Front. Physiol. 2013, 4, 173. [Google Scholar] [CrossRef] [Green Version]
- Oellerich, M.F.; Potente, M. FOXOs and Sirtuins in Vascular Growth, Maintenance, and Aging. Circ. Res. 2012, 110, 1238–1251. [Google Scholar] [CrossRef] [Green Version]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 2006, 4, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 2007, 282, 34356–34364. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-R.; Lai, Y.; Lin, S.; Li, X.; Fu, Y.-C.; Xu, W.-C. SIRT1 interacts with metabolic transcriptional factors in the pancreas of insulin-resistant and calorie-restricted rats. Mol. Biol. Rep. 2013, 40, 3373–3380. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Lu, Y.; Zhang, J.; Jian, F.; Liu, Y.; Li, F.; Li, W.; Wang, X.; Li, G. Activation of SIRT1 protects pancreatic β-cells against palmitate-induced dysfunction. Biochem. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1815–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buteau, J.; Shlien, A.; Foisy, S.; Accili, D. Metabolic diapause in pancreatic beta-cells expressing a gain-of-function mutant of the forkhead protein Foxo1. J. Biol. Chem. 2007, 282, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.C.; Tanabe, K.; Cras-Méneur, C.; Abumrad, N.A.; Bernal-Mizrachi, E.; Permutt, M.A. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 2008, 57, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Vetterli, L.; Brun, T.; Giovannoni, L.; Bosco, D.; Maechler, P. Resveratrol Potentiates Glucose-stimulated Insulin Secretion in INS-1E -Cells and Human Islets through a SIRT1-dependent Mechanism. J. Biol. Chem. 2011, 286, 6049–6060. [Google Scholar] [CrossRef] [Green Version]
- Hughes, K.J.; Meares, G.P.; Hansen, P.A.; Corbett, J.A. FoxO1 and SIRT1 regulate β-cell responses to nitric oxide. J. Biol. Chem. 2011, 286, 8338–8348. [Google Scholar] [CrossRef] [Green Version]
- Bastien-Dionne, P.O.; Valenti, L.; Kon, N.; Gu, W.; Buteau, J. Glucagon-like peptide 1 inhibits the sirtuin deacetylase SirT1 to stimulate pancreatic β-cell mass expansion. Diabetes 2011, 60, 3217–3222. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.-P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.E.; Thai, K.; Advani, S.L.; Cummins, C.L.; Kepecs, D.M.; Schroer, S.A.; Woo, M.; Zhang, Y. SIRT1 activation ameliorates hyperglycaemia by inducing a torpor-like state in an obese mouse model of type 2 diabetes. Diabetologia 2015, 58, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cheng, J.; Zhang, Y.; Li, N.; Chen, L. Effects of caloric restriction on SIRT1 expression and apoptosis of islet beta cells in type 2 diabetic rats. Acta Diabetol. 2010, 47, 177–185. [Google Scholar] [CrossRef]
- Desai, T.; Koulajian, K.; Ivovic, A.; Breen, D.M.; Luu, L.; Tsiani, E.L.; Wheeler, M.B.; Giacca, A. Pharmacologic or genetic activation of SIRT1 attenuates the fat-induced decrease in beta-cell function in vivo. Nutr. Diabetes 2019, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S.I. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, K.M.; Mills, K.F.; Satoh, A.; Imai, S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 2008, 7, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Pinho, A.V.; Bensellam, M.; Wauters, E.; Rees, M.; Giry-Laterriere, M.; Mawson, A.; Ly, L.Q.; Biankin, A.V.; Wu, J.; Laybutt, D.R.; et al. Pancreas-Specific Sirt1-Deficiency in Mice Compromises Beta-Cell Function without Development of Hyperglycemia. PLoS ONE 2015, 10, e0128012. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.-H.; Xu, X.; Kim, H.-S.; Xiao, Z.; Deng, C.-X. SIRT1 Deacetylates FOXA2 and Is Critical for Pdx1 Transcription and β-Cell Formation. Int. J. Biol. Sci. 2013, 9, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Luu, L.; Dai, F.F.; Prentice, K.J.; Huang, X.; Hardy, A.B.; Hansen, J.B.; Liu, Y.; Joseph, J.W.; Wheeler, M.B. The loss of Sirt1 in mouse pancreatic beta cells impairs insulin secretion by disrupting glucose sensing. Diabetologia 2013, 56, 2010–2020. [Google Scholar] [CrossRef]
- Wu, S.Y.; Liang, J.; Yang, B.C.; Leung, P.S. SIRT1 Activation Promotes β-Cell Regeneration by Activating Endocrine Progenitor Cells via AMPK Signaling-Mediated Fatty Acid Oxidation. Stem Cells 2019, 37, 1416–1428. [Google Scholar] [CrossRef]
- Yu, L.; Chen, J.F.; Shuai, X.; Xu, Y.; Ding, Y.; Zhang, J.; Yang, W.; Liang, X.; Su, D.; Yan, C. Artesunate protects pancreatic beta cells against cytokine-induced damage via SIRT1 inhibiting NF-κB activation. J. Endocrinol. Investig. 2016, 39, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Glinka, Y.; Udovyk, O.; Hasilo, C.; Paraskevas, S.; Wang, Q. GABA protects pancreatic beta cells against apoptosis by increasing SIRT1 expression and activity. Biochem. Biophys. Res. Commun. 2014, 452, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sun, F.; Ren, L.; Yang, H.; Tian, N.; Peng, S. Resveratrol controlled the fate of porcine pancreatic stem cells through the Wnt/β-catenin signaling pathway mediated by Sirt1. PLoS ONE 2017, 12, e0187159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.C.; Chen, Y.L.; Hwang, P.A.; Chen, T.H.; Chou, T.C. Fucoidan ameliorates pancreatic β-cell death and impaired insulin synthesis in streptozotocin-treated β cells and mice via a Sirt-1-dependent manner. Mol. Nutr. Food Res. 2017, 61, 1700136. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Caton, P.W.; Richardson, S.J.; Kieswich, J.; Bugliani, M.; Holland, M.L.; Marchetti, P.; Morgan, N.G.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013, 56, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chung, A.C.K.; Fan, R.; Lee, H.M.; Xu, G.; Tomlinson, B.; Chan, J.C.N.; Kong, A.P.S. Sirt3 Deficiency Increased the Vulnerability of Pancreatic Beta Cells to Oxidative Stress-Induced Dysfunction. Antioxid. Redox Signal. 2017, 27, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.S.; Oh, J.E.; Nan, J.; Lee, H.; Jung, H.S.; Chung, S.S.; Park, K.S. SIRT3 Overexpression Attenuates Palmitate-Induced Pancreatic β-Cell Dysfunction. PLoS ONE 2015, 10, e0124744. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Ma, X.-J.; Wu, L.-N.; Zhao, Y.-Y.; Zhang, P.-Y.; Zhang, Y.-H.; Shao, M.-W.; Liu, F.; Li, F.; Qin, G.-J. Sirtuin-3 (SIRT3) protects pancreatic β-cells from endoplasmic reticulum (ER) stress-induced apoptosis and dysfunction. Mol. Cell. Biochem. 2016, 420, 95–106. [Google Scholar] [CrossRef]
- Lee, S.-J.; Choi, S.-E.; Jung, I.-R.; Lee, K.-W.; Kang, Y. Protective effect of nicotinamide on high glucose/palmitate-induced glucolipotoxicity to INS-1 beta cells is attributed to its inhibitory activity to sirtuins. Arch. Biochem. Biophys. 2013, 535, 187–196. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreaticβ Cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Q.; Huan, Y.; Li, R.; Li, C.; Sun, S.; Guo, N.; Yang, M.; Liu, S.; Shen, Z. Sirtuin 5 overexpression attenuates glucolipotoxicity-induced pancreatic β cells apoptosis and dysfunction. Exp. Cell Res. 2018, 371, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fei, X. SIRT5 regulates pancreatic β-cell proliferation and insulin secretion in type 2 diabetes. Exp. Ther. Med. 2018, 16, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Zhang, N.; Zhang, Z.; Nipper, M.; Zhu, Z.; Leighton, J.; Xu, K.; Musi, N.; Wang, P. SIRT6-mediated transcriptional suppression of Txnip is critical for pancreatic beta cell function and survival in mice. Diabetologia 2018, 61, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.-Y.; Wang, J.; Ka, S.-O.; Bae, E.J.; Park, B.-H.; Thorens, B.; Orci, L.; Ahlgren, U.; Jonsson, J.; Jonsson, L.; et al. Insulin secretion impairment in Sirt6 knockout pancreatic β cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci. Rep. 2016, 6, 30321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Wang, G.; Tao, R.; Wu, P.; Kono, T.; Li, K.; Ding, W.-X.; Tong, X.; Tersey, S.A.; Harris, R.A.; et al. Sirtuin 6 regulates glucose-stimulated insulin secretion in mouse pancreatic beta cells. Diabetologia 2016, 59, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.-G.; Hong, D.-F.; Zhang, C.-W.; Sun, X.-D.; Wang, Z.-F.; Shi, Y.; Liu, J.-W.; Shen, G.-L.; Zhang, Y.-B.; Cheng, J.; et al. Sirtuin 1 facilitates chemoresistance of pancreatic cancer cells by regulating adaptive response to chemotherapy-induced stress. Cancer Sci. 2014, 105, 445–454. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1a and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J. Biol. Chem. 2012, 287, 3850–3858. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.H.; Song, S.J.; Kim, A.; Choi, Y.; Seok, J.W.; Kim, H.J.; Lee, Y.J.; Lee, K.S.; Kim, J. Suppression of PPARγ-mediated monoacylglycerol O-acyltransferase 1 expression ameliorates alcoholic hepatic steatosis. Sci. Rep. 2016, 6, 29352. [Google Scholar] [CrossRef] [Green Version]
- Gazit, V.; Huang, J.; Weymann, A.; Rudnick, D.A. Analysis of the role of hepatic PPARγ expression during mouse liver regeneration. Hepatology 2012, 56, 1489–1498. [Google Scholar] [CrossRef]
- Inoue, M.; Ohtake, T.; Motomura, W.; Takahashi, N.; Hosoki, Y.; Miyoshi, S.; Suzuki, Y.; Saito, H.; Kohgo, Y.; Okumura, T. Increased expression of PPARγ in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 2005, 336, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, J.D.; Stewart, A.M.; Saupe, K.W. Downregulation of plasma insulin levels and hepatic PPARγ expression during the first week of caloric restriction in mice. Exp. Gerontol. 2008, 43, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.H.; Kim, H.S.; Xiao, C.; Xu, X.; Gavrilova, O.; Deng, C.X. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J. Clin. Investig. 2011, 121, 4477–4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Rhee, J.; Donovan, J.; Kitamura, Y.; Altomonte, J.; Dong, H. Insulin-regulated hepatic gluconeogenesis through FOXO1—PGC-1 a interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kim, T.H.; Bae, J.S.; Kim, M.Y.; Kim, K.S.; Ahn, Y.H. Role of resveratrol in FOXO1-mediated gluconeogenic gene expression in the liver. Biochem. Biophys. Res. Commun. 2010, 403, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.M.; Schwarz, M.; Coward, P.; Houze, J.; Sawyer, J.K.; Kelley, K.L.; Chai, A.; Rudel, L.L. Effects of peroxisome proliferator-activated receptor alpha/delta agonists on HDL-cholesterol in vervet monkeys. J. Lipid Res. 2005, 46, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Deleye, Y.; Cotte, A.K.; Hannou, S.A.; Hennuyer, N.; Bernard, L.; Derudas, B.; Caron, S.; Legry, V.; Vallez, E.; Dorchies, E.; et al. CDKN2A/p16INK4a suppresses hepatic fatty acid oxidation through the AMPKα2-SIRT1-PPARα signaling pathway. J. Biol. Chem. 2020, 6, jbc.RA120.012543. [Google Scholar] [CrossRef]
- Bantubungi, K.; Hannou, S.A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4aregulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1 α pathway. Diabetes 2014, 63, 3199–3209. [Google Scholar] [CrossRef] [Green Version]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Tsiaousi, E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes, Obes. Metab. 2010, 12, 365–383. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. An Emerging Role of mTOR in Lipid Biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Jeon, S.M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs Inhibit mTORC1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Liu, C.; Hu, K.; Greenberg, A.; Wu, D.; Ausman, L.M.; McBurney, M.W.; Wang, X.-D. Ablation of systemic SIRT1 activity promotes nonalcoholic fatty liver disease by affecting liver-mesenteric adipose tissue fatty acid mobilization. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2783–2790. [Google Scholar] [CrossRef]
- Zhang, E.; Cui, W.; Lopresti, M.; Mashek, M.G.; Najt, C.P.; Hu, H.; Mashek, D.G. Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J. Lipid Res. 2020, 61, 338–350. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Yun, D.; Li, L.; Zhao, W.; Li, Y.; Liu, X.; Liu, Z. Alternate-day fasting alleviates diabetes-induced glycolipid metabolism disorders: Roles of FGF21 and bile acids. J. Nutr. Biochem. 2020, 83, 108403. [Google Scholar] [CrossRef]
- Ishizuka, K.; Kon, K.; Lee-Okada, H.C.; Arai, K.; Uchiyama, A.; Yamashina, S.; Yokomizo, T.; Ikejima, K. Aging exacerbates high-fat diet-induced steatohepatitis through alteration in hepatic lipid metabolism in mice. J. Gastroenterol. Hepatol. 2020, 35, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, K.; Yu, W.; Wang, H.; Liu, L.; Wu, Q.; Li, S.; Guo, J. MiR-181b regulates steatosis in nonalcoholic fatty liver disease via targeting SIRT1. Biochem. Biophys. Res. Commun. 2017, 493, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; An, H.J.; Kim, D.H.; Lee, B.; Lee, H.J.; Ullah, S.; Kim, S.J.; Jeong, H.O.; Moon, K.M.; Lee, E.K.; et al. Novel SIRT1 activator MHY2233 improves glucose tolerance and reduces hepatic lipid accumulation in db/db mice. Bioorg. Med. Chem. Lett. 2018, 28, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-Q.; Chen, L.-L.; Li, N.-X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; He, K.; Li, P.; Gong, J.; Zhu, X.; Ye, S.; Ou, Z.; Ren, G. SIRT1 upregulation protects against liver injury induced by a HFD through inhibiting CD36 and the NF-κB pathway in mouse kupffer cells. Mol. Med. Rep. 2018, 18, 1609–1615. [Google Scholar] [CrossRef]
- Chen, L.; Deng, X.; Li, N. Effects of calorie restriction on SIRT1 expression in liver of nonalcoholic fatty liver disease: Experiment with rats. Chin. Med. J. 2007, 87, 1434–1437. [Google Scholar]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.-J.; Cheng, H.-L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.Z.; He, X.; Mottawea, W.; Seifert, E.L.; Jardine, K.; Dewar-Darch, D.; Cron, G.O.; Harper, M.E.; Stintzi, A.; McBurney, M.W. The SIRT1 deacetylase protects mice against the symptoms of metabolic syndrome. FASEB J. 2014, 28, 1306–1316. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 Deacetylates and Positively Regulates the Nuclear Receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Lang, C.H.; Villegas, K.A.; Tong, M.; Mark, N.M.; De La Monte, S.M.; Wands, J.R. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J. Hepatol. 2011, 54, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-W.; Kao, H.-H.; Wu, C.-H. Exercise training upregulates SIRT1 to attenuate inflammation and metabolic dysfunction in kidney and liver of diabetic db/db mice. Nutr. Metab. 2019, 16, 22. [Google Scholar] [CrossRef]
- Zhou, R.; Yi, L.; Ye, X.; Zeng, X.; Liu, K.; Qin, Y.; Zhang, Q.; Mi, M. Resveratrol Ameliorates Lipid Droplet Accumulation in Liver through a SIRT1/ ATF6-Dependent Mechanism. Cell. Physiol. Biochem. 2018, 51, 2397–2420. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Chen, Y.; Zhang, Y.; Shen, M.; Jia, P.; Ji, S.; Wang, T. Resveratrol Alleviates Endoplasmic Reticulum Stress–Associated Hepatic Steatosis and Injury in Mice Challenged with Tunicamycin. Mol. Nutr. Food Res. 2020, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.Z.; Zhao, Z.H.; Zhang, R.N.; Pan, Q.; Gong, Z.Z.; Sun, C.; Fan, J.G. Folic acid attenuates high-fat diet-induced steatohepatitis via deacetylase SIRT1-dependent restoration of PPAR. World J. Gastroenterol. 2020, 26, 2203–2220. [Google Scholar] [CrossRef]
- Liou, C.-J.; Dai, Y.-W.; Wang, C.-L.; Fang, L.-W.; Huang, W.-C. Maslinic acid protects against obesity-induced nonalcoholic fatty liver disease in mice through regulation of the Sirt1/AMPK signaling pathway. FASEB J. 2019, 33, 11791–11803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dihingia, A.; Ozah, D.; Ghosh, S.; Sarkar, A.; Baruah, P.K.; Kalita, J.; Sil, P.C.; Manna, P. Vitamin K1 inversely correlates with glycemia and insulin resistance in patients with type 2 diabetes (T2D) and positively regulates SIRT1/AMPK pathway of glucose metabolism in liver of T2D mice and hepatocytes cultured in high glucose. J. Nutr. Biochem. 2018, 52, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Luna-Vital, D.; Luzardo-Ocampo, I.; Cuellar-Nuñez, M.L.; Loarca-Piña, G.; Gonzalez de Mejia, E. Maize extract rich in ferulic acid and anthocyanins prevents high-fat-induced obesity in mice by modulating SIRT1, AMPK and IL-6 associated metabolic and inflammatory pathways. J. Nutr. Biochem. 2020, 79, 108343. [Google Scholar] [CrossRef]
- Liou, C.-J.; Wei, C.-H.; Chen, Y.-L.; Cheng, C.-Y.; Wang, C.-L.; Huang, W.-C. Fisetin Protects against Hepatic Steatosis through Regulation of the Sirt1/AMPK and Fatty Acid β-Oxidation Signaling Pathway in High-Fat Diet-Induced Obese Mice. Cell. Physiol. Biochem. 2018, 49, 1870–1884. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, T.; Wang, Z.; Xu, Y.; Zhang, Q.; Luo, D. Low molecular weight fucoidan attenuates liver injury via SIRT1/AMPK/PGC1α axis in db/db mice. Int. J. Biol. Macromol. 2018, 112, 929–936. [Google Scholar] [CrossRef]
- Li, J.; Liu, M.; Yu, H.; Wang, W.; Han, L.; Chen, Q.; Ruan, J.; Wen, S.; Zhang, Y.; Wang, T. Mangiferin Improves Hepatic Lipid Metabolism Mainly Through Its Metabolite-Norathyriol by Modulating SIRT-1/AMPK/SREBP-1c Signaling. Front. Pharmacol. 2018, 9, 201. [Google Scholar] [CrossRef]
- Chen, X.Y.; Cai, C.Z.; Yu, M.L.; Feng, Z.M.; Zhang, Y.W.; Liu, P.H.; Zeng, H.; Yu, C.H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Gu, L.; Cai, N.; Lyu, Y.; Yao, L.; Wang, F.; Xu, H.; Hu, Z.; Li, H.; Xu, X. γ-Mangostin Ameliorates Free Fatty Acid-Induced Lipid Accumulation via the SIRT1/LKB1/AMPK Pathway in HepG2 and L02 Cells. J. Agric. Food Chem. 2019, 67, 13929–13938. [Google Scholar] [CrossRef]
- Li, D.; Liu, F.; Wang, X.; Li, X. Apple Polyphenol Extract Alleviates High-Fat-Diet-Induced Hepatic Steatosis in Male C57BL/6 Mice by Targeting LKB1/AMPK Pathway. J. Agric. Food Chem. 2019, 67, 12208–12218. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Poudel, A.; Welchko, R.; Mekala, N.; Chandramani-Shivalingappa, P.; Rosca, M.G.; Li, L. Liraglutide improves insulin sensitivity in high fat diet induced diabetic mice through multiple pathways. Eur. J. Pharmacol. 2019, 861. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, P.; Shi, L.; Liu, F.; Tang, A.; Xu, S. Phoenixin 14 inhibits high-fat diet-induced non-alcoholic fatty liver disease in experimental mice. Drug Des. Devel. Ther. 2020, 14, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Kim, B.; Lee, C.; Joung, H.; Kim, B.K.; Choi, I.S.; Hyun, C.K. Comprehensive amelioration of high-fat diet-induced metabolic dysfunctions through activation of the PGC-1α pathway by probiotics treatment in mice. PLoS ONE 2020, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Chen, H.; Zaky, A.; Pollock, C.; Saad, S. SIRT1 overexpression attenuates offspring metabolic and liver disorders as a result of maternal high-fat feeding. J. Physiol. 2019, 597, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez, Y.R.; Quiroga, S.; Prochnik, A.; Wald, M.; Tellechea, M.L.; Genaro, A.M.; Burguenõ, A.L. Influence of prenatal stress on metabolic abnormalities induced by postnatal intake of a high-fat diet in BALB/c mice. J. Dev. Orig. Health Dis. 2020, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Saad, S.; Chen, H.; Pollock, C.A. Parental sirt1 overexpression attenuate metabolic disorders due to maternal high-fat feeding. Int. J. Mol. Sci. 2020, 21, 7342. [Google Scholar] [CrossRef]
- Arteaga, M.; Shang, N.; Ding, X.; Yong, S.; Cotler, S.J.; Denning, M.F.; Shimamura, T.; Breslin, P.; Bernhard, L.; Qiu, W. Inhibition of SIRT2 suppresses hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 2, G1155–G1168. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Guo, Y.-Y.; Li, B.-Y.; Peng, W.-Q.; Chang, X.-X.; Gao, X.; Tang, Q.-Q. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 2019, 294, 11805–11816. [Google Scholar] [CrossRef]
- Lemos, V.; de Oliveira, R.M.; Naia, L.; Szegö, É.; Ramos, E.; Pinho, S.; Magro, F.; Cavadas, C.; Rego, A.C.; Costa, V.; et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 2017, 26, 4105–4117. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Inaba, Y.; Kimura, K.; Matsumoto, M.; Kaneko, S.; Kasuga, M.; Inoue, H. Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat. Commun. 2018, 9, 30. [Google Scholar] [CrossRef]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie Restriction and SIRT3 Trigger Global Reprogramming of the Mitochondrial Protein Acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Dittenhafer-Reed, K.E.; Richards, A.L.; Fan, J.; Smallegan, M.J.; Fotuhi Siahpirani, A.; Kemmerer, Z.A.; Prolla, T.A.; Roy, S.; Coon, J.J.; Denu, J.M. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015, 21, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devires, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.K.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xing, D.; Du, X.; Peng, T.; McFadden, J.; Wen, L.; Lei, H.; Dong, W.; Liu, G.; Wang, Z.; et al. Sirtuin 3 improves fatty acid metabolism in response to high nonesterified fatty acids in calf hepatocytes by modulating gene expression. J. Dairy Sci. 2020, 103, 6557–6568. [Google Scholar] [CrossRef]
- Barroso, E.; Rodríguez-Rodríguez, R.; Zarei, M.; Pizarro-Degado, J.; Planavila, A.; Palomer, X.; Villarroya, F.; Vázquez-Carrera, M. SIRT3 deficiency exacerbates fatty liver by attenuating the HIF1α-LIPIN 1 pathway and increasing CD36 through Nrf2. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef]
- Pinterić, M.; Podgorski, I.I.; Hadžija, M.P.; Bujak, I.T.; Dekanić, A.; Bagarić, R.; Farkaš, V.; Sobočanec, S.; Balog, T. Role of sirt3 in differential sex-related responses to a high-fat diet in mice. Antioxidants 2020, 9, 174. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Kong, W.; Zhu, R.; Liang, K.; Kan, Q.; Lou, Y.; Liu, X. Regulation of SIRT3/FOXO1 Signaling Pathway in Rats with Non-alcoholic Steatohepatitis by Salvianolic Acid B. Arch. Med. Res. 2017, 48, 506–512. [Google Scholar] [CrossRef]
- Liu, J.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Amorim, J.A.; Machado, I.F.; Castela, A.C.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P.; Palmeira, C.M. The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function. Int. J. Mol. Sci. 2020, 21, 4896. [Google Scholar] [CrossRef]
- Sharma, G.; Parihar, A.; Parihar, P.; Parihar, M.S. Downregulation of sirtuin 3 by palmitic acid increases the oxidative stress, impairment of mitochondrial function, and apoptosis in liver cells. J. Biochem. Mol. Toxicol. 2019, 33, e22337. [Google Scholar] [CrossRef]
- Chen, M.; Hui, S.; Lang, H.; Zhou, M.; Zhang, Y.; Kang, C.; Zeng, X.; Zhang, Q.; Yi, L.; Mi, M. SIRT3 Deficiency Promotes High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease in Correlation with Impaired Intestinal Permeability through Gut Microbial Dysbiosis. Mol. Nutr. Food Res. 2019, 63, 1800612. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Tang, K.; Chen, R.; Liang, S.; Liang, Y.; Han, L.; Jin, L.; Liang, Z.; Chen, Y.; et al. Berberine Ameliorates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease in Rats via Activation of SIRT3/AMPK/ACC Pathway. Curr. Med. Sci. 2019, 39, 37–43. [Google Scholar] [CrossRef]
- Sun, R.; Kang, X.; Zhao, Y.; Wang, Z.; Wang, R.; Fu, R.; Li, Y.; Hu, Y.; Wang, Z.; Shan, W.; et al. Sirtuin 3-mediated deacetylation of acyl-CoA synthetase family member 3 by protocatechuic acid attenuates non-alcoholic fatty liver disease. Br. J. Pharmacol. 2020, 177, 4166–4180. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Li, Y.; Chen, H.; Wang, C.; Wong, V.K.W.; Jiang, Z.; Zhang, W. Phytotherapy using blueberry leaf polyphenols to alleviate non-alcoholic fatty liver disease through improving mitochondrial function and oxidative defense. Phytomedicine 2020, 69, 153209. [Google Scholar] [CrossRef]
- Lee, D.; Kim, Y.R.; Kim, J.S.; Kim, D.; Kim, S.; Kim, S.Y.; Jang, K.; Lee, J.D.; Yang, C.S. Dietary schizophyllan reduces mitochondrial damage by activating SIRT3 in mice. Arch. Pharm. Res. 2020, 43, 449–461. [Google Scholar] [CrossRef]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Liu, J.; Shen, S.; Tong, Q.; Ma, X.; Lin, L. SIRT3 promotes lipophagy and chaperon-mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death Differ. 2019, 103, 6557–6568. [Google Scholar] [CrossRef] [Green Version]
- Buler, M.; Aatsinki, S.M.; Izzi, V.; Uusimaa, J.; Hakkola, J. SIRT5 is under the control of PGC-1o: And AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014, 28, 3225–3237. [Google Scholar] [CrossRef]
- Li, L.; Zhang, P.; Bao, Z.; Wang, T.; Liu, S.; Huang, F. PGC-1α Promotes Ureagenesis in Mouse Periportal Hepatocytes through SIRT3 and SIRT5 in Response to Glucagon. Sci. Rep. 2016, 6, 24156. [Google Scholar] [CrossRef] [Green Version]
- Haüssinger, D. Nitrogen metabolism in liver: Structural and functional organization and physiological relevance. Biochem. J. 1990, 267, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; de Boer, V.C.J.; Finley, L.W.S.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 Represses Peroxisome Proliferator-Activated Receptor Activity To Suppress Hepatic Fat Oxidation. Mol. Cell. Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yao, W.; He, Q.; Shao, Y.; Zheng, R.; Huang, F. L-leucine stimulates glutamate dehydrogenase activity and glutamate synthesis by regulating mTORC1/SIRT4 pathway in pig liver. Anim. Nutr. 2018, 4, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Assadi-Porter, F.M.; Reiland, H.; Sabatini, M.; Lorenzini, L.; Carnicelli, V.; Rogowski, M.; Alpergin, E.S.S.; Tonelli, M.; Ghelardoni, S.; Saba, A.; et al. Metabolic Reprogramming by 3-Iodothyronamine (T1AM): A New Perspective to Reverse Obesity through Co-Regulation of Sirtuin 4 and 6 Expression. Int. J. Mol. Sci. 2018, 19, 1535. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liu, Y.; Fu, Y.; Liu, X.; Zhou, X. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Du, Y.; Hu, H.; Qu, S.; Wang, J.; Hua, C.; Zhang, J.; Wei, P.; He, X.; Hao, J.; Liu, P.; et al. SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 2018, 36, 347–357. [Google Scholar] [CrossRef]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Kim, H.S.; Xiao, C.; Wang, R.H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Bang, I.H.; Wang, J.; Mao, Y.; Yang, J.D.; Na, S.Y.; Seo, J.K.; Choi, H.S.; Bae, E.J.; Park, B.H. ERRγ suppression by Sirt6 alleviates cholestatic liver injury and fibrosis. JCI Insight 2020, 5, e137566. [Google Scholar] [CrossRef]
- Ka, S.-O.; Bang, I.H.; Bae, E.J.; Park, B.-H. Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J. 2017, 31, 3999–4010. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Huang, M.; Kim, H.G.; Zhang, Y.; Chowdhury, K.; Cai, W.; Saxena, R.; Schwabe, R.F.; Liangpunsakul, S.; Dong, X.C. SIRT6 Protects Against Liver Fibrosis by Deacetylation and Suppression of SMAD3 in Hepatic Stellate Cells. CMGH 2020, 10, 341–364. [Google Scholar] [CrossRef]
- Carreira, M.C.; Izquierdo, A.G.; Amil, M.; Casanueva, F.F.; Crujeiras, A.B. Oxidative Stress Induced by Excess of Adiposity Is Related to a Downregulation of Hepatic SIRT6 Expression in Obese Individuals. Oxid. Med. Cell. Longev. 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Wang, Z.Z.; Liang, Y.; Zhang, L.; Zhang, N.; Liu, Q.; Wang, Z.Z. Phosphodiesterase 4 inhibitor activates AMPK-SIRT6 pathway to prevent aging-related adipose deposition induced by metabolic disorder. Aging 2018, 10, 2394–2406. [Google Scholar] [CrossRef]
- Tao, R.; Xiong, X.; DePinho, R.A.; Deng, C.X.; Dong, X.C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013, 288, 29252–29259. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.G.; Ramadori, G.; Ioris, R.M.; Galiè, M.; Berglund, E.D.; Coate, K.C.; Fujikawa, T.; Pucciarelli, S.; Moreschini, B.; Amici, A.; et al. Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol. Metab. 2015, 4, 846–856. [Google Scholar] [CrossRef]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, Z.; Xu, Z.; Wan, J.; Hua, T.; Sun, Q. Exercise ameliorates insulin resistance and improves SIRT6-mediated insulin signaling transduction in liver of obese rats. Can. J. Physiol. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Karim, M.F.; Sato, Y.; Senokuchi, T.; Miyata, K.; Fukuda, T.; Go, C.; Tasaki, M.; Uchimura, K.; Kadomatsu, T.; et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. 2014, 19, 712–721. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 represses myc activity to suppress er stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Toshikuni, N.; Tsutsumi, M.; Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393–8406. [Google Scholar] [CrossRef]
- Cone, R.D.; Cowley, M.A.; Butler, A.A.; Fan, W.; Marks, D.L.; Low, M.J. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. 2001, 25, S63–S67. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef]
- Sasaki, T.; Kitamura, T. Roles of FoxO1 and Sirt1 in the central regulation of food intake. Endocr. J. 2010, 57, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskandar, K.; Cao, Y.; Hayashi, Y.; Nakata, M.; Takano, E.; Yada, T.; Zhang, C.; Ogawa, W.; Oki, M.; Chua, S.; et al. PDK-1/FoxO1 pathway in POMC neurons regulates Pomc expression and food intake. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E787–E798. [Google Scholar] [CrossRef] [Green Version]
- Çakir, I.; Perello, M.; Lansari, O.; Messier, N.J.; Vaslet, C.A.; Nillni, E.A. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS ONE 2009, 4, e8322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyr, N.E.; Steger, J.S.; Toorie, A.M.; Yang, J.Z.; Stuart, R.; Nillni, E.A. Central Sirt1 regulates body weight and energy expenditure along with the POMC-derived peptide α-MSH and the processing enzyme CPE production in diet-induced obese male rats. Endocrinology 2015, 156, 961–974. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Kikuchi, O.; Shimpuku, M.; Susanti, V.Y.; Yokota-Hashimoto, H.; Taguchi, R.; Shibusawa, N.; Sato, T.; Tang, L.; Amano, K.; et al. Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia 2014, 57, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Brace, C.S.; Ben-Josef, G.; West, T.; Wozniak, D.F.; Holtzman, D.M.; Herzog, E.D.; Imai, S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J. Neurosci. 2010, 30, 10220–10232. [Google Scholar] [CrossRef]
- Ramadori, G.; Fujikawa, T.; Fukuda, M.; Anderson, J.; Morgan, D.A.; Mostoslavsky, R.; Stuart, R.C.; Perello, M.; Vianna, C.R.; Nillni, E.A.; et al. SIRT1 Deacetylase in POMC Neurons Is Required for Homeostatic Defenses against Diet-Induced Obesity. Cell Metab. 2010, 12, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Susanti, V.Y.; Sasaki, T.; Yokota-Hashimoto, H.; Matsui, S.; Lee, Y.S.; Kikuchi, O.; Shimpuku, M.; Kim, H.J.; Kobayashi, M.; Kitamura, T. Sirt1 rescues the obesity induced by insulin-resistant constitutively-nuclear FoxO1 in POMC neurons of male mice. Obesity 2014, 22, 2115–2119. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Sarruf, D.A.; Li, P.; Osborn, O.; Sanchez-Alavez, M.; Talukdar, S.; Chen, A.; Bandyopadhyay, G.; Xu, J.; Morinaga, H.; et al. Neuronal sirt1 deficiency increases insulin sensitivity in both brain and peripheral tissues. J. Biol. Chem. 2013, 288, 10722–10735. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, B.D.; Lau, C.L.; Ryall, J.G.; Thomas, D.S.; Klugmann, M.; Beart, P.M.; Rae, C.D. Silent information regulator 1 modulator resveratrol increases brain lactate production and inhibits mitochondrial metabolism, whereas SRT1720 increases oxidative metabolism. J. Neurosci. Res. 2015, 93, 1147–1156. [Google Scholar] [CrossRef]
- Heyward, F.D.; Gilliam, D.; Coleman, M.A.; Gavin, C.F.; Wang, J.; Kaas, G.; Trieu, R.; Lewis, J.; Moulden, J.; Sweatt, J.D. Obesity Weighs down Memory through a Mechanism Involving the Neuroepigenetic Dysregulation of Sirt1. J. Neurosci. 2016, 36, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Yao, M.; Wei, L.; Ge, S. Obesity caused by a high-fat diet regulates the Sirt1/PGC-1α/FNDC5/BDNF pathway to exacerbate isoflurane-induced postoperative cognitive dysfunction in older mice. Nutr. Neurosci. 2019, 23, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lee, A.J.; Kang, K.S.; Song, J.H.; Zhu, B.T. 4-Hydroxyestrone, an Endogenous Estrogen Metabolite, Can Strongly Protect Neuronal Cells against Oxidative Damage. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Flores-León, M.; Pérez-Domínguez, M.; González-Barrios, R.; Arias, C. Palmitic Acid-Induced NAD+ Depletion is Associated with the Reduced Function of SIRT1 and Increased Expression of BACE1 in Hippocampal Neurons. Neurochem. Res. 2019, 44, 1745–1754. [Google Scholar] [CrossRef]
- Wang, F.; Shang, Y.; Zhang, R.; Gao, X.; Zeng, Q. A SIRT1 agonist reduces cognitive decline in type 2 diabetic rats through antioxidative and anti-inflammatory mechanisms. Mol. Med. Rep. 2018, 19, 1040–1048. [Google Scholar] [CrossRef]
- Wu, D.; Qiu, Y.; Gao, X.; Yuan, X.B.; Zhai, Q. Overexpression of SIRT1 in mouse forebrain impairs lipid/glucose metabolism and motor function. PLoS ONE 2011, 6, e21759. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, D.A.; Martínez, G.; Romero, A.; Vázquez, M.J.; Boit, K.D.; Dopeso-Reyes, I.G.; López, M.; Vidal, A.; Nogueiras, R.; Diéguez, C. The central sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes 2011, 60, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.E.; Supinski, A.M.; Bonkowski, M.S.; Donmez, G.; Guarente, L.P. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009, 23, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Büeler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Ran, M.; Li, Z.; Yang, L.; Tong, L.; Zhang, L.; Dong, H. Calorie restriction attenuates cerebral ischemic injury via increasing SIRT1 synthesis in the rat. Brain Res. 2015, 1610, 61–68. [Google Scholar] [CrossRef]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1 alpha inhibition ameliorates neonatal brain damage after hypoxic-ischemic injury. Acta Neurochir. Suppl. 2008, 102, 395–399. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, T.S.; Sanchez-Mendoza, E.H.; Nascentes, L.M.; Schultz Moreira, A.R.; Sardari, M.; Dzyubenko, E.; Kleinschnitz, C.; Hermann, D.M. Moderate Protein Restriction Protects Against Focal Cerebral Ischemia in Mice by Mechanisms Involving Anti-inflammatory and Anti-oxidant Responses. Mol. Neurobiol. 2019, 56, 8477–8488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koronowski, K.B.; Khoury, N.; Saul, I.; Loris, Z.B.; Cohan, C.H.; Stradecki-Cohan, H.M.; Dave, K.R.; Young, J.I.; Perez-Pinzon, M.A. Neuronal SIRT1 (silent information regulator 2 homologue 1) regulates glycolysis and mediates resveratrol-induced ischemic tolerance. Stroke 2017, 48, 3117–3125. [Google Scholar] [CrossRef]
- Muñoz, A.; Corrêa, C.L.; Lopez-Lopez, A.; Costa-Besada, M.A.; Diaz-Ruiz, C.; Labandeira-Garcia, J.L. Physical Exercise Improves Aging-Related Changes in Angiotensin, IGF-1, SIRT1, SIRT3, and VEGF in the Substantia Nigra. J. Gerontol. Ser. A 2018, 73, 1594–1601. [Google Scholar] [CrossRef]
- Buxton, O.M.; Cain, S.W.; O’Connor, S.P.; Porter, J.H.; Duffy, J.F.; Wang, W.; Czeisler, C.A.; Shea, S.A. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci. Transl. Med. 2012, 4, 129ra43. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-Fat Diet Disrupts Behavioral and Molecular Circadian Rhythms in Mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.E.; Liu, A.C.; Hirota, T.; Miraglia, L.J.; Welch, G.; Pongsawakul, P.Y.; Liu, X.; Atwood, A.; Huss, J.W.; Janes, J.; et al. A Genome-wide RNAi Screen for Modifiers of the Circadian Clock in Human Cells. Cell 2009, 139, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.C.; Hong, H.; Weidemann, B.J.; Ramsey, K.M.; Affinati, A.H.; Schmidt, M.S.; Cedernaes, J.; Omura, C.; Braun, R.; Lee, C.; et al. NAD+ Controls Circadian Reprogramming through PER2 Nuclear Translocation to Counter Aging. Mol. Cell 2020, 78, 835–849.e7. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Drevet, K.; Pévet, P.; Challet, E. Daily meal timing is not necessary for resetting the main circadian clock by calorie restriction. J. Neuroendocrinol. 2008, 20, 251–260. [Google Scholar] [CrossRef]
- Froy, O.; Miskin, R. Effect of feeding regimens on circadian rhythms: Implications for aging and longevity. Aging 2010, 2, 7–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, H.; Genzer, Y.; Cohen, R.; Chapnik, N.; Madar, Z.; Froy, O. Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J. 2012, 26, 3493–3502. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, W.; Ming, R.; Deng, Q.; Zuo, A.; Zhang, S.; Ying, Y.; Zhao, Y.; Ma, J. Noninvasive 40-Hz Light Flicker Rescues Circadian Behavior and Abnormal Lipid Metabolism Induced by Acute Ethanol Exposure via Improving SIRT1 and the Circadian Clock in the Liver-Brain Axis. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Zhang, W.; Gao, X.; Watts, L. Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol. Aging 2015, 36, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Bakshi, V.; Lin, A.L. Early shifts of brain metabolism by caloric restriction preserve white matter integrity and long-term memory in aging mice. Front. Aging Neurosci. 2015, 7, 213. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The Ketogenic Diet Increases Mitochondrial Uncoupling Protein Levels and Activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabke, P.; Das, A.M. Mechanism of action of ketogenic diet treatment: Impact of decanoic acid and beta—hydroxybutyrate on sirtuins and energy metabolism in hippocampal murine neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 2015, 85, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic Diet Improves Motor Performance but Not Cognition in Two Mouse Models of Alzheimer’s Pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Wu, Z. Microglia-aging: Roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav. Brain Res. 2009, 201, 1–7. [Google Scholar] [CrossRef]
- Kim, M.-S.; Pak, Y.K.; Jang, P.-G.; Namkoong, C.; Choi, Y.-S.; Won, J.-C.; Kim, K.-S.; Kim, S.-W.; Kim, H.-S.; Park, J.-Y.; et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Pauli, J.R.; Prada, P.; Cintra, D.E.; Rocha, G.Z.; Moraes, J.C.; Frederico, M.J.S.; da Luz, G.; Pinho, R.A.; Carvalheira, J.B.C.; et al. Inhibition of hypothalamic Foxo1 expression reduced food intake in diet-induced obesity rats. J. Physiol. 2009, 587, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Doan, K.V.; Kinyua, A.W.; Yang, D.J.; Ko, C.M.; Moh, S.H.; Shong, K.E.; Kim, H.; Park, S.; Kim, D.-H.; Kim, I.; et al. FoxO1 in dopaminergic neurons regulates energy homeostasis and targets tyrosine hydroxylase. Nat. Commun. 2016, 7, 12733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef]
- Plum, L.; Lin, H.V.; Dutia, R.; Tanaka, J.; Aizawa, K.S.; Matsumoto, M.; Kim, A.J.; Cawley, N.X.; Paik, J.-H.; Loh, Y.P.; et al. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat. Med. 2009, 15, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.W.; Donato, J.; Berglund, E.D.; Choi, Y.H.; Kohno, D.; Elias, C.F.; De Pinho, R.A.; Elmquist, J.K. FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Investig. 2012, 122, 2578–2589. [Google Scholar] [CrossRef]
- Heinrich, G.; Meece, K.; Wardlaw, S.L.; Accili, D. Preserved energy balance in mice lacking FoxO1 in neurons of Nkx2.1 lineage reveals functional heterogeneity of FoxO1 signaling within the hypothalamus. Diabetes 2014, 63, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, P.; Karthikeyan, A.; Lu, J.; Ling, E.A.; Dheen, S.T. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 2015, 311, 398–414. [Google Scholar] [CrossRef]
- Sanphui, P.; Biswas, S.C. FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 2013, 4, e625. [Google Scholar] [CrossRef]
- Qin, W.; Zhao, W.; Ho, L.; Wang, J.; Walsh, K.; Gandy, S.; Pasinetti, G.M. Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann. N. Y. Acad. Sci. 2008, 1147, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Li, S.; Nielsen, M.; Carcione, T.; Liang, W.S.; Shi, J. Sirtuin 3 attenuates amyloid-β induced neuronal hypometabolism. Aging 2018, 10, 2874–2883. [Google Scholar] [CrossRef] [PubMed]
- Zemva, J.; Schilbach, K.; Stöhr, O.; Moll, L.; Franko, A.; Krone, W.; Wiesner, R.J.; Schubert, M. Central FoxO3a and FoxO6 expression is downregulated in obesity induced diabetes but not in aging. Exp. Clin. Endocrinol. Diabetes 2012, 120, 340–350. [Google Scholar] [CrossRef]
- Salih, D.A.M.; Rashid, A.J.; Colas, D.; de la Torre-Ubieta, L.; Zhu, R.P.; Morgan, A.A.; Santo, E.E.; Ucar, D.; Devarajan, K.; Cole, C.J.; et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev. 2012, 26, 2780–2801. [Google Scholar] [CrossRef] [Green Version]
- Sajan, M.; Hansen, B.; Ivey, R.; Sajan, J.; Ari, C.; Song, S.; Braun, U.; Leitges, M.; Farese-Higgs, M.; Farese, R.V. Brain Insulin Signaling Is Increased in Insulin-Resistant States and Decreases in FOXOs and PGC-1α and Increases in Aβ1-40/42 and Phospho-Tau May Abet Alzheimer Development. Diabetes 2016, 65, 1892–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chu, X.; Yin, M.; Liu, X.; Yuan, H.; Niu, Y.; Fu, L. MTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav. Brain Res. 2014, 264, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dong, W.; Wang, R.; Li, Y.; Xu, B.; Zhang, J.; Zhao, Z.; Wang, Y. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res. Bull. 2015, 116, 67–72. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kemball, C.C.; Flynn, C.T.; Wood, M.R.; Whitton, J.L.; Kiosses, W.B. Short-term fasting induces profound neuronal autophagy. Autophagy 2010, 6, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Taïb, B.; Bouyakdan, K.; Hryhorczuk, C.; Rodaros, D.; Fulton, S.; Alquier, T. Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J. Biol. Chem. 2013, 288, 37216–37229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocai, A.; Lam, T.K.T.; Obici, S.; Gutierrez-Juarez, R.; Muse, E.D.; Arduini, A.; Rossetti, L. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Investig. 2006, 116, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-Y.; Cai, Y.-B.; Liu, Z. Activation of AMPK improves lipopolysaccharide-induced dysfunction of the blood-brain barrier in mice. Brain Inj. 2015, 9052, 1–8. [Google Scholar] [CrossRef]
- Yang, T.T.; Shih, Y.S.; Chen, Y.W.; Kuo, Y.M.; Lee, C.W. Glucose regulates amyloid β production via AMPK. J. Neural Transm. 2015, 122, 1381–1390. [Google Scholar] [CrossRef]
- Huang, J.; Wang, X.; Zhu, Y.; Li, Z.; Zhu, Y.; Wu, J.; Qin, Z.; Xiang, M.; Lin, F. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci. Ther. 2019, 25, 796–807. [Google Scholar] [CrossRef]
- Jiang, N.; Lv, J.; Wang, H.; Huang, H.; Wang, Q.; Lu, C.; Zeng, G.; Liu, X. min Ginsenoside Rg1 ameliorates chronic social defeat stress-induced depressive-like behaviors and hippocampal neuroinflammation. Life Sci. 2020, 252, 117669. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, A.; Dey, C.S. SIRT2 regulates insulin sensitivity in insulin resistant neuronal cells. Biochem. Biophys. Res. Commun. 2016, 474, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Chen, H.; Han, J.; Hong, Y.; Ma, Y.; Xia, W.; Ying, W. Silencing of SIRT2 induces cell death and a decrease in the intracellular ATP level of PC12 cells. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 65–70. [Google Scholar]
- Lantier, L.; Williams, A.S.; Hughey, C.C.; Bracy, D.P.; James, F.D.; Ansari, M.A.; Gius, D.; Wasserman, D.H. SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PLoS ONE 2018, 13, e0208634. [Google Scholar] [CrossRef]
- Xie, X.Q.; Zhang, P.; Tian, B.; Chen, X.Q. Downregulation of NAD-Dependent Deacetylase SIRT2 Protects Mouse Brain against Ischemic Stroke. Mol. Neurobiol. 2016, 54, 7251–7261. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Bang, Y.; Choi, H.J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 2012, 61, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Balabadra, U.; Xiang, Z.; Woodman, B.; Meade, S.; Amore, A.; Maxwell, M.M.; Reeves, S.; Bates, G.P.; Luthi-Carter, R.; et al. A Brain-Permeable Small Molecule Reduces Neuronal Cholesterol by Inhibiting Activity of Sirtuin 2 Deacetylase. ACS Chem. Biol. 2011, 6, 540–546. [Google Scholar] [CrossRef]
- Bobrowska, A.; Donmez, G.; Weiss, A.; Guarente, L.; Bates, G. SIRT2 Ablation Has No Effect on Tubulin Acetylation in Brain, Cholesterol Biosynthesis or the Progression of Huntington’s Disease Phenotypes in vivo. PLoS ONE 2012, 7, e34805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, N.; Bittner, S.; Ellwardt, E. Role of the epigenetic factor Sirt7 in neuroinflammation and neurogenesis. Neurosci. Res. 2018, 131, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L.; Chua, C.E.L. SIRT2, tubulin deacetylation, and oligodendroglia differentiation. Cell Motil. Cytoskelet. 2008, 65, 179–182. [Google Scholar] [CrossRef]
- Pandithage, R.; Lilischkis, R.; Harting, K.; Wolf, A.; Jedamzik, B.; Lüscher-Firzlaff, J.; Vervoorts, J.; Lasonder, E.; Kremmer, E.; Knöll, B.; et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 2008, 180, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Pais, T.F.; Szego, É.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; De Oliveira, R.M.H.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, L.; Li, C.Y.; Pei, Z.; Zhou, M.; Li, N. SIRT3 protects cells from hypoxia via PGC-1α- and MnSOD-dependent pathways. Neuroscience 2015, 286, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Shi, L.; Liang, F.; Xu, W.; Li, T.; Gao, L.; Sun, Z.; Yu, J.; Zhang, J. Sirt3 Ameliorates Oxidative Stress and Mitochondrial Dysfunction After Intracerebral Hemorrhage in Diabetic Rats. Front. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Docrat, T.F.; Nagiah, S.; Naicker, N.; Baijnath, S.; Singh, S.; Chuturgoon, A.A. The protective effect of metformin on mitochondrial dysfunction and endoplasmic reticulum stress in diabetic mice brain. Eur. J. Pharmacol. 2020, 875, 173059. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, A.; Li, Y.-J.; Yang, Y.; Kishimoto, Y.; Zhang, S.; Wang, Y.; Wan, R.; Raefsky, S.M.; Lu, D.; et al. SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Commun. 2019, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, J.; Tselykh, T.V.; Kukkonen, J.P.; Eriksson, O.; Korhonen, L.T.; Lindholm, D. Peroxisome proliferator-activated receptor-γ (PPARγ) agonist is neuroprotective and stimulates PGC-1α expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 2016, 102, 266–275. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-T.; Teng, L.; Lin, Y.; Miao, L.-H.; Ge, X.-P.; Hao, J.; Huang, X.; Liu, B. Molecular and functional characterization of sirt4 and sirt6 in Megalobrama amblycephala under high glucose metabolism. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2019, 231, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Yalçın, G.D.; Colak, M. SIRT4 prevents excitotoxicity via modulating glutamate metabolism in glioma cells. Hum. Exp. Toxicol. 2020, 39, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Drews, L.; Zimmermann, M.; Westhoff, P.; Brilhaus, D.; Poss, R.E.; Bergmann, L.; Wiek, C.; Brenneisen, P.; Piekorz, R.P.; Mettler-Altmann, T.; et al. Ammonia inhibits energy metabolism in astrocytes in a rapid and glutamate dehydrogenase 2-dependent manner. DMM Dis. Model. Mech. 2020, 13, dmm047134. [Google Scholar] [CrossRef]
- Geng, Y.-Q.; Li, T.-T.; Liu, X.-Y.; Li, Z.-H.; Fu, Y.-C. SIRT1 and SIRT5 activity expression and behavioral responses to calorie restriction. J. Cell. Biochem. 2011, 112, 3755–3761. [Google Scholar] [CrossRef] [PubMed]
- Morris-Blanco, K.C.; Dave, K.R.; Saul, I.; Koronowski, K.B.; Stradecki, H.M.; Perez-Pinzon, M.A. Protein Kinase C Epsilon Promotes Cerebral Ischemic Tolerance Via Modulation of Mitochondrial Sirt5. Sci. Rep. 2016, 6, 29790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koronowski, K.B.; Khoury, N.; Morris-Blanco, K.C.; Stradecki-Cohan, H.M.; Garrett, T.J.; Perez-Pinzon, M.A. Metabolomics based identification of SIRT5 and protein kinase C epsilon regulated pathways in brain. Front. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.H.; Kim, J.; Kim, J.M.; Lee, H.; Kim, E.H.; Bae, S.K.; Choi, Y.; Nam, H.S.; Heo, J.H. Decreased expression of sirtuin 6 is associated with release of high mobility group box-1 after cerebral ischemia. Biochem. Biophys. Res. Commun. 2013, 438, 388–394. [Google Scholar] [CrossRef]
- Kanfi, Y.; Shalman, R.; Peshti, V.; Pilosof, S.N.; Gozlan, Y.M.; Pearson, K.J.; Lerrer, B.; Moazed, D.; Marine, J.-C.; de Cabo, R.; et al. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 2008, 582, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, A.; de Stefano, M.C.; Mollinari, C.; Racaniello, M.; Garaci, E.; Merlo, D. Biochemical Characterization of Sirtuin 6 in the Brain and Its Involvement in Oxidative Stress Response. Neurochem. Res. 2014, 40, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Yang, X.; Liu, T.; Zhang, T.; Xie, Q.R.; Xia, W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 2016, 7, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Favero, G.; Rezzani, R.; Rodella, L.F. Sirtuin 6 nuclear localization at cortical brain level of young diabetic mice: An immunohistochemical study. Acta Histochem. 2014, 116, 272–277. [Google Scholar] [CrossRef]
- Schwer, B.; Schumacher, B.; Lombard, D.B.; Xiao, C.; Kurtev, M.V.; Gao, J.; Schneider, J.I.; Chai, H.; Bronson, R.T.; Tsai, L.-H.; et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 21790–21794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, S.; Di Rocco, G.; Toietta, G.; Russo, M.A.; Petrangeli, E.; Salvatori, L. Sirtuins 1–7 expression in human adipose-derived stem cells from subcutaneous and visceral fat depots: Influence of obesity and hypoxia. Endocrine 2016, 57, 455–463. [Google Scholar] [CrossRef]
- Wei, X.; Jia, R.; Wang, G.; Hong, S.; Song, L.; Sun, B.; Chen, K.; Wang, N.; Wang, Q.; Luo, X.; et al. Depot-specific regulation of NAD+/SIRTs metabolism identified in adipose tissue of mice in response to high-fat diet feeding or calorie restriction. J. Nutr. Biochem. 2020, 80, 108377. [Google Scholar] [CrossRef]
- Hammes, T.O.; Costa, C. dos S.; Rohden, F.; Margis, R.; Almeida, J.C. de; Padoin, A.V.; Mottin, C.C.; Guaragna, R.M. Parallel Down-Regulation of FOXO1, PPARγ and Adiponectin mRNA Expression in Visceral Adipose Tissue of Class III Obese Individuals. Obes. Facts 2012, 5, 452–459. [Google Scholar] [CrossRef]
- Serrano-Marco, L.; Chacón, M.R.; Maymó-Masip, E.; Barroso, E.; Salvadó, L.; Wabitsch, M.; Garrido-Sánchez, L.; Tinahones, F.J.; Palomer, X.; Vendrell, J.; et al. TNF-α inhibits PPARβ/δ activity and SIRT1 expression through NF-κB in human adipocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Lee, S.K.; Jang, Y.J.; Park, H.S.; Kim, J.-H.; Lee, Y.J.; Heo, Y.-S. Association between low SIRT1 expression in visceral and subcutaneous adipose tissues and metabolic abnormalities in women with obesity and type 2 diabetes. Diabetes Res. Clin. Pract. 2013, 101, 341–348. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, T.; Peng, J.; Zhou, Z.; Wei, H.; Zhou, R.; Jiang, S.; Peng, J. SIRT1 suppresses adipogenesis by activating Wnt/β-catenin signaling in vivo and in vitro. Oncotarget 2016, 7, 77707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, H.; Jin, X.; Zhang, Y.; Kang, X.; Zhang, Z.; Xu, M.; Qian, Z.; Ma, Z.; Gao, X.; et al. Adipose-specific knockdown of Sirt1 results in obesity and insulin resistance by promoting exosomes release. Cell Cycle 2019, 18, 2067–2082. [Google Scholar] [CrossRef]
- Nawaz, A.; Mehmood, A.; Kanatani, Y.; Kado, T.; Igarashi, Y.; Takikawa, A.; Yamamoto, S.; Okabe, K.; Nakagawa, T.; Yagi, K.; et al. Sirt1 activator induces proangiogenic genes in preadipocytes to rescue insulin resistance in diet-induced obese mice. Sci. Rep. 2018, 8, 11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.J.; Park, U.-H.; Kim, J.W.; Choi, H.; Um, S.-J.; Kim, E.-J. CACUL1 reciprocally regulates SIRT1 and LSD1 to repress PPARγ and inhibit adipogenesis. Cell Death Dis. 2017, 8, 3201. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Cai, Y.; Fan, P.; Bai, B.; Chen, J.; Deng, H.B.; Che, C.M.; Xu, A.; Vanhoutte, P.M.; Wang, Y. Calorie restriction prevents metabolic aging caused by abnormal sirt1 function in adipose tissues. Diabetes 2015, 64, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Liu, G.; Chen, Y.; Wu, T.; Feng, F.; Sun, C. Sirt1 decreased adipose inflammation by interacting with Akt2 and inhibiting mTOR/S6K1 pathway in mice. J. Lipid Res. 2016, 57, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Escalona-Garrido, C.; Vázquez, P.; Mera, P.; Zagmutt, S.; García-Casarrubios, E.; Montero-Pedrazuela, A.; Rey-Stolle, F.; Guadaño-Ferraz, A.; Rupérez, F.J.; Serra, D.; et al. Moderate SIRT1 overexpression protects against brown adipose tissue inflammation. Mol. Metab. 2020, 42, 101097. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. High-Fat Diet Triggers Inflammation-Induced Cleavage of SIRT1 in Adipose Tissue To Promote Metabolic Dysfunction. Cell Metab. 2012, 16, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutant, M.; Joffraud, M.; Kulkarni, S.S.; García-Casarrubios, E.; García-Roves, P.M.; Ratajczak, J.; Fernández-Marcos, P.J.; Valverde, A.M.; Serrano, M.; Cantó, C. SIRT1 enhances glucose tolerance by potentiating brown adipose tissue function. Mol. Metab. 2015, 4, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zheng, X.; Lin, B.; Liang, H.; Cai, M.; Cao, H.; Ye, J.; Weng, J. Diet-induced obesity and insulin resistance are associated with brown fat degeneration in SIRT1-deficient mice. Obesity 2016, 24, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown Remodeling of White Adipose Tissue by SirT1-Dependent Deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.H.; Koh, Y.C.; Lee, T.L.; Wang, B.; Chen, W.K.; Nagabhushanam, K.; Ho, C.T. Resveratrol and Oxyresveratrol Activate Thermogenesis via Different Transcriptional Coactivators in High-Fat Diet-Induced Obese Mice. J. Agric. Food Chem. 2019, 67, 13605–13616. [Google Scholar] [CrossRef]
- Andrade, J.M.O.; Frade, A.C.M.; Guimarães, J.B.; Freitas, K.M.; Lopes, M.T.P.; Guimarães, A.L.S.; de Paula, A.M.B.; Coimbra, C.C.; Santos, S.H.S. Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur. J. Nutr. 2014, 53, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Ryu, R.; Choi, J.-Y.; Choi, M.-S. Platycodon grandiflorus Root Ethanol Extract Induces Lipid Excretion, Lipolysis, and Thermogenesis in Diet-Induced Obese Mice. J. Med. Food 2019, 22, 1100–1109. [Google Scholar] [CrossRef]
- Kim, S.N.; Ahn, S.Y.; Song, H.D.; Kwon, H.J.; Saha, A.; Son, Y.; Cho, Y.K.; Jung, Y.S.; Jeong, H.W.; Lee, Y.H. Antiobesity effects of coumestrol through expansion and activation of brown adipose tissue metabolism. J. Nutr. Biochem. 2020, 76. [Google Scholar] [CrossRef]
- Paraíso, A.F.; Sousa, J.N.; Andrade, J.M.O.; Mangabeira, E.S.; Lelis, D.F.; de Paula, A.M.B.; Martins, A.M.E.B.L.; Lima, W.J.N.; Guimarães, A.L.S.; Melo, G.A.; et al. Oral gallic acid improves metabolic profile by modulating SIRT1 expression in obese mice brown adipose tissue: A molecular and bioinformatic approach. Life Sci. 2019, 237, 116914. [Google Scholar] [CrossRef]
- Costa, C. dos S.; Rohden, F.; Hammes, T.O.; Margis, R.; Bortolotto, J.W.; Padoin, A.V.; Mottin, C.C.; Guaragna, R.M. Resveratrol Upregulated SIRT1, FOXO1, and Adiponectin and Downregulated PPARγ1–3 mRNA Expression in Human Visceral Adipocytes. Obes. Surg. 2011, 21, 356–361. [Google Scholar] [CrossRef]
- Qiao, L.; Shao, J. SIRT1 Regulates Adiponectin Gene Expression through Foxo1-C/Enhancer-binding Protein Transcriptional Complex. J. Biol. Chem. 2006, 281, 39915–39924. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, Z.; Ke, L.; Sun, Y.; Li, W.; Feng, X.; Zhu, W.; Chen, S. Resveratrol promotes white adipocytes browning and improves metabolic disorders in Sirt1-dependent manner in mice. FASEB J. 2020, 34, 4527–4539. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gu, M.; Ma, Y.; Peng, Y. LncRNA TUG1 reduces inflammation and enhances insulin sensitivity in white adipose tissue by regulating miR-204/SIRT1 axis in obesity mice. Mol. Cell. Biochem. 2020, 475, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, Y.; Gu, M.; Peng, Y. lncRNA TUG1 promotes the brown remodeling of white adipose tissue by regulating miR-204-targeted SIRT1 in diabetic mice. Int. J. Mol. Med. 2020, 46, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, M.; Cao, Y.; Ma, L.; Shen, Y.; Velikanova, A.A.; Li, X.; Sun, C.; Zhao, Y. miR-34a regulates lipid metabolism by targeting SIRT1 in non-alcoholic fatty liver disease with iron overload. Arch. Biochem. Biophys. 2020, 695, 108642. [Google Scholar] [CrossRef] [PubMed]
- Briones-Espinoza, M.J.; Cortés-García, J.D.; Vega-Cárdenas, M.; Uresti-Rivera, E.U.; Gómez-Otero, A.; López-López, N.; Mejía-Torres, M.; Portales-Pérez, D.P. Decreased levels and activity of Sirt1 are modulated by increased miR-34a expression in adipose tissue mononuclear cells from subjects with overweight and obesity: A pilot study. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.-G.; Choung, S.-Y. Anti-obesity effects of Spirulina maxima in high fat diet induced obese rats via the activation of AMPK pathway and SIRT1. Food Funct. 2018, 9, 4906–4915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, X.; Sheng, Y.; Xu, J.; Yang, C.; Zheng, S.; Liu, J.; Li, H.; Ge, J.; Yang, M.; et al. Allicin Regulates Energy Homeostasis through Brown Adipose Tissue. iScience 2020, 23, 101113. [Google Scholar] [CrossRef]
- Yuan, J.; Shi, Q.; Chen, J.; Lu, J.; Wang, L.; Qiu, M.; Liu, J. Effects of 23-epi-26-deoxyactein on adipogenesis in 3T3-L1 preadipocytes and diet-induced obesity in C57BL/6 mice. Phytomedicine 2020, 76, 153264. [Google Scholar] [CrossRef]
- Parida, I.S.; Takasu, S.; Ito, J.; Ikeda, R.; Yamagishi, K.; Kimura, T.; Eitsuka, T.; Nakagawa, K. Supplementation of: Bacillus amyloliquefaciens AS385 culture broth powder containing 1-deoxynojirimycin in a high-fat diet altered the gene expressions related to lipid metabolism and insulin signaling in mice epididymal white adipose tissue. Food Funct. 2020, 11, 3926–3940. [Google Scholar] [CrossRef]
- Martínez-Jiménez, V.; Cortez-Espinosa, N.; Rodríguez-Varela, E.; Vega-Cárdenas, M.; Briones-Espinoza, M.; Ruíz-Rodríguez, V.M.; López-López, N.; Briseño-Medina, A.; Turiján-Espinoza, E.; Portales-Pérez, D.P. Altered levels of sirtuin genes (SIRT1, SIRT2, SIRT3 and SIRT6) and their target genes in adipose tissue from individual with obesity. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Perrini, S.; Porro, S.; Nigro, P.; Cignarelli, A.; Caccioppoli, C.; Genchi, V.A.; Martines, G.; De Fazio, M.; Capuano, P.; Natalicchio, A.; et al. Reduced SIRT1 and SIRT2 expression promotes adipogenesis of human visceral adipose stem cells and associates with accumulation of visceral fat in human obesity. Int. J. Obes. 2019. [Google Scholar] [CrossRef]
- Wang, F.; Nguyen, M.; Qin, F.X.-F.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Wang, F.; Tong, Q. SIRT2 Suppresses Adipocyte Differentiation by Deacetylating FOXO1 and Enhancing FOXO1′s Repressive Interaction with PPAR. Mol. Biol. Cell 2008, 20, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Armoni, M.; Harel, C.; Karni, S.; Chen, H.; Bar-Yoseph, F.; Ver, M.R.; Quon, M.J.; Karnieli, E. FOXO1 Represses Peroxisome Proliferator-activated Receptor-1 and -2 Gene Promoters in Primary Adipocytes: A Novel Paradigm to Increase Insulin Sensitivity. J. Biol. Chem. 2006, 281, 19881–19891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, J.; Danzer, C.; Simka, T.; Ukropec, J.; Walter, K.M.; Kumpf, S.; Mirtschink, P.; Ukropcova, B.; Gasperikova, D.; Pedrazzini, T.; et al. Dietary obesity-associated Hif1 activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012, 26, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Takeda, K.; Narita, T.; Nagai, K.; Okita, N.; Sudo, Y.; Miura, Y.; Tsumoto, H.; Nakagawa, Y.; Shimano, H.; et al. Mitochondrial intermediate peptidase is a novel regulator of sirtuin-3 activation by caloric restriction. FEBS Lett. 2017, 591, 4067–4073. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a Mitochondrial Sirtuin Deacetylase, Regulates Mitochondrial Function and Thermogenesis in Brown Adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebaa, R.; Johnson, J.; Pileggi, C.; Norgren, M.; Xuan, J.; Sai, Y.; Tong, Q.; Krystkowiak, I.; Bondy-Chorney, E.; Davey, N.E.; et al. SIRT3 controls brown fat thermogenesis by deacetylation regulation of pathways upstream of UCP1. Mol. Metab. 2019, 25, 35–49. [Google Scholar] [CrossRef]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Díaz-Delfín, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome Proliferator-activated Receptor-γ Coactivator-1α Controls Transcription of the Sirt3 Gene, an Essential Component of the Thermogenic Brown Adipocyte Phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L.; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Zamora-Mendoza, R.; Rosas-Vargas, H.; Ramos-Cervantes, M.T.; Garcia-Zuniga, P.; Perez-Lorenzana, H.; Mendoza-Lorenzo, P.; Perez-Ortiz, A.C.; Estrada-Mena, F.J.; Miliar-Garcia, A.; Lara-Padilla, E.; et al. Dysregulation of mitochondrial function and biogenesis modulators in adipose tissue of obese children. Int. J. Obes. 2018, 42, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Akindehin, S.; Jung, Y.-S.; Kim, S.-N.; Son, Y.-H.; Lee, I.; Seong, J.; Jeong, H.; Lee, Y.-H. Myricetin Exerts Anti-Obesity Effects through Upregulation of SIRT3 in Adipose Tissue. Nutrients 2018, 10, 1962. [Google Scholar] [CrossRef] [Green Version]
- García-Fuentes, E.; Santiago-Fernández, C.; Gutiérrez-Repiso, C.; Mayas, M.D.; Oliva-Olivera, W.; Coín-Aragüez, L.; Alcaide, J.; Ocaña-Wilhelmi, L.; Vendrell, J.; Tinahones, F.J.; et al. Hypoxia is associated with a lower expression of genes involved in lipogenesis in visceral adipose tissue. J. Transl. Med. 2015, 13, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, L.C.; Franczyk, M.P.; Pietka, T.; Yamaguchi, S.; Lin, J.B.; Sasaki, Y.; Verdin, E.; Apte, R.S.; Yoshino, J. NAD+-dependent deacetylase SIRT3 in adipocytes is dispensable for maintaining normal adipose tissue mitochondrial function and whole body metabolism. Am. J. Physiol. Metab. 2018, 315, E520–E530. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-T.; Chi, K.-T.; Lan, Y.-W.; Chan, J.-C.; Ma, Y.-S.; Wei, Y.-H. Depletion of Sirt3 leads to the impairment of adipogenic differentiation and insulin resistance via interfering mitochondrial function of adipose-derived human mesenchymal stem cells. Free Radic. Res. 2018, 52, 1398–1415. [Google Scholar] [CrossRef]
- Zhang, T.; Fang, Z.; Linghu, K.G.; Liu, J.; Gan, L.; Lin, L. Small molecule-driven SIRT3-autophagy-mediated NLRP3 inflammasome inhibition ameliorates inflammatory crosstalk between macrophages and adipocytes. Br. J. Pharmacol. 2020, 177, 4645–4665. [Google Scholar] [CrossRef] [PubMed]
- de Pinho, L.; Andrade, J.M.O.; Paraíso, A.; Filho, A.B.M.; Feltenberger, J.D.; Guimarães, A.L.S.; de Paula, A.M.B.; Caldeira, A.P.; de Carvalho Botelho, A.C.; Campagnole-Santos, M.J.; et al. Diet composition modulate expression of sirtuins and Renin-Angiotensin system components in adipose tissue. Obesity 2013, 21, 1830–1835. [Google Scholar] [CrossRef] [Green Version]
- Shuai, L.; Zhang, L.-N.; Li, B.-H.; Tang, C.-L.; Wu, L.-Y.; Li, J.; Li, J.-Y. SIRT5 Regulates Brown Adipocyte Differentiation and Browning of Subcutaneous White Adipose Tissue. Diabetes 2019, 68, 1449–1461. [Google Scholar] [CrossRef]
- Wang, G.X.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e7. [Google Scholar] [CrossRef]
- Yu, J.; Sadhukhan, S.; Noriega, L.G.; Moullan, N.; He, B.; Weiss, R.S.; Lin, H.; Schoonjans, K.; Auwerx, J. Metabolic characterization of a Sirt5 deficient mouse model. Sci. Rep. 2013, 3, 2806. [Google Scholar] [CrossRef] [PubMed]
- Jukarainen, S.; Heinonen, S.; Rämö, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity Is Associated with Low NAD + /SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Uppala, R.; Verdin, E.; Gibson, B.W.; Goetzman, E.S. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain Acyl-CoA dehydrogenase. PLoS ONE 2015, 10, e0122297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Onofrio, N.; Pieretti, G.; Ciccarelli, F.; Gambardella, A.; Passariello, N.; Rizzo, M.R.; Barbieri, M.; Marfella, R.; Nicoletti, G.; Balestrieri, M.L.; et al. Abdominal Fat SIRT6 Expression and Its Relationship with Inflammatory and Metabolic Pathways in Pre-Diabetic Overweight Patients. Int. J. Mol. Sci. 2019, 20, 1153. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Peshti, V.; Gil, R.; Naiman, S.; Nahum, L.; Levin, E.; Kronfeld-Schor, N.; Cohen, H.Y. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 2010, 9, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Mohammadkhani, R.; Khaledi, N.; Rajabi, H.; Salehi, I.; Komaki, A. Influence of the maternal high-intensityinterval- training on the cardiac Sirt6 and lipid profile of the adult male offspring in rats. PLoS ONE 2020, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Mei, C.; Abbas Raza, S.H.; Khan, R.; Cheng, G.; Zan, L. SIRT6 cooperates with SIRT5 to regulate bovine preadipocyte differentiation and lipid metabolism via the AMPKα signaling pathway. Arch. Biochem. Biophys. 2020, 681, 108260. [Google Scholar] [CrossRef]
- Jung, T.W.; Park, J.; Sun, J.L.; Ahn, S.H.; Abd El-Aty, A.M.; Hacimuftuoglu, A.; Kim, H.C.; Shim, J.H.; Shin, S.S.; Jeong, J.H. Administration of kynurenic acid reduces hyperlipidemia-induced inflammation and insulin resistance in skeletal muscle and adipocytes. Mol. Cell. Endocrinol. 2020, 518, 110928. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Hung, C.-M.; Hildebrand, S.R.; Sanchez-Gurmaches, J.; Martinez-Pastor, B.; Gengatharan, J.M.; Wallace, M.; Mukhopadhyay, D.; Martinez Calejman, C.; Luciano, A.K.; et al. Non-canonical mTORC2 Signaling Regulates Brown Adipocyte Lipid Catabolism through SIRT6-FoxO1. Mol. Cell 2019, 75, 807–822.e8. [Google Scholar] [CrossRef]
- Wang, Q.; Pan, Y.; Zhao, B.; Qiao, L.; Liu, J.; Liang, Y.; Liu, W. MiR-33a inhibits the adipogenic differentiation of ovine adipose–derived stromal vascular fraction cells by targeting SIRT6. Domest. Anim. Endocrinol. 2021, 74, 106513. [Google Scholar] [CrossRef]
- Kurylowicz, A.; Owczarz, M.; Polosak, J.; Jonas, M.I.; Lisik, W.; Jonas, M.I.; Chmura, A.; Puzianowska-Kuznicka, M. SIRT1 and SIRT7 expression in adipose tissues of obese and normal-weight individuals is regulated by microRNAs but not by methylation status. Int. J. Obes. 2016, 40, 1635–1642. [Google Scholar] [CrossRef]
- Bober, E.; Fang, J.; Smolka, C.; Ianni, A.; Vakhrusheva, O.; Krüger, M.; Braun, T. Sirt7 promotes adipogenesis by binding to and inhibiting Sirt1. BMC Proc. 2012, 6, P57. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Vallespinos-Serrano, M.; Trabulo, S.M.; Fernandez-Marcos, P.J.; Firment, A.N.; Vazquez, B.N.; Vieira, C.R.; Mulero, F.; Camara, J.A.; Cronin, U.P.; et al. MiR-93 Controls Adiposity via Inhibition of Sirt7 and Tbx3. Cell Rep. 2015, 12, 1594–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Ianni, A.; Smolka, C.; Vakhrusheva, O.; Nolte, H.; Krüger, M.; Wietelmann, A.; Simonet, N.G.; Adrian-Segarra, J.M.; Vaquero, A.; et al. Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proc. Natl. Acad. Sci. USA 2017, 114, E8352–E8361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappou, E.; Jukarainen, S.; Rinnankoski-Tuikka, R.; Kaye, S.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Saunavaara, V.; Rissanen, A.; et al. Weight Loss Is Associated With Increased NAD + /SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue. J. Clin. Endocrinol. Metab. 2016, 101, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Zhang, P.; Jiang, Q.; Xiong, Y.; Wang, Y.; Kuang, S. Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia 2016, 59, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, P.-G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Cote, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPAR-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaue, H.; Ogawa, W.; Matsumoto, M.; Kuroda, S.; Takata, M.; Sugimoto, T.; Spiegelman, B.M.; Kasuga, M. Posttranscriptional control of adipocyte differentiation through activation of phosphoinositide 3-kinase. J. Biol. Chem. 1998, 273, 28945–28952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Bedmar, R.; Lorenzo, M.; Fernández-Veledo, S. Adenosine 5′-Monophosphate-Activated Protein Kinase-Mammalian Target of Rapamycin Cross Talk Regulates Brown Adipocyte Differentiation. Endocrinology 2010, 151, 980–992. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liang, X.; Yang, Q.; Fu, X.; Rogers, C.J.; Zhu, M.; Rodgers, B.D.; Jiang, Q.; Dodson, M.V.; Du, M. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) α1. Int. J. Obes. 2015, 39, 967–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceddia, R.B. The role of AMP-activated protein kinase in regulating white adipose tissue metabolism. Mol. Cell. Endocrinol. 2013, 366, 194–203. [Google Scholar] [CrossRef]
- Xu, L.; Panel, V.; Ma, X.; Du, C.; Hugendubler, L.; Gavrilova, O.; Liu, A.; McLaughlin, T.; Kaestner, K.H.; Mueller, E. The Winged Helix Transcription Factor Foxa3 Regulates Adipocyte Differentiation and Depot-Selective Fat Tissue Expansion. Mol. Cell. Biol. 2013, 33, 3392–3399. [Google Scholar] [CrossRef] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draznin, B.; Wang, C.; Adochio, R.; Leitner, J.; Cornier, M.-A. Effect of Dietary Macronutrient Composition on AMPK and SIRT1 Expression and Activity in Human Skeletal Muscle. Horm. Metab. Res. 2012, 44, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Gurd, B.J.; Perry, C.G.R.; Heigenhauser, G.J.F.; Spriet, L.L.; Bonen, A. High-intensity interval training increases SIRT1 activity in human skeletal muscle. Appl. Physiol. Nutr. Metab. 2010, 35, 350–357. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Rathbone, C.R.; Booth, F.W.; Lees, S.J. Sirt1 increases skeletal muscle precursor cell proliferation. Eur. J. Cell Biol. 2009, 88, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-C.; Wang, T.; Tung, Y.-T.; Lin, W.-T. Effect of Exercise Training on Skeletal Muscle SIRT1 and PGC-1α Expression Levels in Rats of Different Age. Int. J. Med. Sci. 2016, 13, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, K.; Tahvilian, S.; Martins, V.F.; Dent, J.R.; Lemanek, A.; Barooni, N.; Greyslak, K.; McCurdy, C.E.; Schenk, S. Combined overexpression of SIRT1 and knockout of GCN5 in adult skeletal muscle does not affect glucose homeostasis or exercise performance in mice. Am. J. Physiol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Shiota, A.; Shimabukuro, M.; Fukuda, D.; Soeki, T.; Sato, H.; Uematsu, E.; Hirata, Y.; Kurobe, H.; Maeda, N.; Sakaue, H.; et al. Telmisartan ameliorates insulin sensitivity by activating the AMPK/SIRT1 pathway in skeletal muscle of obese db/db mice. Cardiovasc. Diabetol. 2012, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Wang, W.; Fang, C.; Ni, C.; Zhou, J.; Wang, X.; Zhang, L.; Xu, X.; Cao, R.; Lang, H.; et al. Vitamin K2 Alleviates Insulin Resistance in Skeletal Muscle by Improving Mitochondrial Function Via SIRT1 Signaling. Antioxid. Redox Signal. 2020, 34, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Abu Bakar, M.H.; Shariff, K.A.; Tan, J.S.; Lee, L.K. Celastrol attenuates inflammatory responses in adipose tissues and improves skeletal muscle mitochondrial functions in high fat diet-induced obese rats via upregulation of AMPK/SIRT1 signaling pathways. Eur. J. Pharmacol. 2020, 883, 173371. [Google Scholar] [CrossRef]
- Ahn, J.; Ha, T.Y.; Ahn, J.; Jung, C.H.; Seo, H.D.; Kim, M.J.; Kim, Y.S.; Jang, Y.J. Undaria pinnatifida extract feeding increases exercise endurance and skeletal muscle mass by promoting oxidative muscle remodeling in mice. FASEB J. 2020, 34, 8068–8081. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, F.; Della-Morte, D.; Piermarini, F.; Arriga, R.; Scioli, M.G.; Capuani, B.; Pastore, D.; Coppola, A.; Rea, S.; Donadel, G.; et al. Prdx6 plays a main role in the crosstalk between aging and metabolic sarcopenia. Antioxidants 2020, 9, 329. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Ma, X.-J.; Wu, L.-N.; Zhao, Y.-Y.; Zhang, P.-Y.; Zhang, Y.-H.; Shao, M.-W.; Liu, F.; Li, F.; Qin, G.-J. SIRT1 attenuates high glucose-induced insulin resistance via reducing mitochondrial dysfunction in skeletal muscle cells. Exp. Biol. Med. 2015, 240, 557–565. [Google Scholar] [CrossRef] [Green Version]
- White, A.T.; McCurdy, C.E.; Philp, A.; Hamilton, D.L.; Johnson, C.D.; Schenk, S. Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia 2013, 56, 1629–1637. [Google Scholar] [CrossRef]
- Brandon, A.E.; Tid-Ang, J.; Wright, L.E.; Stuart, E.; Suryana, E.; Bentley, N.; Turner, N.; Cooney, G.J.; Ruderman, N.B.; Kraegen, E.W. Overexpression of SIRT1 in rat skeletal muscle does not alter glucose induced insulin resistance. PLoS ONE 2015, 10, e0121959. [Google Scholar] [CrossRef] [PubMed]
- White, A.T.; Philp, A.; Fridolfsson, H.N.; Schilling, J.M.; Murphy, A.N.; Hamilton, D.L.; McCurdy, C.E.; Patel, H.H.; Schenk, S. High-fat diet-induced impairment of skeletal muscle insulin sensitivity is not prevented by SIRT1 overexpression. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E764–E772. [Google Scholar] [CrossRef] [Green Version]
- Fritzen, A.M.; Lundsgaard, A.-M.; Jeppesen, J.F.; Sjøberg, K.A.; Høeg, L.D.; Deleuran, H.H.; Wojtaszewski, J.F.P.; Richter, E.A.; Kiens, B. Fatty acid type–specific regulation of SIRT1 does not affect insulin sensitivity in human skeletal muscle. FASEB J. 2019, 33, 5510–5519. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; McCurdy, C.E.; Philp, A.; Chen, M.Z.; Holliday, M.J.; Bandyopadhyay, G.K.; Osborn, O.; Baar, K.; Olefsky, J.M. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Investig. 2011, 121, 4281–4288. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.P.; Lee, H.Y.; Lau, D.P.; Supaat, W.; Chan, Y.H.; Koh, A.F.Y. Effects of resveratrol in patients with type 2 diabetes mellitus on skeletal muscle SIRT1 expression and energy expenditure. Int. J. Sport Nutr. Exerc. Metab. 2014, 24, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-C.; Chen, J.-Y.; Kuo, Y.-T.; Chen, W.-L.; Lee, H.-M.; Wang, S.-G. Camptothecin activates SIRT1 to promote lipid catabolism through AMPK/FoxO1/ATGL pathway in C2C12 myogenic cells. Arch. Pharm. Res. 2019, 42, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hatazawa, Y.; Hirose, Y.; Ono, Y.; Kamei, Y. FOXO1 delays skeletal muscle regeneration and suppresses myoblast proliferation. Biosci. Biotechnol. Biochem. 2016, 8451, 1531–1535. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.A.; Sandesara, P.B.; Senf, S.M.; Judge, A.R. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 2012, 26, 987–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Goldberg, A.L. SIRT1 Protein, by Blocking the Activities of Transcription Factors FoxO1 and FoxO3, Inhibits Muscle Atrophy and Promotes Muscle Growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Ochi, E.; Song, H.; Nakazato, K. Activation of AMP-activated protein kinase induce expression of FoxO1, FoxO3a, and myostatin after exercise-induced muscle damage. Biochem. Biophys. Res. Commun. 2015, 466, 289–294. [Google Scholar] [CrossRef]
- Mounier, R.; Lantier, L.; Leclerc, J.; Sotiropoulos, A.; Pende, M.; Daegelen, D.; Sakamoto, K.; Foretz, M.; Viollet, B. Important role for AMPK 1 in limiting skeletal muscle cell hypertrophy. FASEB J. 2009, 23, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.L.; Butler, D.C.; Alway, S.E. AMPK inhibits myoblast differentiation through a PGC-1 -dependent mechanism. AJP Endocrinol. Metab. 2009, 297, E304–E314. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [Green Version]
- Stefanowicz, M.; Nikołajuk, A.; Matulewicz, N.; Karczewska-Kupczewska, M. Adipose tissue, but not skeletal muscle, sirtuin 1 expression is decreased in obesity and related to insulin sensitivity. Endocrine 2018, 60, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bétry, C.; Meugnier, E.; Pflieger, M.; Grenet, G.; Hercberg, S.; Galan, P.; Kesse-Guyot, E.; Vidal, H.; Laville, M. High expression of CPT1b in skeletal muscle in metabolically healthy older subjects. Diabetes Metab. 2019, 45, 152–159. [Google Scholar] [CrossRef]
- Arora, A.; Dey, C.S. SIRT2 negatively regulates insulin resistance in C2C12 skeletal muscle cells. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1372–1378. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Song, C.; Lu, H.; Jia, L.; Yang, G.; Shi, X.; Sun, S. Sirt2 induces C2C12 myoblasts proliferation by activation of the ERK1/2 pathway. Acta Biochim. Biophys. Sin. 2014, 46, 342–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Chen, K.; Khalek, W.A.; Ward, J.L.; Yang, H.; Chabi, B.; Wrutniak-Cabello, C.; Tong, Q. Regulation of Skeletal Muscle Oxidative Capacity and Muscle Mass by SIRT3. PLoS ONE 2014, 9, e85636. [Google Scholar] [CrossRef]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marcos, P.J.; Jeninga, E.H.; Canto, C.; Harach, T.; de Boer, V.C.J.; Andreux, P.; Moullan, N.; Pirinen, E.; Yamamoto, H.; Houten, S.M.; et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci. Rep. 2012, 2, 425. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 Regulates Metabolic Flexibility of Skeletal Muscle Through Reversible Enzymatic Deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.S.; Koves, T.R.; Davidson, M.T.; Crown, S.B.; Fisher-Wellman, K.H.; Torres, M.J.; Draper, J.A.; Narowski, T.M.; Slentz, D.H.; Lantier, L.; et al. Disruption of Acetyl-Lysine Turnover in Muscle Mitochondria Promotes Insulin Resistance and Redox Stress without Overt Respiratory Dysfunction. Cell Metab. 2020, 31, 131–147. [Google Scholar] [CrossRef]
- Song, Y.; Shi, J.; Wu, Y.; Han, C.; Zou, J.; Shi, Y.; Liu, Z. Metformin ameliorates insulin resistance in L6 rat skeletal muscle cells through upregulation of SIRT3. Chin. Med. J. 2014, 127, 1523–1529. [Google Scholar]
- Vargas-Ortiz, K.; Perez-Vazquez, V.; Diaz-Cisneros, F.J.; Figueroa, A.; Jiménez-Flores, L.M.; Rodriguez-DelaRosa, G.; Macias, M.H. Aerobic Training Increases Expression Levels of SIRT3 and PGC-1α in Skeletal Muscle of Overweight Adolescents Without Change in Caloric Intake. Pediatr. Exerc. Sci. 2015, 27, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Edgett, B.A.; Bonafiglia, J.T.; Baechler, B.L.; Quadrilatero, J.; Gurd, B.J. The effect of acute and chronic sprint-interval training on LRP130, SIRT3, and PGC-1 α expression in human skeletal muscle. Physiol. Rep. 2016, 4, e12879. [Google Scholar] [CrossRef]
- Vargas-Ortiz, K.; Pérez-Vázquez, V.; Figueroa, A.; Díaz, F.J.; Montaño-Ascencio, P.G.; Macías-Cervantes, M.H. Aerobic training but no resistance training increases SIRT3 in skeletal muscle of sedentary obese male adolescents. Eur. J. Sport Sci. 2018, 18, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Koltai, E.; Bori, Z.; Osvath, P.; Ihasz, F.; Peter, S.; Toth, G.; Degens, H.; Rittweger, J.; Boldogh, I.; Radak, Z. Master athletes have higher miR-7, SIRT3 and SOD2 expression in skeletal muscle than age-matched sedentary controls. Redox Biol. 2018, 19, 46–51. [Google Scholar] [CrossRef]
- Gurd, B.J.; Holloway, G.P.; Yoshida, Y.; Bonen, A. In mammalian muscle, SIRT3 is present in mitochondria and not in the nucleus; and SIRT3 is upregulated by chronic muscle contraction in an adenosine monophosphate-activated protein kinase–independent manner. Metabolism 2012, 61, 733–741. [Google Scholar] [CrossRef]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects Against Severe Insulin Resistance in High-Fat–Fed Mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Zhang, T.; Zhou, Y.; Zeng, X.; Ran, L.; Zhang, Q.; Zhu, J.; Mi, M. Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3 signaling pathway. Endocrine 2015, 50, 378–389. [Google Scholar] [CrossRef]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging 2013, 5, 835–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acs, Z.; Bori, Z.; Takeda, M.; Osvath, P.; Berkes, I.; Taylor, A.W.; Yang, H.; Radak, Z. High altitude exposure alters gene expression levels of DNA repair enzymes, and modulates fatty acid metabolism by SIRT4 induction in human skeletal muscle. Respir. Physiol. Neurobiol. 2014, 196, 33–37. [Google Scholar] [CrossRef]
- Lensu, S.; Pekkala, S.P.; Mäkinen, A.; Karstunen, N.; Turpeinen, A.T.; Hulmi, J.J.; Silvennoinen, M.M.; Ma, H.; Kujala, U.M.; Karvinen, S.; et al. Beneficial effects of running and milk protein supplements on Sirtuins and risk factors of metabolic disorders in rats with low aerobic capacity. Metab. Open 2019, 4, 100019. [Google Scholar] [CrossRef] [PubMed]
- Roichman, A.; Kanfi, Y.; Glazz, R.; Naiman, S.; Amit, U.; Landa, N.; Tinman, S.; Stein, I.; Pikarsky, E.; Leor, J.; et al. SIRT6 Overexpression Improves Various Aspects of Mouse Healthspan. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 72, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Kim, H.-S.; Lahusen, T.; Wang, R.-H.; Xu, X.; Gavrilova, O.; Jou, W.; Gius, D.; Deng, C.-X. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 2010, 285, 36776–36784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Yao, L.; Yang, X.; Gao, Y.; Fang, F.; Zhang, J.; Wang, Q.; Chang, Y. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E493–E505. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.A.; Kanwal, A.; Pillai, V.B.; Bao, R.; Gupta, M.P. The histone deacetylase SIRT6 blocks myostatin expression and development of muscle atrophy. Sci. Rep. 2017, 7, 11877. [Google Scholar] [CrossRef] [Green Version]
- Koltai, E.; Szabo, Z.; Atalay, M.; Boldogh, I.; Naito, H.; Goto, S.; Nyakas, C.; Radak, Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 2010, 131, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin resistance and sarcopenia: Mechanistic links between common co-morbidities. J. Endocrinol. 2016, 229, R67–R81. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef]
- Wronska, A.; Lawniczak, A.; Wierzbicki, P.M.; Kmiec, Z. Age-Related Changes in Sirtuin 7 Expression in Calorie-Restricted and Refed Rats. Gerontology 2016, 62, 304–310. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L. Leptin gene transfer regulates fibromuscular development and lipid deposition in muscles via SIRT1, FOXO3a and PGC-1α in mice in vivo. Int. J. Mol. Med. 2011, 28, 617–623. [Google Scholar] [CrossRef]
- García-Carrizo, F.; Nozhenko, Y.; Palou, A.; Rodríguez, A.M. Leptin Effect on Acetylation and Phosphorylation of Pgc1α in Muscle Cells Associated With Ampk and Akt Activation in High-Glucose Medium. J. Cell. Physiol. 2016, 231, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Adhihetty, P.J.; Hood, D.A. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genom. 2009, 37, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 Improves Aerobic Exercise Capacity and Augments Skeletal Muscle Mitochondrial DNA Content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1α’. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Ahn, N.; Park, J.; Kim, K. Effects of 8 Weeks Calorie Reduction and Resistance Exercise on Traf2-NFkB-mTOR and SIRT1-FoxO1 Signal Expression of Cardiac Muscle in High-fat Induced Obese Middle-Aged Rats. Exerc. Sci. 2018, 27, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Qin, J.; Chen, C.; Fu, Y.; Wang, W. Moderate calorie restriction attenuates age-associated alterations and improves cardiac function by increasing SIRT1 and SIRT3 expression. Mol. Med. Rep. 2018, 18, 4087–4094. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Alcendor, R.; Zhai, P.; Park, J.Y.; Shao, D.; Cho, J.; Yamamoto, T.; Tian, B.; Sadoshima, J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the err transcriptional pathway. Cell Metab. 2011, 14, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef] [PubMed]
- Kalliora, C.; Kyriazis, I.D.; Oka, S.; Lieu, M.J.; Yue, Y.; Area-Gomez, E.; Pol, C.J.; Tian, Y.; Mizushima, W.; Chin, A.; et al. Dual PPARα/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight 2019, 5, e129556. [Google Scholar] [CrossRef] [Green Version]
- Elahi, M.M.; Kong, Y.X.; Matata, B.M. Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell. Longev. 2009, 2, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Haneda, M.; Kanasaki, K.; Sugimoto, T.; Araki, S.I.; Isono, M.; Isshiki, K.; Uzu, T.; Kashiwagi, A.; Koya, D. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006, 40, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Van Der Horst, A.; Tertoolen, L.G.J.; De Vries-Smits, L.M.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M.T. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047. [Google Scholar] [CrossRef] [Green Version]
- Paiva, M.A.; Rutter-Locher, Z.; Gonçalves, L.M.; Providência, L.A.; Davidson, S.M.; Yellon, D.M.; Mocanu, M.M. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2123–H2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol. 2012, 8, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Shang, X.; Wu, H.; Gautam, S.C.; Al-Holou, S.; Li, C.; Kuo, J.; Zhang, L.; Chopp, M. Resveratrol downregulates PI3K/Akt/mTOR signaling pathways in human U251 glioma cells. J. Exp. Ther. Oncol. 2009, 8, 25–33. [Google Scholar]
- Hudlicka, O.; Brown, M.; Egginton, S. Angiogenesis in skeletal and cardiac muscle. Physiol. Rev. 1992, 72, 369–400. [Google Scholar] [CrossRef]
- Shiojima, I.; Sato, K.; Izumiya, Y.; Schiekofer, S.; Ito, M.; Liao, R.; Colucci, W.S.; Walsh, K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 2005, 115, 2108–2118. [Google Scholar] [CrossRef] [Green Version]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is required for physiological cardiac growth. Circulation 2006, 113, 2097–2104. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The Deacetylase SIRT1 Promotes Membrane Localization and Activation of Akt and PDK1 During Tumorigenesis and Cardiac Hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, N.; Koitabashi, N.; Matsui, H.; Sunaga, H.; Umbarawan, Y.; Syamsunarno, M.R.A.A.; Yamaguchi, A.; Obokata, M.; Hanaoka, H.; Yokoyama, T.; et al. DPP-4 inhibitor induces FGF21 expression via sirtuin 1 signaling and improves myocardial energy metabolism. Heart Vessel. 2020, 36, 136–146. [Google Scholar] [CrossRef]
- Sunaga, H.; Koitabashi, N.; Iso, T.; Matsui, H.; Obokata, M.; Kawakami, R.; Murakami, M.; Yokoyama, T.; Kurabayashi, M. Activation of cardiac AMPK-FGF21 feed-forward loop in acute myocardial infarction: Role of adrenergic overdrive and lipolysis byproducts. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Liu, W.; Liu, B.; Zhang, B.; Li, W.; Xu, Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: An insight into endoplasmic reticulum stress response mechanism. Int. J. Cardiol. 2015, 191, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Najafipour, H.; Rostamzadeh, F.; Yeganeh-Hajahmadi, M.; Joukar, S. Improvement of cardiac function in rats with myocardial infarction by low- to moderate-intensity endurance exercise is associated with normalization of Klotho and SIRT1. J. Cardiovasc. Pharmacol. 2021, 77, 79–86. [Google Scholar] [CrossRef]
- Liu, J.; Ai, Y.; Niu, X.; Shang, F.; Li, Z.; Liu, H.; Li, W.; Ma, W.; Chen, R.; Wei, T.; et al. Taurine protects against cardiac dysfunction induced by pressure overload through SIRT1–p53 activation. Chem. Biol. Interact. 2020, 317, 108972. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.T.; Nithiyanantham, S.; Hsieh, D.J.Y.; Chen, R.J.; Day, C.H.; Liao, J.Y.; Kuo, C.H.; Mahalakshmi, B.; Kuo, W.W.; Huang, C.Y. Bioactive peptides attenuate cardiac apoptosis in spontaneously hypertensive rat hearts through activation of autophagy and mitochondrial biogenesis pathway. Environ. Toxicol. 2020, 35, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Hamawaki, M.; Narishige, T.; Sato, H.; Nemoto, S.; DeFreyte, G.; Zile, M.R.; Cooper, G.; Carabello, B.A. Microtubule Depolymerization Normalizes in vivo Myocardial Contractile Function in Dogs With Pressure-Overload Left Ventricular Hypertrophy. Circulation 2000, 102, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Zhan, L.; Zhou, Q.Y.; Zhang, L.L.; Chen, X.M.; Hu, X.M.; Yuan, X.C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef]
- Tang, X.; Chen, X.-F.; Wang, N.-Y.; Wang, X.-M.; Liang, S.-T.; Zheng, W.; Lu, Y.-B.; Zhao, X.; Hao, D.-L.; Zhang, Z.-Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- Sarikhani, M.; Maity, S.; Mishra, S.; Jain, A.; Tamta, A.K.; Ravi, V.; Kondapalli, M.S.; Desingu, P.A.; Khan, D.; Kumar, S.; et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. J. Biol. Chem. 2018, 293, 5281–5294. [Google Scholar] [CrossRef] [Green Version]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell. Physiol. Biochem. 2019, 53, 465–479. [Google Scholar] [CrossRef]
- Kanwal, A.; Pillai, V.B.; Samant, S.; Gupta, M.; Gupta, M.P. The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J. 2019, 33, 10872–10888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Jeevanandam, V.; Gupta, M.P. Mitochondrial SIRT3 and heart disease. Cardiovasc. Res. 2010, 88, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.G.; Wang, N.; Cao, D.J.; Sachan, N.; Morris, D.J.; Gerard, R.D.; Kuro-O, M.; Rothermel, B.A.; Hill, J.A. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. USA 2007, 104, 20517–20522. [Google Scholar] [CrossRef] [Green Version]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 1–20. [Google Scholar] [CrossRef]
- Palomer, X.; Román-Azcona, M.S.; Pizarro-Delgado, J.; Planavila, A.; Villarroya, F.; Valenzuela-Alcaraz, B.; Crispi, F.; Sepúlveda-Martínez, Á.; Miguel-Escalada, I.; Ferrer, J.; et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, L.; Cheng, S.; Guo, J.; Zhu, S.; Wang, Y.; Gao, H. Hypoxia exacerbates cardiomyocyte injury via upregulation of Wnt3a and inhibition of Sirt3. Cytokine 2020, 136, 155237. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.N.; Li, J.L.; Xu, T.; Yao, H.Q.; Chen, G.H.; Hu, J. Effects of Sirt3-autophagy and resveratrol activation on myocardial hypertrophy and energy metabolism. Mol. Med. Rep. 2020, 22, 1342–1350. [Google Scholar] [CrossRef]
- Alrob, O.A.; Sankaralingam, S.; Ma, C.; Wagg, C.S.; Fillmore, N.; Jaswal, J.S.; Sack, M.N.; Lehner, R.; Gupta, M.P.; Michelakis, E.D.; et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. 2014, 103, 485–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Vaka, V.R.; He, X.; Booz, G.W.; Chen, J.-X. High-fat diet induces cardiac remodelling and dysfunction: Assessment of the role played by SIRT3 loss. J. Cell. Mol. Med. 2015, 19, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-F.; Huang, C.-C.; Xiao, Y.-F.; Guan, X.-H.; Wang, X.-N.; Cao, Q.; Liu, Y.; Huang, X.; Deng, L.-B.; Deng, K.-Y.; et al. CD38 Deficiency Protects Heart from High Fat Diet-Induced Oxidative Stress Via Activating Sirt3/FOXO3 Pathway. Cell. Physiol. Biochem. 2018, 48, 2350–2363. [Google Scholar] [CrossRef]
- Bagul, P.; Katare, P.; Bugga, P.; Dinda, A.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Xue, R.-Q.; Lu, Y.; Yong, S.-Y.; Wu, Q.; Cui, Y.-L.; Zuo, X.-T.; Yu, X.-J.; Zhao, M.; Zang, W.-J. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc. Res. 2019, 115, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.; Dong, B.; Xing, F.; Huang, H.; Yao, F.; Ma, Y.; He, J.; Dong, Y. AMPK attenuates ventricular remodeling and dysfunction following aortic banding in mice via the Sirt3/Oxidative stress pathway. Eur. J. Pharmacol. 2017, 814, 335–342. [Google Scholar] [CrossRef]
- Zhang, M.; Lin, J.; Wang, S.; Cheng, Z.; Hu, J.; Wang, T.; Man, W.; Yin, T.; Guo, W.; Gao, E.; et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J. Pineal Res. 2017, 63, e12418. [Google Scholar] [CrossRef]
- Guo, R.; Liu, N.; Liu, H.; Zhang, J.; Zhang, H.; Wang, Y.; Baruscotti, M.; Zhao, L.; Wang, Y. High content screening identifies licoisoflavone A as a bioactive compound of Tongmaiyangxin Pills to restrain cardiomyocyte hypertrophy via activating Sirt3. Phytomedicine 2020, 68, 153171. [Google Scholar] [CrossRef]
- Luo, Y.-X.; Tang, X.; An, X.-Z.; Xie, X.-M.; Chen, X.-F.; Zhao, X.; Hao, D.-L.; Chen, H.-Z.; Liu, D.-P. Sirt4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 2016, 38, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Che, W.; Xue, J.; Zheng, C.; Tang, K.; Zhang, J.; Wen, J.; Xu, Y. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell. Physiol. Biochem. 2013, 32, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Liu, H.; Wang, H. Amelioration of myocardial ischemia-reperfusion injury by SIRT4 involves mitochondrial protection and reduced apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 15–21. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 201519858. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, K.A.; Abraham, D.M.; Martin, A.S.; Mao, L.; Liu, J.; Gu, H.; Locasale, J.W.; Hirschey, M.D. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 2017, 292, 19767–19781. [Google Scholar] [CrossRef] [Green Version]
- Zou, R.; Shi, W.; Tao, J.; Li, H.; Lin, X.; Yang, S.; Hua, P. SIRT5 and post-translational protein modifications: A potential therapeutic target for myocardial ischemia-reperfusion injury with regard to mitochondrial dynamics and oxidative metabolism. Eur. J. Pharmacol. 2018, 818, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, X.L.; Tong, M.M.; Gan, L.; Chen, H.; Wu, S.S.; Chen, J.X.; Li, R.L.; Wu, Y.; Zhang, H.Y.; et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3a-dependent antioxidant defense mechanisms. Basic Res. Cardiol. 2016, 111, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Vasudevan, P.; Zhong, L.; Kim, G.; Samant, S.; Parekh, V.; Pillai, V.B.; Ravindra, P.V.; Gupta, M.; Jeevanandam, V.; et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 2012, 18, 1643–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Sun, D.; Liu, Z.; Li, M.; Hong, H.; Liu, C.; Gao, S.; Li, H.; Cai, Y.; Chen, S.; et al. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl. Res. 2016, 172, 96–112. [Google Scholar] [CrossRef]
- Yu, S.S.; Cai, Y.; Ye, J.T.; Pi, R.B.; Chen, S.R.; Liu, P.Q.; Shen, X.Y.; Ji, Y. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-κB-dependent transcriptional activity. Br. J. Pharmacol. 2013, 168, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.; Sarikhani, M.; Dasgupta, S.; Maniyadath, B.; Pandit, A.S.; Mishra, S.; Ahamed, F.; Dubey, A.; Fathma, N.; Atreya, H.S.; et al. SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart. J. Cell. Physiol. 2018, 233, 5478–5489. [Google Scholar] [CrossRef]
- Araki, S.; Izumiya, Y.; Rokutanda, T.; Ianni, A.; Hanatani, S.; Kimura, Y.; Onoue, Y.; Senokuchi, T.; Yoshizawa, T.; Yasuda, O.; et al. Sirt7 Contributes to Myocardial Tissue Repair by Maintaining Transforming Growth Factor-β Signaling Pathway. Circulation 2015, 132, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Zhou, H.F.; Lin, R.B.; Fu, Y.C.; Wang, W. Short-term calorie restriction activates SIRT1-4 and -7 in cardiomyocytes in vivo and in vitro. Mol. Med. Rep. 2014, 9, 1218–1224. [Google Scholar] [CrossRef] [Green Version]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, P.B.; Yacoub, M.H.; Barton, P.J. Cyclin-dependent kinase inhibitor expression in human heart failure. A comparison with fetal development. Eur. Heart J. 1999, 20, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Nozato, T.; Ito, H.; Watanabe, M.; Ono, Y.; Adachi, S.; Tanaka, H.; Hiroe, M.; Sunamori, M.; Marum, F. Overexpression of cdk Inhibitor p16INK4a by adenovirus vector inhibits cardiac hypertrophy in vitro and in vivo: A novel strategy for the gene therapy of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2001, 33, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [Green Version]


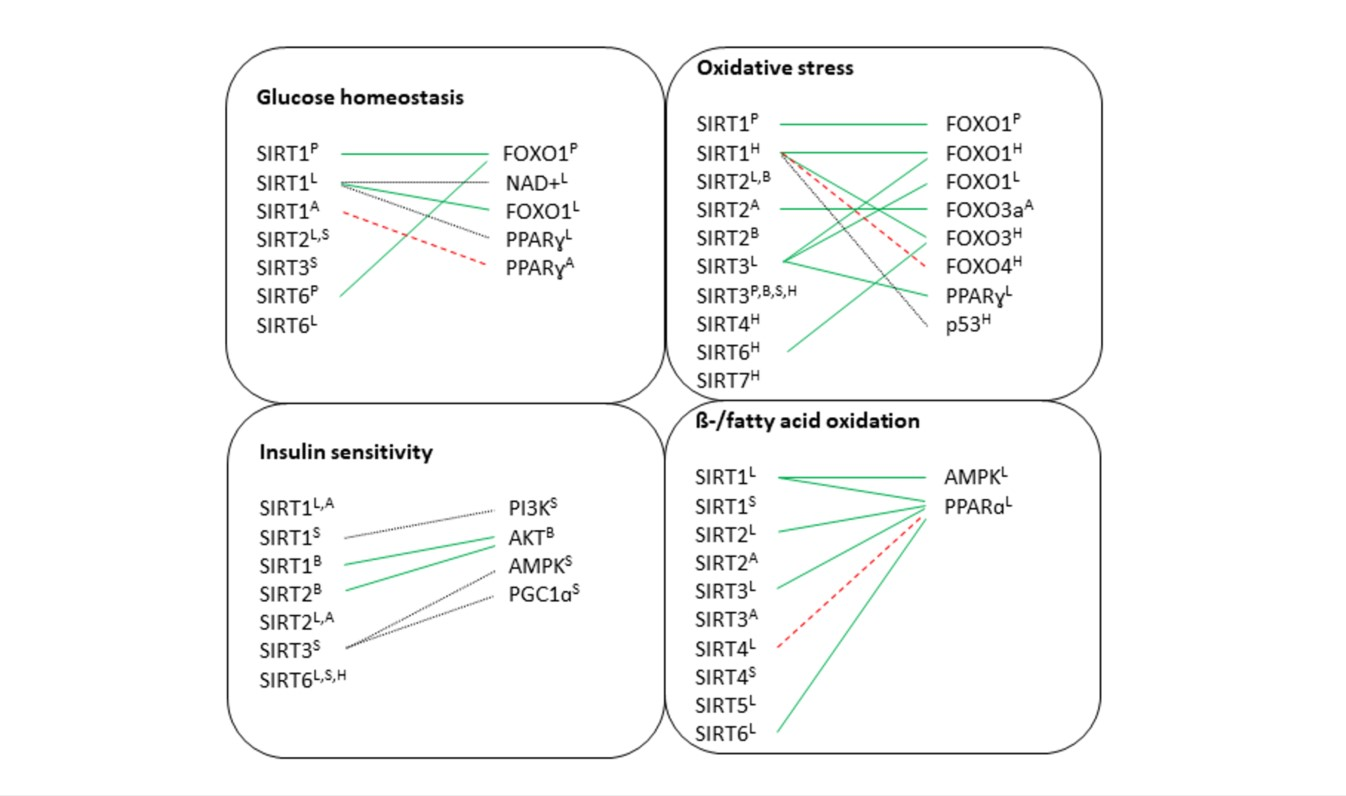{kind=link}
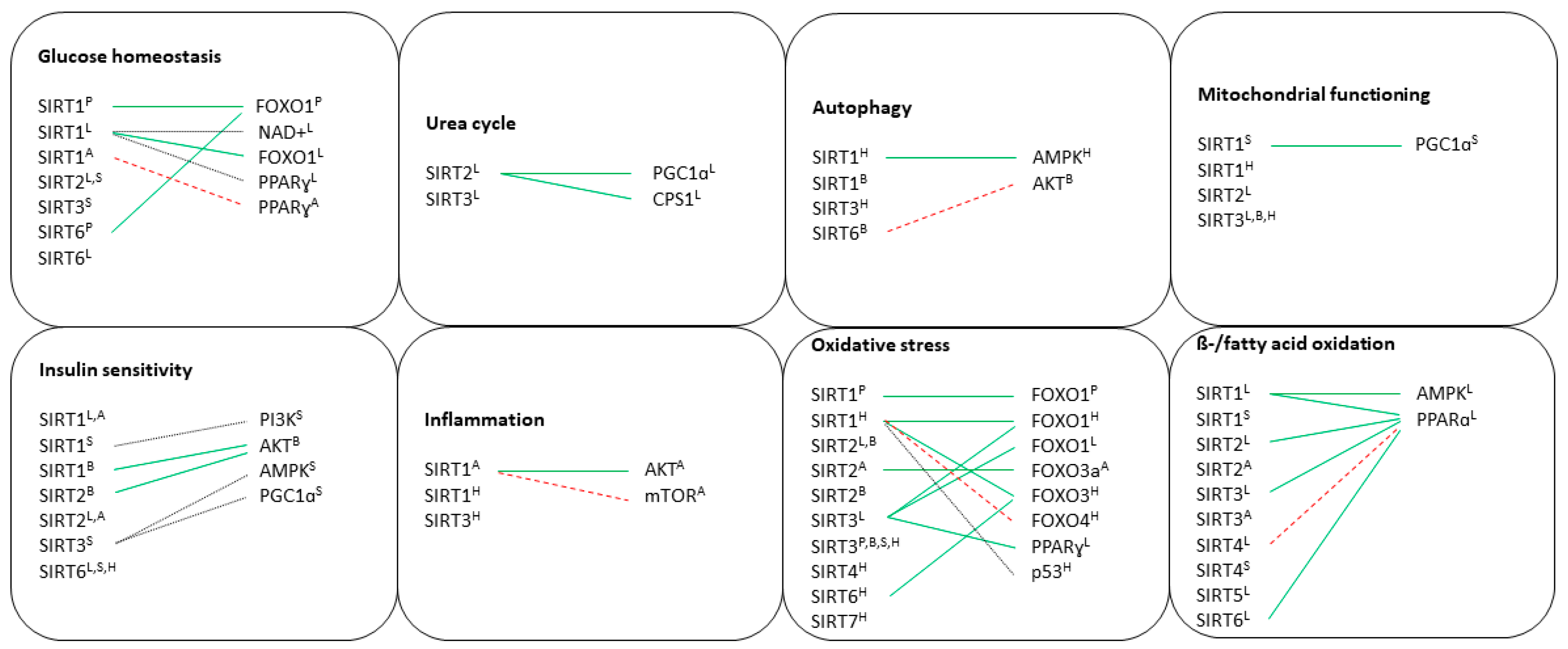{kind=link}
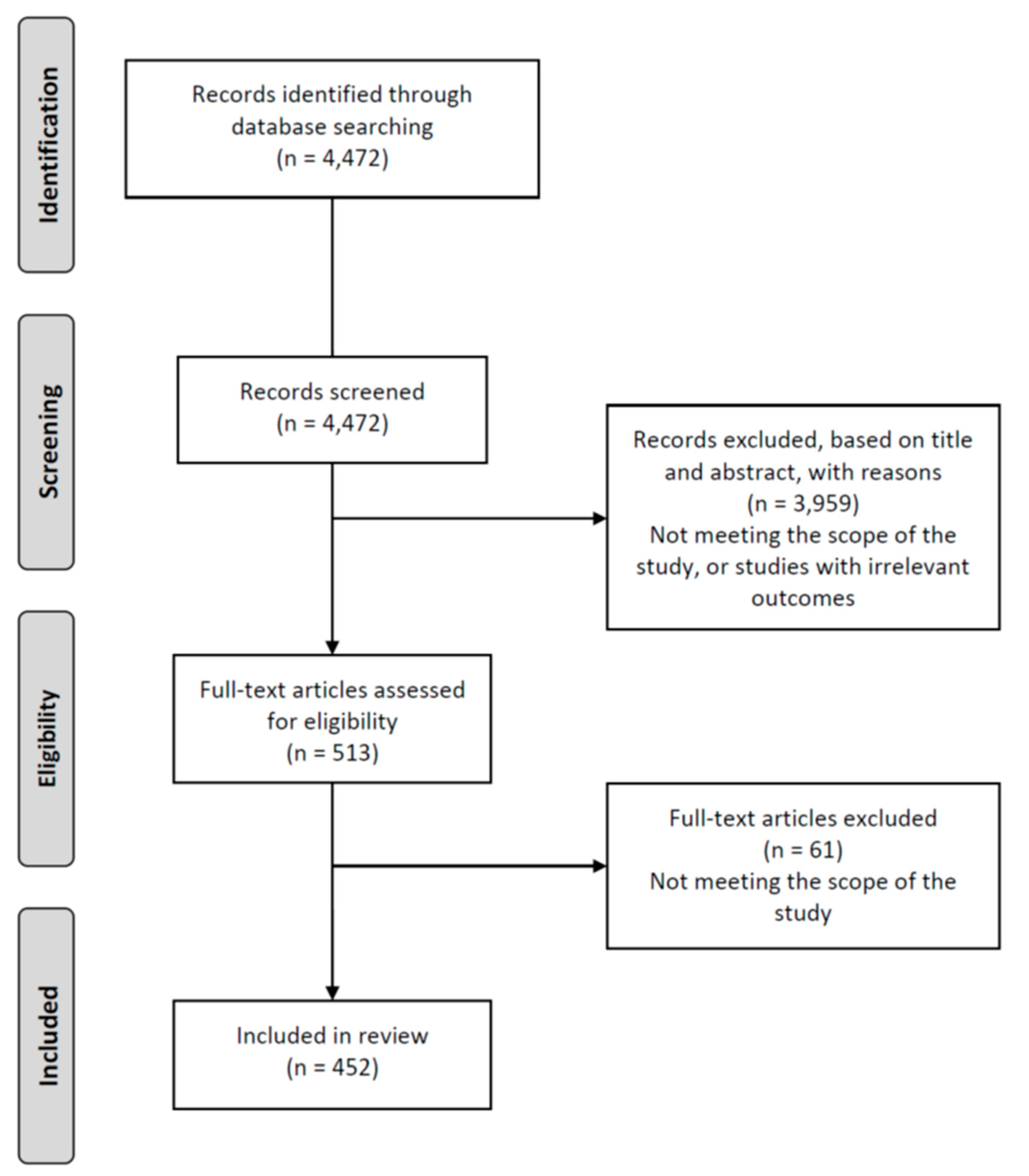{kind=link}
| Sirtuin | Expression and Localization | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on Sirtuin |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes, β-cells | PGC1α (N/A) FOXO1 (↓) FOXA2 (↓) FOXO3a (N/A) PPARγ (N/A) AMPK (↑) | Glucose tolerance (↑) Insulin secretion (↑) β-cells proliferation (↑) Oxidative stress (↓) | T2DM | Positive | CR (↑) HFD (↓) | Resveratrol (↑) Artesunate (↑) GABA (↑) Fucoidan (↑) |
| Sirt2 | Yes, RNA and cancer cells | N/A | N/A | N/A | N/A | N/A | N/A |
| Sirt3 | Yes, β-cells | N/A | Inflammation (↓) Insulin secretion (↑) Oxidative stress (↓) | T2DM | Ambiguous | N/A | N/A |
| Sirt4 | Yes, β-cells | N/A | Insulin secretion (↓) | N/A | Negative | CR (↓) | N/A |
| Sirt5 | Yes, β-cells | N/A | Insulin signaling (↑↓) β-cell maintenance (↑↓) | T2DM | Ambiguous | N/A | N/A |
| Sirt6 | Yes, β-cells | FOXO1 (↓) | Glucose tolerance (↑) Insulin signaling (↑) | N/A | Positive | N/A | N/A |
| Sirt7 | Yes, exocrine glands and islets of Langerhans | N/A | N/A | N/A | N/A | N/A | N/A |
| Sirtuin | Expression | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on Sirtuin |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes | PGC1α (↑) PPARγ (N/A) FOXO1 (↑) AMPK (↑) PPARα (↑) | Gluconeogenesis (↑) Lipid metabolism (↑) Inflammation (↓) Insulin sensitivity (↑) β-oxidation (↑) | T2DM NAFLD | Positive | HFD (↑↓) CR (↓) Fasting (↑) | Metformin (↑) Resveratrol (↑) Maslinic acid (↑) Vitamin K (↑) Fisetin (↑) Fucoidan (↑) Mangiferin (↑) LB100 (↑) APE (↑) γ-mangostin (↑) Liraglutide (↑) |
| Sirt2 | Yes | N/A | Insulin sensitivity (↑) Glucose uptake (↑) Glucose tolerance (↑) | N/A | Positive | N/A | N/A |
| Sirt3 | Yes | PPARγ (N/A) FOXO1 (N/A) PPARα (N/A) PGC1α (N/A) | ROS scavenging (↑) Mitochondrial integrity (↑) | NAFLD | Positive | CR (↑) Ketogenic (↑) Fasting (↑) | Salvianolic acid B (↑) Berberine (↑) |
| Sirt4 | Yes | SIRT1 (↓) PPARα (↓) mTOR (N/A) | Fatty acid oxidation (↓) Mitochondrial gene expression (↓) | N/A | Negative | N/A | 3-Iodothyronamine (↓) |
| Sirt5 | Yes | PGC1α (N/A) PPARα (N/A) AMPK (N/A) | Ketogenesis (↑) β-oxidation (↑) | NAFLD | Positive | Fasting (↑) High protein (↑) | N/A |
| Sirt6 | Yes | SIRT1 (N/A) FOXO3a (N/A) PPARα (↑) ERRγ (↓) | Glycolysis (↓) Triglyceride synthesis (↓) Lipid metabolism (↓) Insulin sensitivity (↑) β-oxidation (↑) | NAFLD | Positive | N/A | Rolipram (↑) 3-Iodothyronamine (↑) |
| Sirt7 | Yes | N/A | Triglyceride synthesis (↓) | NAFLD | Ambiguous | N/A | N/A |
| Sirtuin | Expression and Localization | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on SIRTUIN |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes, hypothalamus (POMS and AgRP neurons) and hippocampus | p53 (N/A) AMPK (N/A) PPARγ (↑) PGC1α (↑) mTOR (↓) FOXO3a (N/A) | Insulin sensitivity (↓) Systemic fat accumulation (↑) Inflammation (↓) | TD2M Ischemic injury | Negative | Fasting (↑) CR (↓) Ketosis (↑) Low protein (↑) | Resveratrol (↑) SRT1720 (↑) |
| Sirt2 | Yes | N/A | Insulin sensitivity (↓) Ischemic injury (↓) | N/A | Negative | N/A | N/A |
| Sirt3 | Yes, microglia, hippocampus and substantia nigra | FOXO3a (↑) PPARγ (↑) PGC1α (↑) | Mitochondrial biogenesis (↑) ROS reduction (↑) | Ischemic injury TD2M | Positive | Ketosis (↑) | Honokiol (↑) |
| Sirt4 | Yes | Inhibited by mTOR | N/A | N/A | N/A | Glucose (↑) | N/A |
| Sirt5 | Yes | N/A | Ischemic injury (↓) | N/A | Positive | CR (↑) | N/A |
| Sirt6 | Yes, uniformly expressed | N/A | Apoptosis and cell survival (↑) | N/A | Ambiguous | CR (↑) Glucose (↑) | N/A |
| Sirt7 | Yes | N/A | Neurogenesis (↑) | N/A | N/A | N/A | N/A |
| Sirtuin | Expression and Localization | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on Sirtuin |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes, brown and white adipose tissues | PPARδ (N/A) PPARγ (↓) mTOR (↓) FOXO1 (↑) Adiponectin (↑) AMPK (N/A) | Insulin resistance (↓) Adipogenesis (↓) Lipolysis (↑) Inflammation (↓) Mitochondrial function (↑) Glucose homeostasis (↑) Thermogenesis (↑) | Obesity | Positive | CR (↑) HFD (↓) | Resveratrol (↑) Spirulina-maxima (↑) Gallic acid (↑) Coumestrol (↑) P. grandifloras (↑) |
| Sirt2 | Yes, brown and white adipose tissues | FOXO3a (↑) FOXO1 (↑) PGC1α (↑) | Adipogenesis (↓) ROS levels (↓) Lipolysis (↑) | Obesity | Positive | CR (↑) Fasting (↑) | N/A |
| Sirt3 | Yes, brown and white adipose tissues | PGC1α (N/A) AMPK (N/A) PPARγ (N/A) | Insulin resistance (↓) Thermogenesis (↑) Cellular respiration (↑) ROS levels (↓) Lipogenesis (↓) | Obesity NALFD | Positive | CR (↑) Fasting (↑) | P. grandifloras (↑) |
| Sirt4 | Yes, white adipose tissue | N/A | Lipogenesis (↑) | Obesity | Negative | HFD (↑) | N/A |
| Sirt5 | Yes | N/A | N/A | Obesity | Ambiguous | N/A | N/A |
| Sirt6 | Yes, brown and white adipose tissues | PPARγ (↓) FOXO1 (↑) AMPK (↑) | Glucose tolerance (↑) Insulin secretion (↑) | Obesity Lipidemia | N/A | N/A | Metformin (↑) Rolipram (↑) 3-Iodothyronamine (↑) |
| Sirt7 | Yes | SIRT1 (↓) | Adipogenesis (↑) | Obesity | Ambiguous | N/A | N/A |
| Sirtuin | Expression | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on Sirtuin |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes | PGC1α (↑) SIRT3 (↑) AMPK (N/A) FOXO1 (↓) FOXO3 (↓) PI3K (↑) | Mitochondrial activity (↑) Aerobic capacity (↑) Insulin resistance (↓) Fatty acid oxidation (↑) | T2DM | Positive | CR (↑) HFD (↓) | Resveratrol (↑) Telmisartan (↑) |
| Sirt2 | Yes | N/A | Glucose uptake (↓) | N/A | Negative | N/A | N/A |
| Sirt3 | Yes | AMPK (N/A) PGC1α (N/A) FOXO1 (↑) | Energy expenditure (↑) Respiratory rates (↓) Insulin resistance (↓) ROS levels (↓) Glucose uptake (↑) | T2DM | Positive | CR (↑) Fasting (↑) HFD (↓) | Metformin (↑) |
| Sirt4 | Yes | AMPK (↓) | ATP levels (↑) Fatty acid oxidation (↓) Cellular respiration (↓) | N/A | Negative | N/A | N/A |
| Sirt5 | Yes | N/A | N/A | N/A | N/A | N/A | N/A |
| Sirt6 | Yes | AMPK (↑) | Insulin sensitivity (?) | N/A | Ambiguous | N/A | N/A |
| Sirt7 | Yes | N/A | N/A | N/A | N/A | CR (↓) | N/A |
| Sirtuin | Expression | Effect of Sirtuin on Target Genes | Effect of Sirtuin on Local Processes | Involvement in Metabolic Diseases | Function in MetS (Effect on the Organism) | Effect of Dietary Influence on Sirtuin | Effect of Modulating Drugs on Sirtuin |
|---|---|---|---|---|---|---|---|
| Sirt1 | Yes | PGC1α (↑) FOXO1 (↑) FOXO3 (↑) FOXO4 (↑) p53 (↓) AMPK (↑) PGC1α (↑) PPARγ (↑) PPARα (↓) | Inflammation (↓) Oxidative stress (↓) Mitochondrial function (↓) Apoptosis (↓) Autophagy (↓) | Hypertrophy | Positive | CR (↑) | Resveratrol (↑) |
| Sirt2 | Yes | AMPK (↑) | Microtubule activity (↓) | Hypertrophy T2DM | Positive | CR (↑) | N/A |
| Sirt3 | Yes | AMPK (↑) SIRT6 (↑) | Oxidative stress (↓) Inflammation (↓) Apoptosis (↓) Mitochondrial function (↑) Autophagy | Hypertrophy | Positive | CR (↑) HFD (↓) | Melatonin (↑) Resveratrol (↑) |
| Sirt4 | Yes | N/A | Oxidative stress (↑) Apoptosis (↓) | Hypertrophy | Ambiguous | CR (↑) | N/A |
| Sirt5 | Yes | N/A | N/A | Hypertrophy | Positive | N/A | N/A |
| Sirt6 | Yes | AMPK (↑) SIRT3 (↑) | Oxidative stress (↓) Insulin resistance (↓) | Ischemia T2DM Hypertrophy | Positive | HFD (↓) | N/A |
| Sirt7 | Yes | p53 (↓) | Apoptosis (↓) Oxidative stress (↓) | Hypertrophy | Positive | CR (↑) | N/A |
| Sirtuin | Effect in Organs and Disease Progression (P, Positive; N, Negative; N/A, Not Available; AM, Ambiguous) | Effect of Dietary Influence and Organs | Effect of Modulating Drugs | Role in Metabolic Diseases |
|---|---|---|---|---|
| Sirt1 | Pancreas (P) Liver (P) Adipose tissue (P) Brain (N) Muscle (P) Heart (P) | CR (↑)—heart, muscle, adipose tissue, pancreas HFD (↓)—muscle, adipose tissue, pancreas HFD (↑↓)—liver CR (↑↓)—liver Fasting (↑)—brain CR (↓)—brain Ketosis (↑)—brain | Resveratrol (↑) Artesunate (↑) GABA (↑) Metformin (↑) Maslinic acid (↑) Vitamin K (↑) Fisetin (↑) Fucoidan (↑) Mangiferin (↑) Spirulina-maxima (↑) SRT1720 (↑) Telmisartan (↑) | Hypertrophy Obesity NAFLD T2DM Ischemic injury |
| Sirt2 | Pancreas (N/A) Liver (P) Adipose tissue (P) Brain (N) Muscle (N) Heart (P) | CR (↑)—heart, adipose tissue Fasting (↑)—adipose tissue | N/A | Obesity T2DM Hypertrophy |
| Sirt3 | Pancreas (AM) Liver (P) Adipose tissue (P) Brain (P) Muscle (P) Heart (P) | CR (↑)—heart, muscle, adipose tissue, liver, pancreas HFD (↓)—heart, muscle, pancreas Fasting (↑)—muscle, adipose tissue, liver Ketosis (↑)—brain, liver | Melatonin (↑) Resveratrol (↑) Metformin (↑) Honokiol (↑) Salvianolic acid B (↑) Berberine (↑) | Hypertrophy Obesity NAFLD T2DM Ischemic injury |
| Sirt4 | Pancreas (N) Liver (N) Adipose tissue (N) Brain (N/A) Muscle (N) Heart (AM) | HFD (↑)—adipose tissue Glucose (↑)—brain CR (↓)—pancreas | 3-Iodothyronamine (↓) | Hypertrophy Obesity |
| Sirt5 | Pancreas (AM) Liver (P) Adipose tissue (AM) Brain (P) Muscle (N/A) Hear (P) | Fasting (↑)—liver High protein (↑)—liver CR (↑)—brain | N/A | Hypertrophy Obesity NAFLD T2DM |
| Sirt6 | Pancreas (P) Liver (N/A) Adipose tissue (N/A) Brain (AM) Muscle (AM) Heart (P) | HFD (↓)—heart CR (↑)—brain Glucose (↑)—brain | Metformin (↑) Rolipram (↑) 3-Iodothyronamine (↑) | Ischemia T2DM Hypertrophy Obesity Lipidemia NAFLD |
| Sirt7 | Pancreas (N/A) Liver (AM) Adipose (N/A) Brain (N/A) Muscle (N/A) Heart (P) | CR (↓)—muscle CR (↑)—heart | N/A | Hypertrophy Obesity NAFLD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review. Biology 2021, 10, 194. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030194
Maissan P, Mooij EJ, Barberis M. Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review. Biology. 2021; 10(3):194. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030194
Chicago/Turabian StyleMaissan, Parcival, Eva J. Mooij, and Matteo Barberis. 2021. "Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review" Biology 10, no. 3: 194. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030194