
Identification and Characterization of MicroRNAs Involving in Initial Sex Differentiation of Chlamys farreri Gonads


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
2.2. RNA Extraction, Small RNA Library Construction, and Sequencing
2.3. Processing of Sequencing Data
2.4. Analysis of Differentially Expressed miRNAs
2.5. Prediction of Potential Target Genes of DEMs
2.6. Verification of DEMs Using RT-qPCR
2.7. Validation of miRNA–mRNA Interaction by Dual-Luciferase Reporter Assay
2.8. Statistical Analysis
3. Results
3.1. Overview of Small RNA Sequencing Statistics in C. farreri Gonads
3.2. Identification of miRNAs
3.3. DEMs Identified between Ovary and Testis
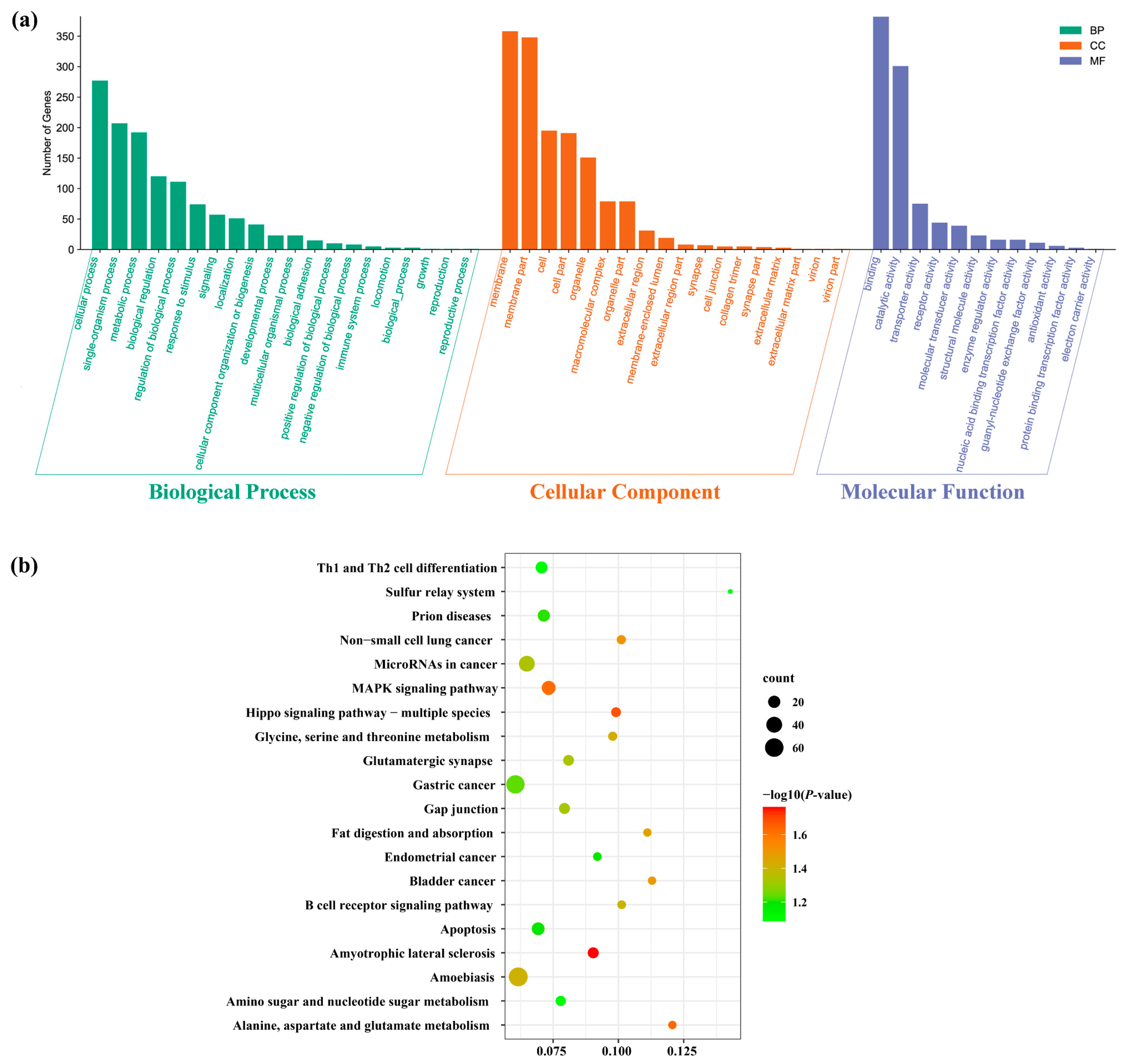
3.4. Target Gene Prediction and Functional Enrichment Analysis of DEMs
3.5. Validation of DEMs Using RT-qPCR
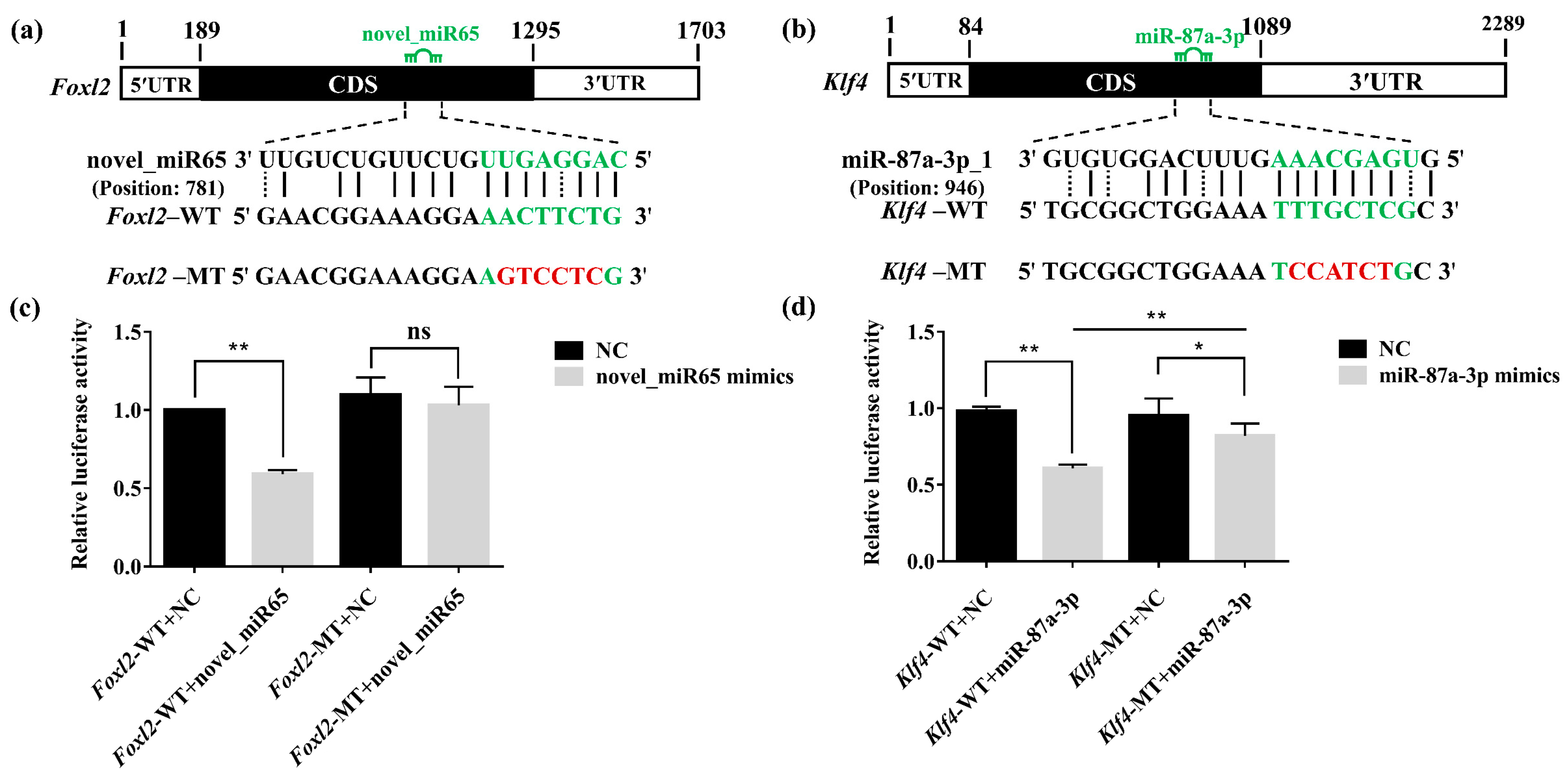
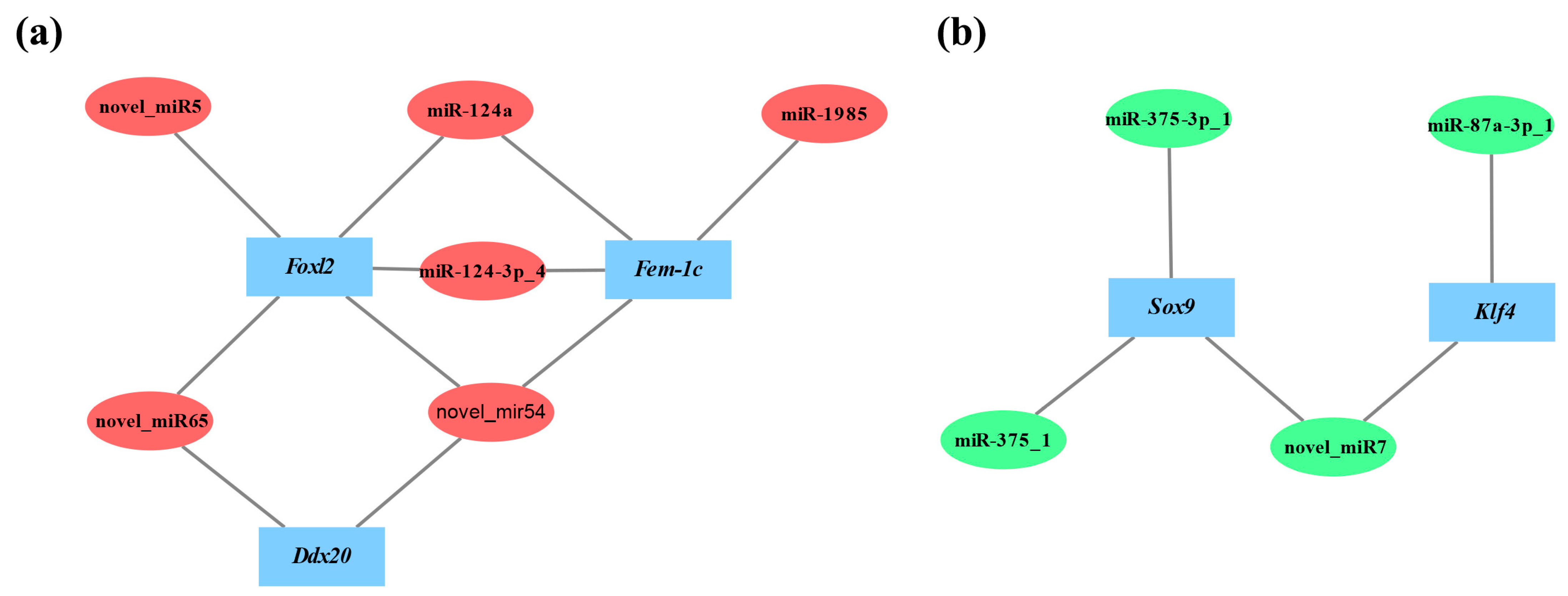
3.6. Validation of the Interaction between DEMs and Target Genes by Dual-Luciferase Reporter Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eggers, S.; Sinclair, A. Mammalian sex determination-insights from humans and mice. Chromosome Res. 2012, 20, 215–238. [Google Scholar] [CrossRef] [Green Version]
- Robert, H.D.; Yoshitaka, N. Sex determination and sex differentiation in fish: An overview of genetic, physiological, and environmental influences. Aquaculture 2002, 208, 191–364. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef]
- Wienholds, E.; Kloosterman, W.P.; Miska, E.; Alvarez-Saavedra, E.; Berezikov, E.; de-Bruijn, E.; Horvitz, H.R.; Kauppinen, S.; Plasterk, R.H. MicroRNA expression in zebrafish embryonic development. Science 2005, 309, 310–311. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, L.; Song, L.; Liu, R.; Zhang, H.; Huang, M.; Chen, H. The identification and characteristics of immune-related microRNAs in haemocytes of oyster Crassostrea gigas. PLoS ONE 2014, 9, e88397. [Google Scholar] [CrossRef]
- Real, F.M.; Sekido, R.; Lupiáñez, D.G.; Lovell-Badge, R.; Jiménez, R.; Burgos, M. A microRNA (mmu-miR-124) prevents Sox9 expression in developing mouse ovarian cells. Biol. Reprod. 2013, 89, 1–11. [Google Scholar] [CrossRef]
- Peng, W.; Yu, S.; Handler, A.M.; Tu, Z.; Saccone, G.; Xi, Z.; Zhang, H. miRNA-1-3p is an early embryonic male sex-determining factor in the Oriental fruit fly Bactrocera dorsalis. Nat. Commun. 2020, 11, 932. [Google Scholar] [CrossRef] [Green Version]
- McJunkin, K.; Ambros, V. A microRNA family exerts maternal control on sex determination in C. elegans. Genes Dev. 2017, 31, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Yue, C.Y.; Li, Q.; Yu, H. Integrated analysis of miRNA and mRNA expression profiles identifies potential regulatory interactions during sexual development of Pacific oyster Crassostrea gigas. Aquaculture 2022, 546, 737294. [Google Scholar] [CrossRef]
- Wei, P.; He, P.; Zhang, X.; Li, W.; Zhang, L.; Guan, J.; Chen, X.; Lin, Y.; Zhuo, X.; Li, Q.; et al. Identification and characterization of microRNAs in the gonads of Crassostrea hongkongensis using high-throughput sequencing. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 31, 100606. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Duan, S.H.; Wang, G.L.; Li, J.L. Integrated mRNA and miRNA expression profile analysis of female and male gonads in Hyriopsis cumingii. Sci. Rep. 2021, 11, 665. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, T.; Zhang, X.; Hu, S.; Xiang, J. Sequencing and analysis of four BAC clones containing innate immune genes from the Zhikong scallop (Chlamys farreri). Gene 2012, 502, 9–15. [Google Scholar] [CrossRef]
- Liu, X.L.; Li, Y.; Liu, J.G.; Cui, L.B.; Zhang, Z.F. Gonadogenesis in scallop Chlamys farreri and Cf-foxl2 expression pattern during gonadal sex differentiation. Aquac. Res. 2014, 47, 1605–1611. [Google Scholar] [CrossRef]
- Li, X.X. Molecular Identification of Early Gonadal Sex Differentiation Time in Chlamys farreri. Master’s Thesis, Ocean University of China, Qingdao, China, 2020. [Google Scholar]
- Li, R.J.; Zhang, L.L.; Li, W.R.; Zhang, Y.; Li, Y.P.; Zhang, M.M.; Zhao, L.; Hu, X.L.; Wang, S.; Bao, Z.M. FOXL2 and DMRT1L Are Yin and Yang Genes for Determining Timing of Sex Differentiation in the Bivalve Mollusk Patinopecten yessoensis. Front. Physiol. 2018, 9, 1166. [Google Scholar] [CrossRef]
- Li, W.R. Dynamics of FOXL2, DMRT1L and Sex Steroids on Gonadal Development in the Bay Scallops, Argopecten irradians. Master’s Thesis, Ocean University of China, Qingdao, China, 2019. [Google Scholar]
- Liu, X.L.; Zhang, Z.F.; Shao, M.Y.; Liu, J.G.; Muhammad, F. Sexually dimorphic expression of foxl2 during gametogenesis in scallop Chlamys farreri, conserved with vertebrates. Dev. Genes Evol. 2012, 222, 279–286. [Google Scholar] [CrossRef]
- Yang, D.D.; Zhang, Z.F.; Liang, S.S.; Yang, Q.K.; Wang, Y.R.; Qin, Z.K. A novel role of Krüppel-like factor 4 in Zhikong scallop Chlamys farreri during spermatogenesis. PLoS ONE 2017, 12, e0180351. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.L.; Zhang, X.H.; Li, J.; Liu, P. Identification and comparative profiling of ovarian and testicular microRNAs in the swimming crab Portunus trituberculatus. Gene 2018, 640, 6–13. [Google Scholar] [CrossRef]
- Li, S.Z.; Lin, G.M.; Fang, W.Y.; Gao, D.; Huang, J.; Xie, J.G.; Lu, J.G. Identification and Comparison of microRNAs in the Gonad of the Yellowfin Seabream (Acanthopagrus Latus). Int. J. Mol. Sci. 2020, 21, 5690. [Google Scholar] [CrossRef]
- Mi, X.; Wei, Z.L.; Zhou, Z.C.; Liu, X.L. Identification and profiling of sex-biased microRNAs from sea urchin Strongylocentrotus nudus gonad by Solexa deep sequencing. Comp. Biochem. Physiol. Part D Genom. Proteom. 2014, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.S.; Liu, M.; Zhou, C.F.; You, X.B.; Su, T.; Yang, Y.B.; Xu, D.Q. Integrated analysis of miRNA and mRNA expression profiles in testes of Duroc and Meishan boars. BMC Genom. 2020, 21, 686. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.Y.; Xiong, X.Y.; Liu, X.; He, X.Y.; Qiu, G.F. Identification and characterization of sex-biased and differentially expressed miRNAs in gonadal developments of the Chinese mitten crab, Eriocheir sinensis. Mol. Reprod. Dev. 2021, 88, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.W.; Liu, Q.; Jiang, J.M.; Shen, X.F.; Zhang, L.; Yuan, Z.; Wu, Y.M.; Liu, Y. Identification of sex differentiation-related microRNA and long non-coding RNA in Takifugu rubripes gonads. Sci. Rep. 2021, 11, 7459. [Google Scholar] [CrossRef]
- Wang, F.; Jia, Y.F.; Wang, P.; Yang, Q.W.; Du, Q.Y.; Chang, Z.J. Identification and profiling of Cyprinus carpio microRNAs during ovary differentiation by deep sequencing. BMC Genom. 2017, 18, 333. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.B.; Wang, G.Z.; Xie, Z.J.; Zhang, C.L.; Wang, J.M. Identification and characterization of microRNA in the dairy goat (Capra hircus) mammary gland by Solexa deep-sequencing technology. Mol. Biol. Rep. 2012, 39, 9361–9371. [Google Scholar] [CrossRef]
- Gonen, N.; Lovell-Badge, R. The regulation of Sox9 expression in the gonad. Curr. Top. Dev. Biol. 2019, 134, 223–252. [Google Scholar] [CrossRef]
- Jiang, T.; Hou, C.C.; She, Z.Y.; Yang, W.X. The SOX gene family: Function and regulation in testis determination and male fertility maintenance. Mol. Biol. Rep. 2013, 40, 2187–2194. [Google Scholar] [CrossRef]
- Zheng, J.B.; Chen, L.R.; Jia, Y.Y.; Chi, M.L.; Li, F.; Cheng, S.; Liu, S.L.; Liu, Y.N.; Gu, Z.M. Genomic structure, expression, and functional characterization of the Fem-1 gene family in the redclaw crayfish, Cherax quadricarinatus. Gen. Comp. Endocrinol. 2022, 316, 113961. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Wu, C.D.; Guo, P.F.; Wang, G.L.; Li, J.L. Molecular characterization and expression of the feminization-1c (fem-1c) in the freshwater mussel (Hyriopsis cumingii). Aquac. Fish. 2018, 3, 6–13. [Google Scholar] [CrossRef]
- Song, C.W.; Cui, Z.X.; Hui, M.; Liu, Y.; Li, Y.D. Molecular characterization and expression profile of three Fem-1 genes in Eriocheir sinensis provide a new insight into crab sex-determining mechanism. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2015, 189, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Mouillet, J.F.; Yan, X.M.; Ou, Q.L.; Jin, L.L.; Muglia, L.J.; Crawford, P.A.; Sadovsky, Y. DEAD-box protein-103 (DP103, Ddx20) is essential for early embryonic development and modulates ovarian morphology and function. Endocrinology 2008, 149, 2168–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Bloedite, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Ni, F.D.; Hao, S.L.; Yang, W.X. Multiple signaling pathways in Sertoli cells: Recent findings in spermatogenesis. Cell Death Dis. 2019, 10, 541. [Google Scholar] [CrossRef] [Green Version]
- Li, M.W.; Mruk, D.D.; Cheng, C.Y. Mitogen-activated protein kinases in male reproductive function. Trends Mol. Med. 2009, 15, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, F.Y.; Huang, J.; Zheng, L.P.; Zheng, Y.H. Research Progress on Hippo Signaling Pathway in Female Reproductive System. Chin. J. Cell Biol. 2014, 36, 1689–1694. [Google Scholar] [CrossRef]
- Yu, J.Z.; John, P.; Huang, Y.C.; Deng, W.M. The hippo pathway promotes Notch signaling in regulation of cell differentiation, proliferation, and oocyte polarity. PLoS ONE 2008, 3, e1761. [Google Scholar] [CrossRef] [Green Version]
- Cocquet, J.; De-Baere, E.; Gareil, M.; Pannetier, M.; Xia, X.; Fellous, M.; Veitia, R.A. Structure, evolution and expression of the FOXL2 transcription unit. Cytogenet. Genome Res. 2003, 101, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Bertho, S.; Herpin, A.; Branthonne, A.; Jouanno, E.; Yano, A.; Nicol, B.; Muller, T.; Pannetier, M.; Pailhoux, E.; Miwa, M.; et al. The unusual rainbow trout sex determination gene hijacked the canonical vertebrate gonadal differentiation pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 12781–12786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.B.; Li, M.R.; Ma, H.; Liu, X.Y.; Shi, H.J.; Li, M.H.; Wang, D.S. Mutation of Foxl2 or Cyp19a1a results in female to male sex reversal in XX Nile Tilapia. Endocrinology 2017, 158, 2634–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulanger, L.; Pannetier, M.; Gall, L.; Allais-Bonnet, A.; Elzaiat, M.; Le Bourhis, D.; Daniel, N.; Richard, C.; Cotinot, C.; Ghyselinck, N.; et al. FOXL2 is a Female Sex-determining gene in the goat. Curr. Biol. 2014, 24, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Xu, F.; Guo, X. Genomic analysis of the Pacific oyster (Crassostrea gigas) reveals possible conservation of vertebrate sex determination in a mollusc. G3 Genes Genom. Genet. 2014, 4, 2207–2217. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, J.; Liu, Y.; Zhang, Z.; Duan, S. MicroRNA-937 inhibits cell proliferation and metastasis in gastric cancer cells by downregulating FOXL2. Cancer Biomark. 2017, 21, 105–116. [Google Scholar] [CrossRef]
- Sun, S.D.; Su, W.J. Effect of miR-133b targeted regulation of FOXL2 on migration and proliferation of COV434 cells. J. Tissue Eng. Reconstr. Surg. 2021, 17, 205–211. [Google Scholar] [CrossRef]
- Behr, R.; Deller, C.; Godmann, M.; Müller, T.; Bergmann, M.; Ivell, R.; Steger, K. Kruppel-like factor 4 expression in normal and pathological human testes. Mol. Hum. Reprod. 2007, 13, 815–820. [Google Scholar] [CrossRef] [Green Version]
- Godmann, M.; Katz, J.P.; Guillou, F.; Simoni, M.; Kaestner, K.H.; Behr, R. Krüppel-like factor 4 is involved in functional differentiation of testicular Sertoli cells. Dev. Biol. 2008, 315, 552–566. [Google Scholar] [CrossRef] [Green Version]
- Yucel, P.A.; Ayva, E.S.; Gurdal, H.; Ozdemir, B.H.; Gur, D.B. MiR-25 and KLF4 relationship has early prognostic significance in the development of cervical cancer. Pathol. Res. Pract. 2021, 222, 153435. [Google Scholar] [CrossRef]
- Dong, X.; Wang, F.; Xue, Y.; Lin, Z.; Song, W.; Yang, N.; Li, Q. MicroRNA-9-5p downregulates Klf4 and influences the progression of hepatocellular carcinoma via the AKT signaling pathway. Int. J. Mol. Med. 2019, 43, 1417–1429. [Google Scholar] [CrossRef]
- Marín, R.M.; Sulc, M.; Vanícek, J. Searching the coding region for microRNA targets. RNA 2013, 19, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Zhang, X.R.; Cai, Z.Q.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.G.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Low Quality | Invalid Adapter | Poly N Reads | Short Valid Length | Clean Reads | Mapped Reads | Mapped Percentage | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| F1 | 30,119,160 | 102,088 | 279,083 | 921 | 394,152 | 29,342,916 | 23,187,887 | 79.02 | 99.0 | 96.8 |
| F2 | 29,055,638 | 95,864 | 969,435 | 5444 | 1,406,610 | 26,578,285 | 20,211,226 | 76.04 | 98.9 | 96.8 |
| F3 | 30,063,513 | 123,937 | 872,204 | 1424 | 1,350,206 | 27,715,742 | 21,220,447 | 76.56 | 98.7 | 96.2 |
| M1 | 29,785,617 | 105,133 | 356,592 | 1123 | 793,287 | 28,529,482 | 22,027,953 | 77.21 | 98.9 | 96.5 |
| M2 | 29,832,065 | 98,316 | 411,700 | 1059 | 2,013,570 | 27,307,420 | 18,620,412 | 68.19 | 98.9 | 96.7 |
| M3 | 30,537,297 | 129,186 | 243,137 | 975 | 139,038 | 30,024,961 | 22,272,516 | 74.18 | 98.8 | 96.4 |
| Species Name | miR-124a | miR-124-3p_4 | miR-1985 | miR-87a-3p_1 | miR-375_1 | miR-375-3p_1 |
|---|---|---|---|---|---|---|
| C. farreri | T | T | T | O | O | O |
| C. gigas | - | - | T | - | - | - |
| C. hongkongensis | O | - | - | - | - | - |
| H. cumingii | - | - | - | T | O | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Lin, S.; Fan, S.; Huang, X.; Zhang, Z.; Qin, Z. Identification and Characterization of MicroRNAs Involving in Initial Sex Differentiation of Chlamys farreri Gonads. Biology 2022, 11, 456. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11030456
Li X, Lin S, Fan S, Huang X, Zhang Z, Qin Z. Identification and Characterization of MicroRNAs Involving in Initial Sex Differentiation of Chlamys farreri Gonads. Biology. 2022; 11(3):456. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11030456
Chicago/Turabian StyleLi, Xixi, Siyu Lin, Shutong Fan, Xiaoting Huang, Zhifeng Zhang, and Zhenkui Qin. 2022. "Identification and Characterization of MicroRNAs Involving in Initial Sex Differentiation of Chlamys farreri Gonads" Biology 11, no. 3: 456. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11030456