
Genomic Features and Molecular Function of a Novel Stress-Tolerant Bacillus halotolerans Strain Isolated from an Extreme Environment

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Samples and Isolation of Bacteria
2.2. DNA Isolation and gyrB Gene Sequence Analysis
2.3. Genome Sequencing and Analysis
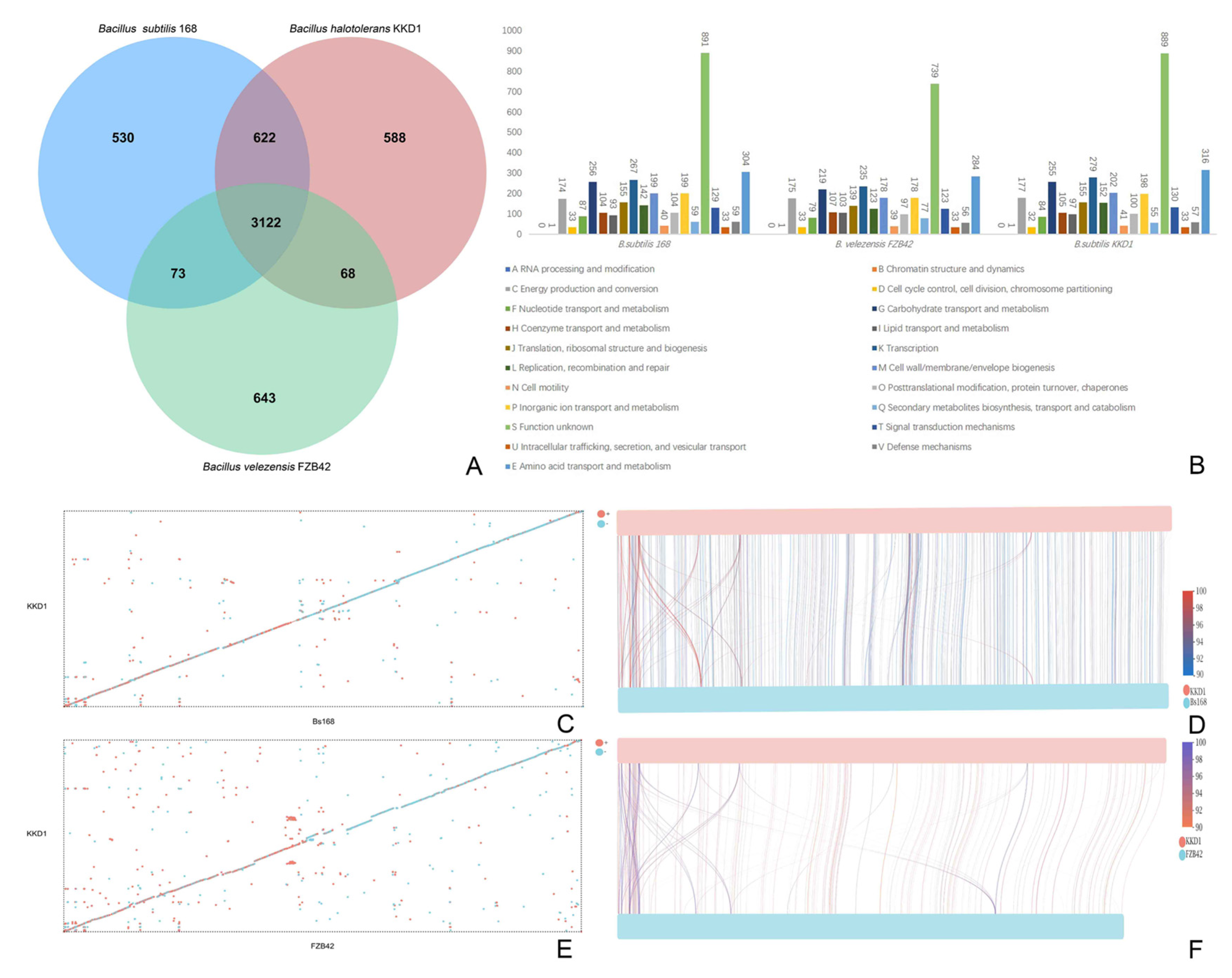
2.4. Comparative Genome Analysis
2.5. Inhibition of Phytopathogenic Fungi
2.6. Biocontrol Insights into KKD1 In Vitro Responses to Infection in Wheat
2.7. Extraction of Lipopeptides
2.8. MALDI-TOF-MS
2.9. HPLC Analysis
2.10. Environmental Stress
2.11. Quantitative Real-Time PCR
2.12. Primers, Sequence Alignment, RNA Analysis, and Statistical Analysis
3. Results and Discussion
3.1. Isolation and Identification of the Stress-Tolerant Bacillus spp. from the QTP
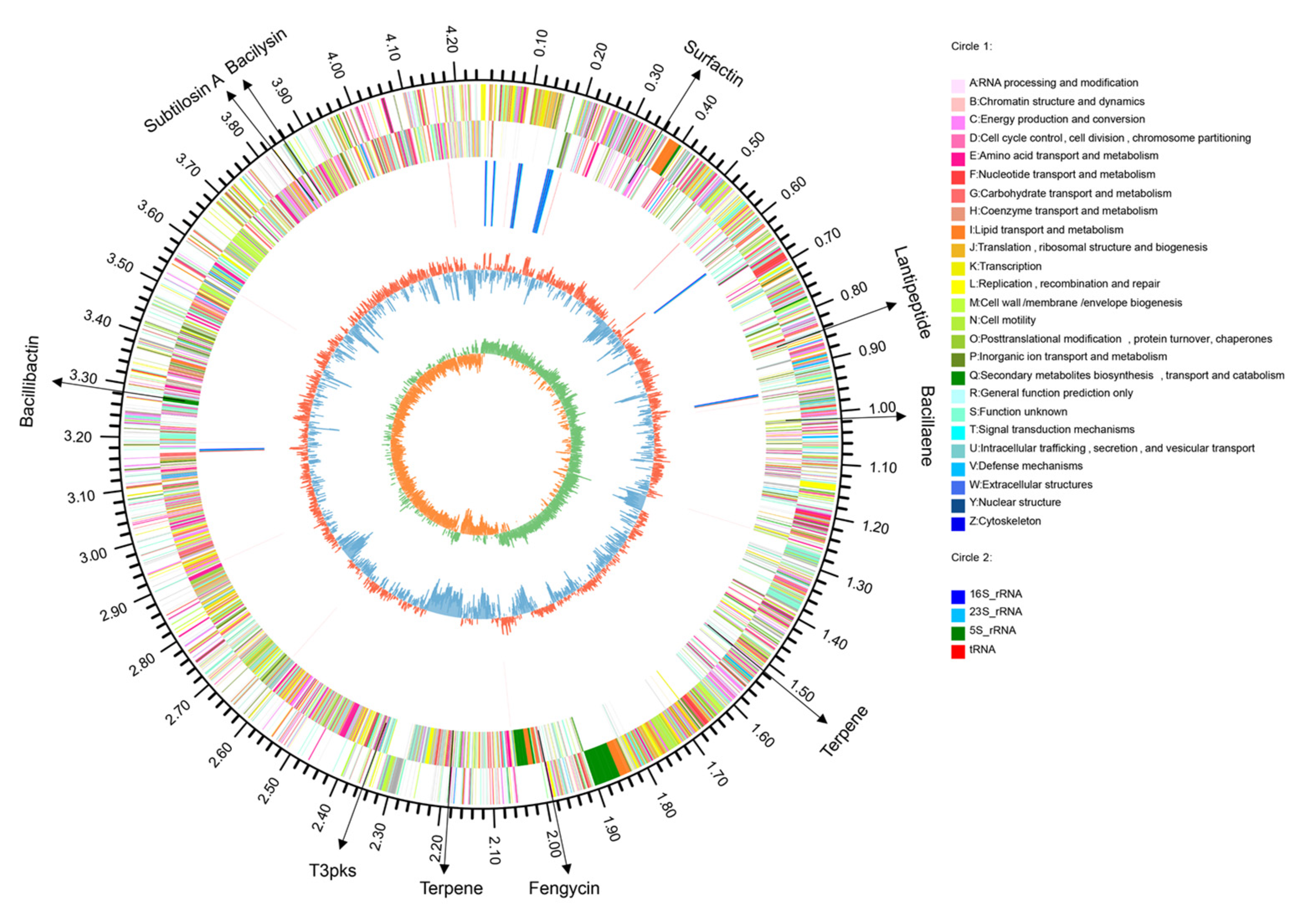
3.2. The Genome of B. halotolerans KKD1
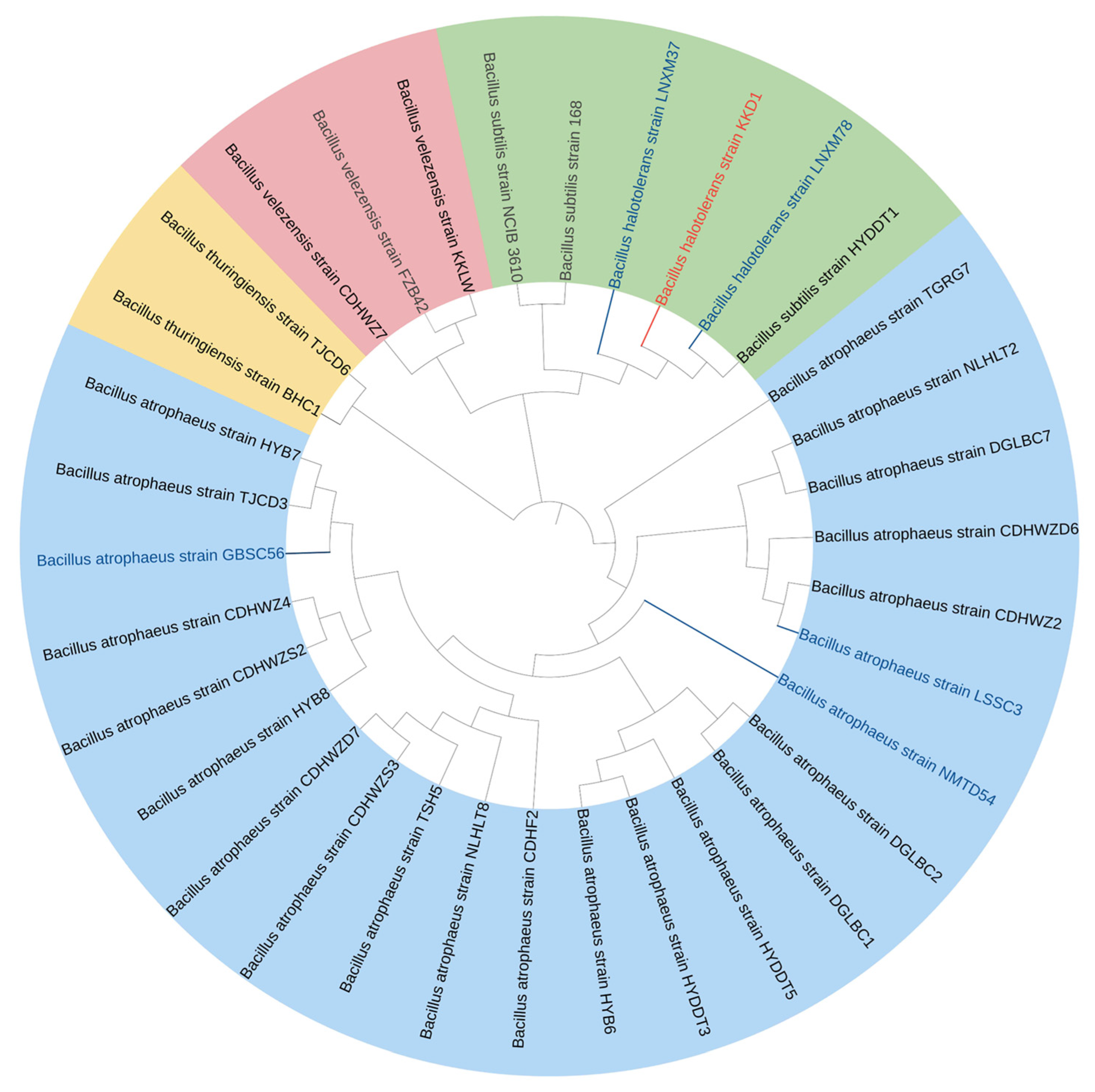
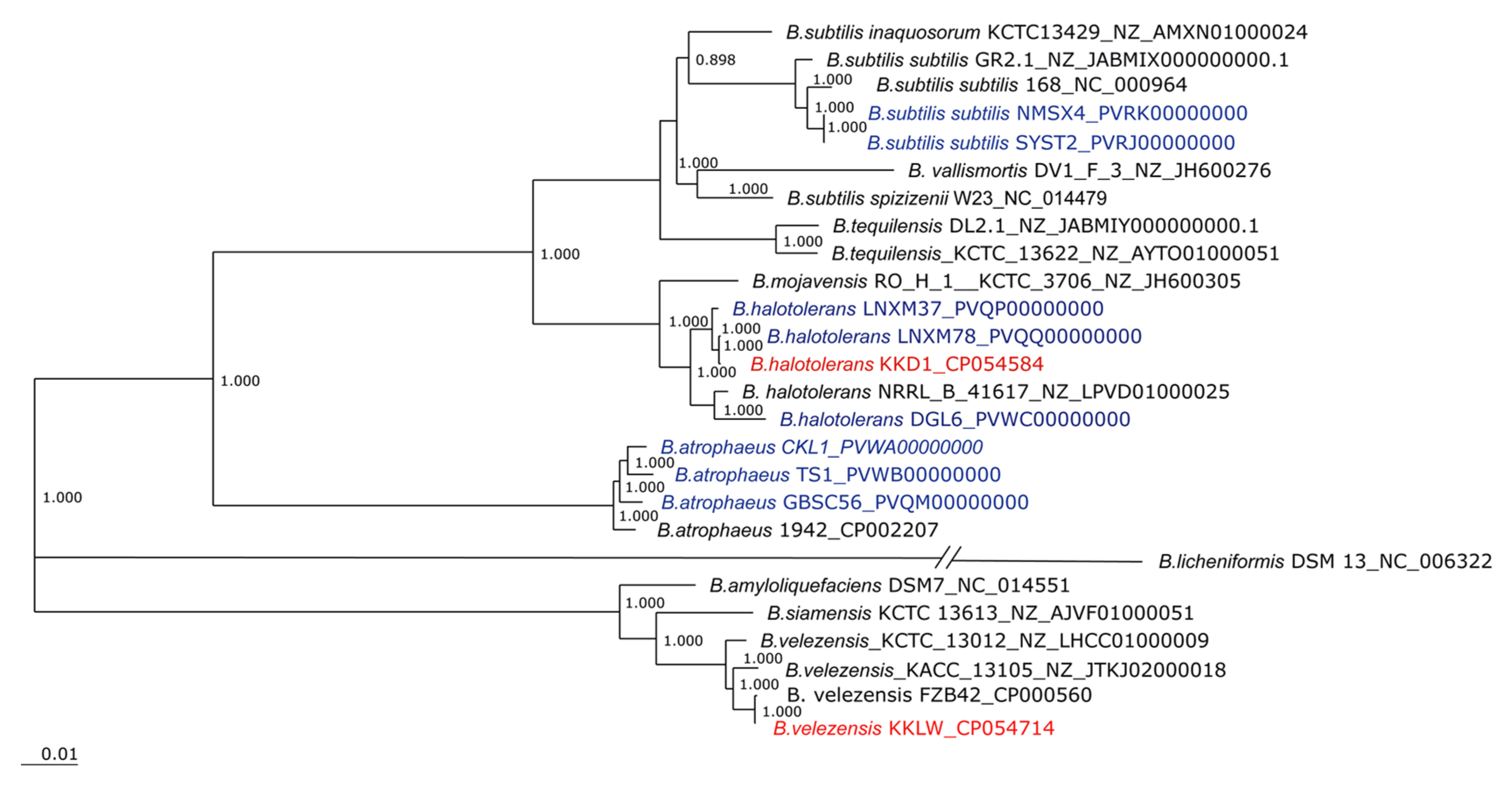
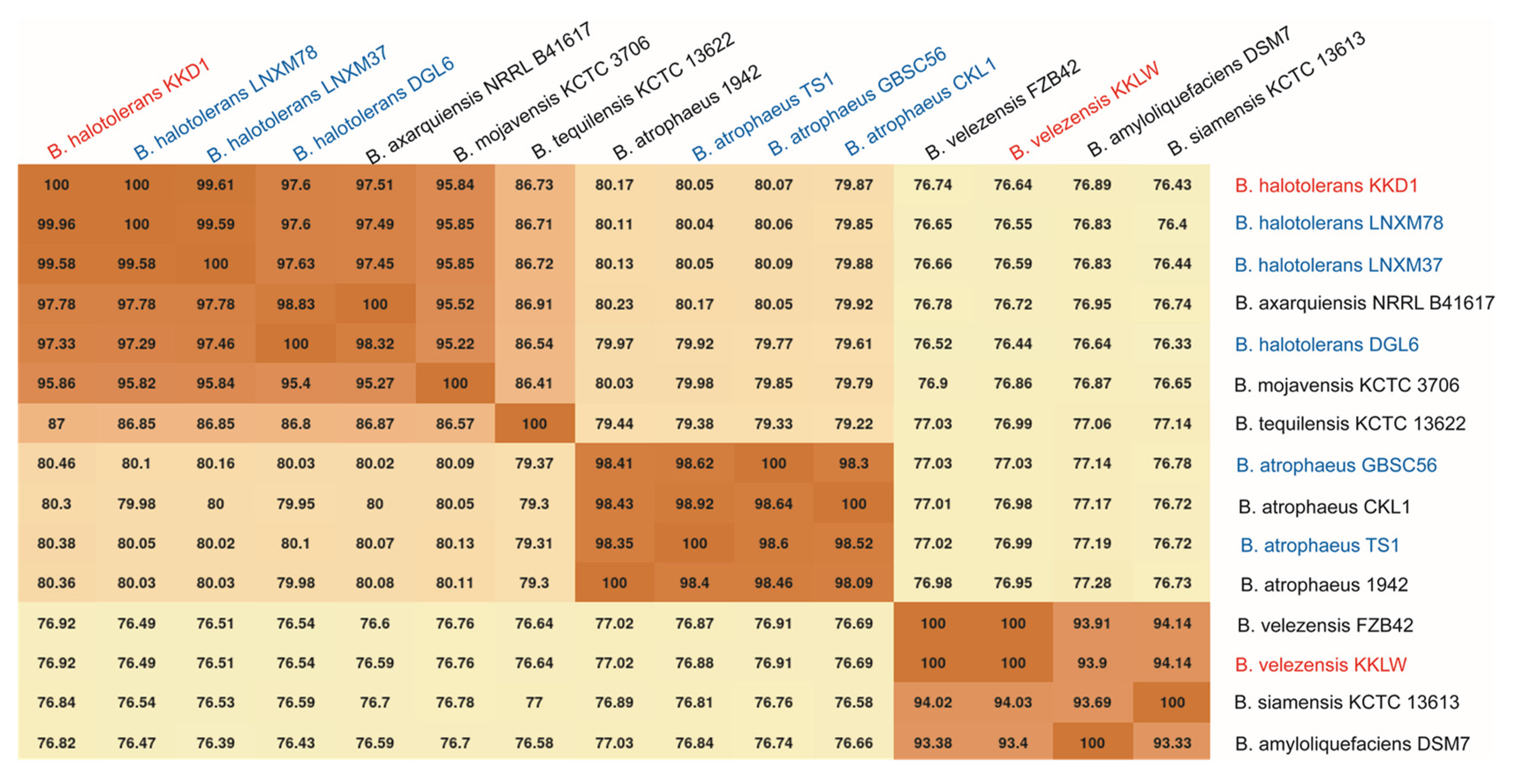
3.3. Taxonomical Assignment of Bacillus halotolerans KKD1 by Its Core Genome
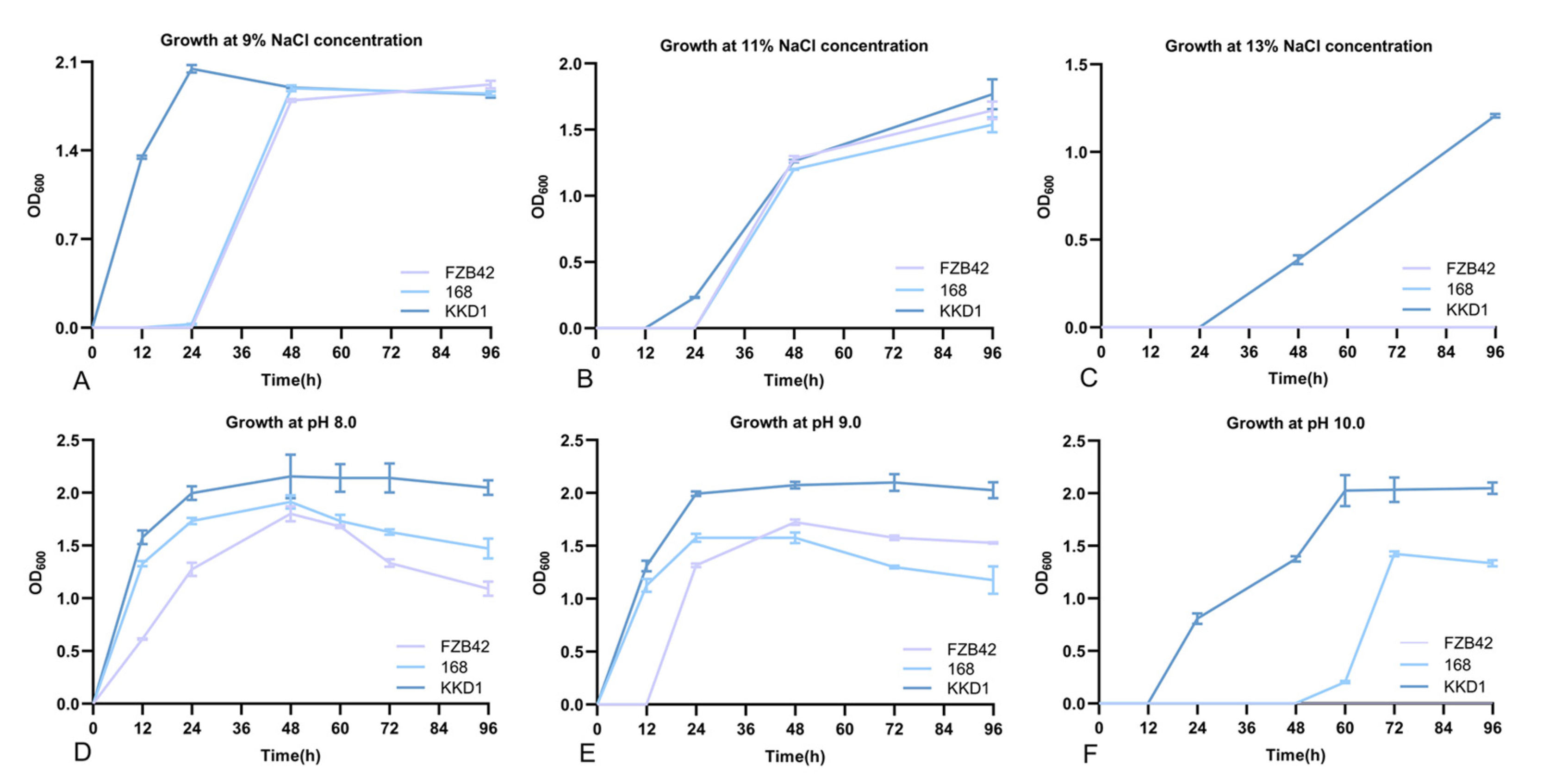
3.4. B. halotolerans KKD1 Tolerates Different Environmental Stress Conditions
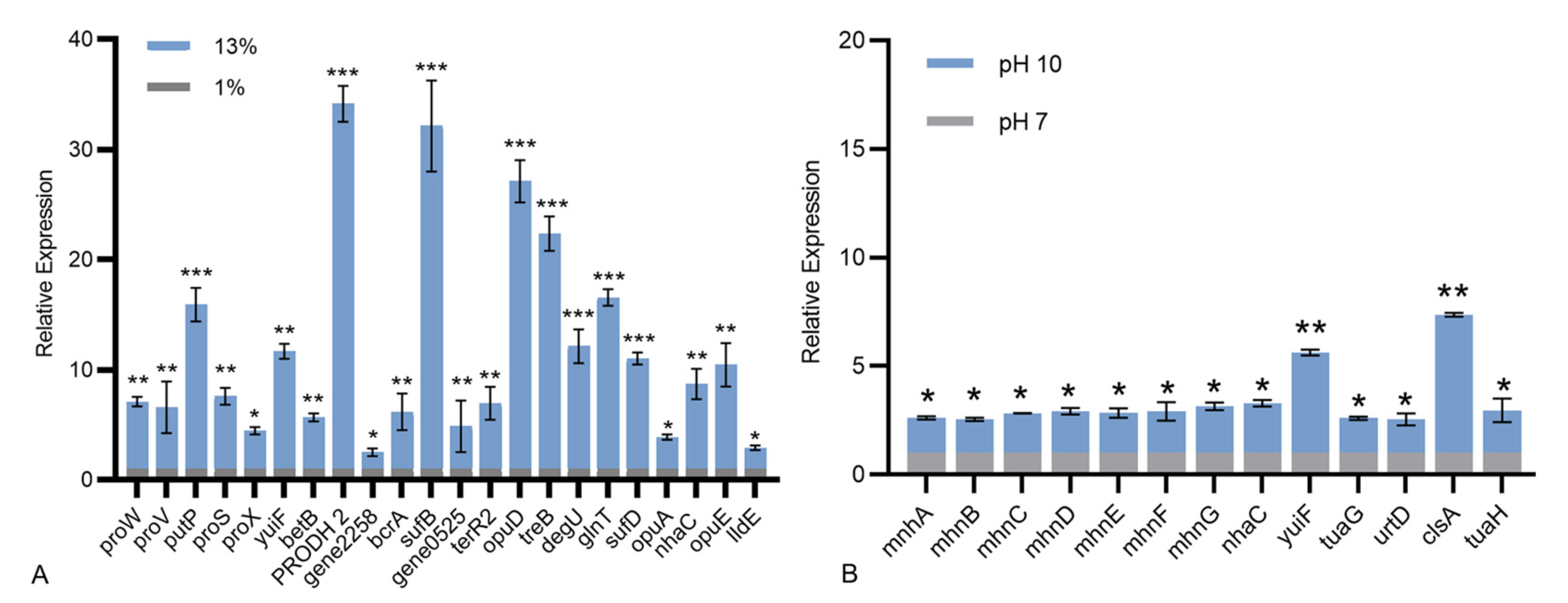
3.5. Genome Mining for Genes Possibly Involved in Tolerance to Stress Conditions
3.5.1. Signal Transduction Pathways Involved in Salt Tolerance
3.5.2. Alkaline Resistance
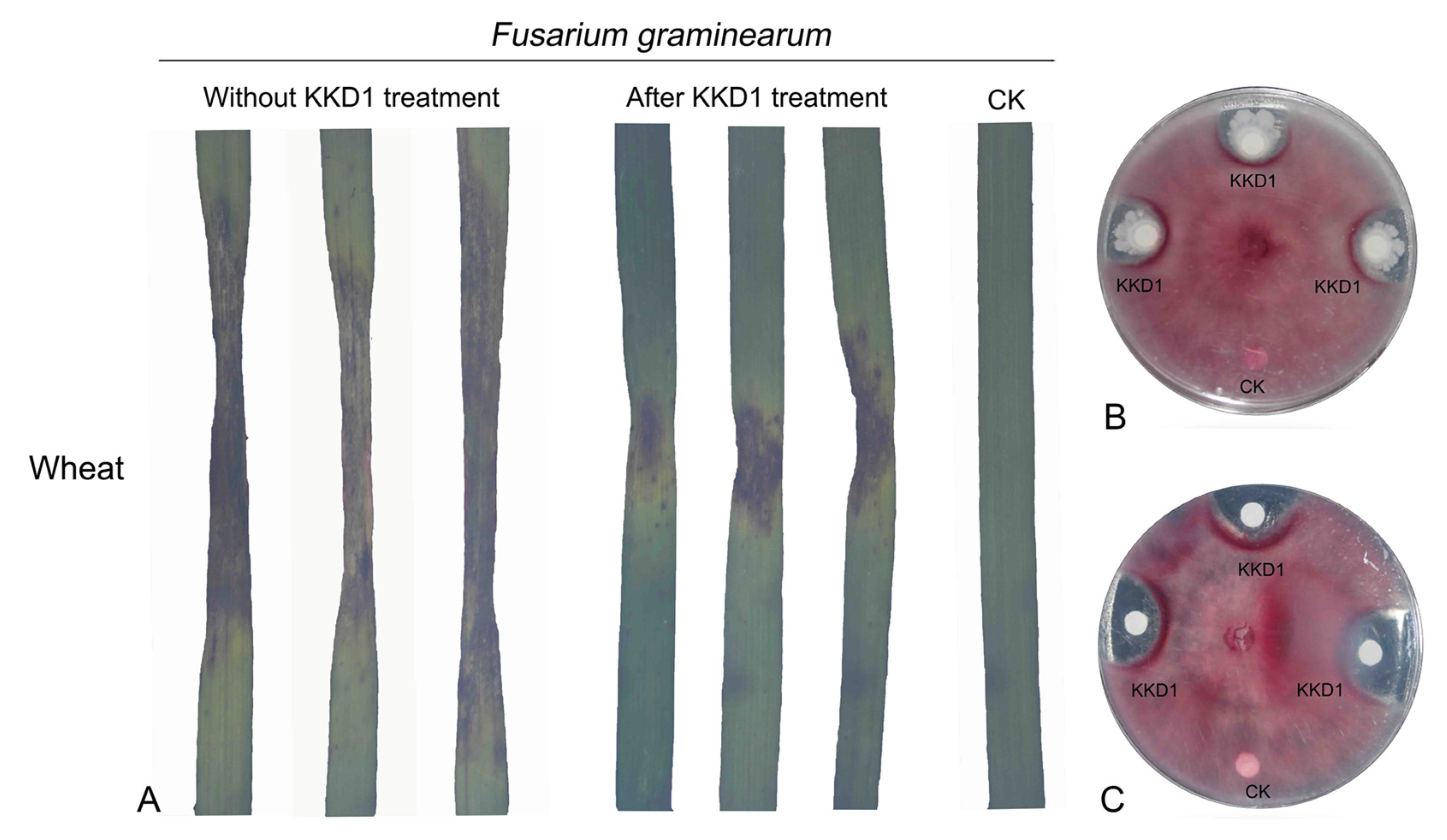
3.6. Biocontrol of Plant Pathogens In Vitro
3.7. Genome Mining for Gene Clusters Devoted to the Synthesis of Antimicrobial Secondary Metabolites
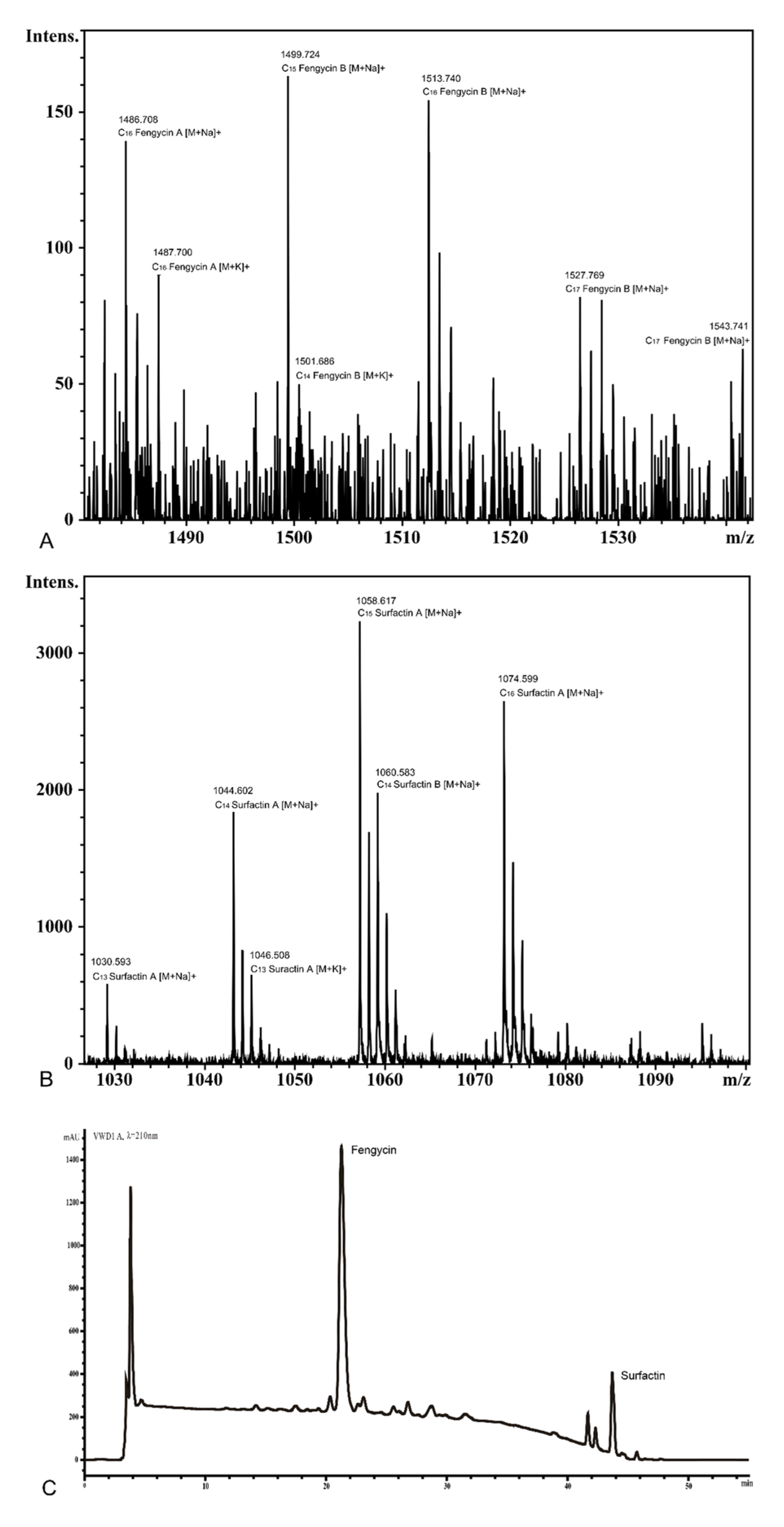
3.8. Detection of Surfactin and Fengycin in the Supernatant of KKD1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mipam, T.-D.; Zhong, L.-L.; Liu, J.-Q.; Miehe, G.; Tian, L.-M. Productive Overcompensation of Alpine Meadows in Response to Yak Grazing in the Eastern Qinghai-Tibet Plateau. Front. Plant Sci. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Niu, D.; Ma, P.; Wang, Y.; Wang, Z.; Fu, H.; Elser, J.J. Bacterial Communities in Stream Biofilms in a Degrading Grassland Watershed on the Qinghai-Tibet Plateau. Front. Microbiol. 2020, 11, 1021. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Gu, Q.; Xie, Y.; Lou, Z.; Xue, P.; Fang, L.; Yu, C.; Jia, D.; Huang, G.; Zhu, B.; et al. Cold-adapted Bacilli isolated from the Qinghai-Tibetan Plateau are able to promote plant growth in extreme environments. Environ. Microbiol. 2019. [Google Scholar] [CrossRef]
- Chu, T.N.; Bui, L.V.; Hoang, M.T.T. Pseudomonas PS01 Isolated from Maize Rhizosphere Alters Root System Architecture and Promotes Plant Growth. Microorganisms 2020, 8, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.H.; Ahmed, A.; Razzaq, A.; Hildebrandt, D.; Liu, X.; Park, Y.-K. Incorporation of solar-thermal energy into a gasification process to co-produce bio-fertilizer and power. Environ. Pollut. 2020, 266, 115103. [Google Scholar] [CrossRef]
- Zubair, M.; Hanif, A.; Farzand, A.; Sheikh, T.M.M.; Khan, A.R.; Suleman, M.; Ayaz, M.; Gao, X. Genetic Screening and Expression Analysis of Psychrophilic Bacillus spp. Reveal Their Potential to Alleviate Cold Stress and Modulate Phytohormones in Wheat. Microorganisms 2019, 7, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azmat, A.; Yasmin, H.; Hassan, M.N.; Nosheen, A.; Naz, R.; Sajjad, M.; Ilyas, N.; Akhtar, M.N. Co-application of bio-fertilizer and salicylic acid improves growth, photosynthetic pigments and stress tolerance in wheat under drought stress. PeerJ 2020, 8, e9960. [Google Scholar] [CrossRef]
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Eisenreich, A.; Schneider, K.; Heinemeyer, I.; Morgenstern, B.; Voss, B.; Hess, W.R.; Reva, O.; et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 2007, 25, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Chen, Y.; Liu, X.; Wang, X.; Wang, X.; Li, X.; Zhao, Y.; Wei, L. Engineered biosynthesis of cyclic lipopeptide locillomycins in surrogate host Bacillus velezensis FZB42 and derivative strains enhance antibacterial activity. Appl. Microbiol. Biotechnol. 2019, 103, 4467–4481. [Google Scholar] [CrossRef]
- Massawe, V.C.; Hanif, A.; Farzand, A.; Mburu, D.K.; Ochola, S.O.; Wu, L.; Tahir, H.A.S.; Gu, Q.; Wu, H.; Gao, X. Volatile Compounds of Endophytic Bacillus spp. have Biocontrol Activity against Sclerotinia sclerotiorum. Phytopathology 2018, 108, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Hanif, A.; Zhang, F.; Li, P.; Li, C.; Xu, Y.; Zubair, M.; Zhang, M.; Jia, D.; Zhao, X.; Liang, J.; et al. Fengycin Produced by Bacillus amyloliquefaciens FZB42 Inhibits Fusarium graminearum Growth and Mycotoxins Biosynthesis. Toxins 2019, 11, 295. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Tahir, H.A.S.; Zhang, H.; Huang, H.; Ji, T.; Sun, X.; Wu, L.; Wu, H.; Gao, X. Involvement of FvSet1 in Fumonisin B1 Biosynthesis, Vegetative Growth, Fungal Virulence, and Environmental Stress Responses in Fusarium verticillioides. Toxins 2017, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Wu, H.; Chen, L.; Yu, X.; Borriss, R.; Gao, X. Difficidin and bacilysin from Bacillus amyloliquefaciens FZB42 have antibacterial activity against Xanthomonas oryzae rice pathogens. Sci. Rep. 2015, 5, 12975. [Google Scholar] [CrossRef] [PubMed]
- Reeve, M.A.; Bachmann, D. MALDI-TOF MS protein fingerprinting of mixed samples. Biol. Methods Protoc. 2019, 4, bpz013. [Google Scholar] [CrossRef] [PubMed]
- Farzand, A.; Moosa, A.; Zubair, M.; Khan, A.R.; Massawe, V.C.; Tahir, H.A.S.; Sheikh, T.M.M.; Ayaz, M.; Gao, X. Suppression of Sclerotinia sclerotiorum by the Induction of Systemic Resistance and Regulation of Antioxidant Pathways in Tomato Using Fengycin Produced by Bacillus amyloliquefaciens FZB42. Biomolecules 2019, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Aliyu, H.; Lebre, P.; Blom, J.; Cowan, D.; De Maayer, P. Phylogenomic re-assessment of the thermophilic genus Geobacillus. Syst. Appl. Microbiol. 2016, 39, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Blom, J.; Klenk, H.-P.; Borriss, R. Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. amyloliquefaciens” within the B. subtilis Species Complex. Front. Microbiol. 2017, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Fritze, D. Taxonomy of the genus bacillus and related genera: The aerobic endospore-forming bacteria. Phytopathology 2004, 94, 1245–1248. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Ara, K.; Ozaki, K.; Nakamura, K.; Yamane, K.; Sekiguchi, J.; Ogasawara, N. Bacillus minimum genome factory: Effective utilization of microbial genome information. Biotechnol. Appl. Biochem. 2007, 46, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; von Blohn, C.; Stanek, A.; Moses, S.; Barzantny, H.; Bremer, E. Synthesis, release, and recapture of compatible solute proline by osmotically stressed Bacillus subtilis cells. Appl. Environ. Microbiol. 2012, 78, 5753–5762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, T.; Bleisteiner, M.; Sappa, P.K.; Steil, L.; Mäder, U.; Völker, U.; Bremer, E. Synthesis of the compatible solute proline by Bacillus subtilis: Point mutations rendering the osmotically controlled proHJ promoter hyperactive. Environ. Microbiol. 2017, 19, 3700–3720. [Google Scholar] [CrossRef]
- El-Esawi, M.A.; Alaraidh, I.A.; Alsahli, A.A.; Alamri, S.A.; Ali, H.M.; Alayafi, A.A. Bacillus firmus (SW5) augments salt tolerance in soybean (Glycine max L.) by modulating root system architecture, antioxidant defense systems and stress-responsive genes expression. Plant Physiol. Biochem. PPB 2018, 132, 375–384. [Google Scholar] [CrossRef]
- Bürklen, L.; Schöck, F.; Dahl, M.K. Molecular analysis of the interaction between the Bacillus subtilis trehalose repressor TreR and the tre operator. Mol. Gen. Genet. 1998, 260, 48–55. [Google Scholar] [CrossRef]
- Servet, C.; Ghelis, T.; Richard, L.; Zilberstein, A.; Savoure, A. Proline dehydrogenase: A key enzyme in controlling cellular homeostasis. Front. Biosci. (Landmark Ed.) 2012, 17, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Laye, V.J.; DasSarma, S. An Antarctic Extreme Halophile and Its Polyextremophilic Enzyme: Effects of Perchlorate Salts. Astrobiology 2018, 18, 412–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, M.; Kosono, S.; Kudo, T. The Na+/H+ antiporter of alkaliphilic Bacillus sp. Extremophiles 2000, 4, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Crowe-McAuliffe, C.; Graf, M.; Huter, P.; Takada, H.; Abdelshahid, M.; Nováček, J.; Murina, V.; Atkinson, G.C.; Hauryliuk, V.; Wilson, D.N. Structural basis for antibiotic resistance mediated by the Bacillus subtilis ABCF ATPase VmlR. Proc. Natl. Acad. Sci. USA 2018, 115, 8978–8983. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Meng, Y.; Cui, Y.; Li, C.; Tao, F.; Yin, H.; Yang, C.; Xu, P. Alkaline Response of a Halotolerant Alkaliphilic Halomonas Strain and Functional Diversity of Its Na+(K+)/H+ Antiporters. J. Biol. Chem. 2016, 291, 26056–26065. [Google Scholar] [CrossRef] [Green Version]
- Mirouze, N.; Ferret, C.; Cornilleau, C.; Carballido-López, R. Antibiotic sensitivity reveals that wall teichoic acids mediate DNA binding during competence in Bacillus subtilis. Nat. Commun. 2018, 9, 5072. [Google Scholar] [CrossRef] [PubMed]
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Alloni, G.; Azevedo, V.; Bertero, M.G.; Bessières, P.; Bolotin, A.; Borchert, S.; et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [Green Version]
- Keswani, C.; Singh, H.B.; Hermosa, R.; García-Estrada, C.; Caradus, J.; He, Y.-W.; Mezaache-Aichour, S.; Glare, T.R.; Borriss, R.; Vinale, F.; et al. Antimicrobial secondary metabolites from agriculturally important fungi as next biocontrol agents. Appl. Microbiol. Biotechnol. 2019, 103, 9287–9303. [Google Scholar] [CrossRef]
- Ali, S.; Hameed, S.; Shahid, M.; Iqbal, M.; Lazarovits, G.; Imran, A. Functional characterization of potential PGPR exhibiting broad-spectrum antifungal activity. Microbiol. Res. 2020, 232, 126389. [Google Scholar] [CrossRef]
- Zhao, H.; Shao, D.; Jiang, C.; Shi, J.; Li, Q.; Huang, Q.; Rajoka, M.S.R.; Yang, H.; Jin, M. Biological activity of lipopeptides from Bacillus. Appl. Microbiol. Biotechnol. 2017, 101, 5951–5960. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, S.; Mukherjee, A.; Rastogi, R.P.; Verma, J.P. Salt-tolerant plant growth-promoting Bacillus pumilus strain JPVS11 to enhance plant growth attributes of rice and improve soil health under salinity stress. Microbiol. Res. 2021, 242, 126616. [Google Scholar] [CrossRef]
- Moldenhauer, J.; Götz, D.C.G.; Albert, C.R.; Bischof, S.K.; Schneider, K.; Süssmuth, R.D.; Engeser, M.; Gross, H.; Bringmann, G.; Piel, J. The final steps of bacillaene biosynthesis in Bacillus amyloliquefaciens FZB42: Direct evidence for beta, gamma dehydration by a trans-acyltransferase polyketide synthase. Angew. Chem. Int. Ed. Engl. 2010, 49, 1465–1467. [Google Scholar] [CrossRef]
- Chatterjee, C.; Paul, M.; Xie, L.; van der Donk, W.A. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 2005, 105, 633–684. [Google Scholar] [CrossRef]
- Putkaradze, N.; Litzenburger, M.; Abdulmughni, A.; Milhim, M.; Brill, E.; Hannemann, F.; Bernhardt, R. CYP109E1 is a novel versatile statin and terpene oxidase from Bacillus megaterium. Appl. Microbiol. Biotechnol. 2017, 101, 8379–8393. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Chen, X.-H.; Vater, J.; Franke, P.; Nicholson, G.; Borriss, R.; Süssmuth, R.D. Macrolactin is the polyketide biosynthesis product of the pks2 cluster of Bacillus amyloliquefaciens FZB42. J. Nat. Prod. 2007, 70, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Scholz, R.; Molohon, K.J.; Nachtigall, J.; Vater, J.; Markley, A.L.; Süssmuth, R.D.; Mitchell, D.A.; Borriss, R. Plantazolicin, a novel microcin B17/streptolysin S-like natural product from Bacillus amyloliquefaciens FZB42. J. Bacteriol. 2011, 193, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Wu, H.; Wang, R.; Wang, Z.; Zhang, Y.; Gu, Q.; Farzand, A.; Yang, X.; Semenov, M.; Borriss, R.; et al. Genomic Features and Molecular Function of a Novel Stress-Tolerant Bacillus halotolerans Strain Isolated from an Extreme Environment. Biology 2021, 10, 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101030
Wu X, Wu H, Wang R, Wang Z, Zhang Y, Gu Q, Farzand A, Yang X, Semenov M, Borriss R, et al. Genomic Features and Molecular Function of a Novel Stress-Tolerant Bacillus halotolerans Strain Isolated from an Extreme Environment. Biology. 2021; 10(10):1030. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101030
Chicago/Turabian StyleWu, Xiaohui, Huijun Wu, Ruoyi Wang, Zhengqi Wang, Yaming Zhang, Qin Gu, Ayaz Farzand, Xue Yang, Mikhail Semenov, Rainer Borriss, and et al. 2021. "Genomic Features and Molecular Function of a Novel Stress-Tolerant Bacillus halotolerans Strain Isolated from an Extreme Environment" Biology 10, no. 10: 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101030