
Innovative Hybrid-Alignment Annotation Method for Bioinformatics Identification and Functional Verification of a Novel Nitric Oxide Synthase in Trichomonas vaginalis

 , , , , , , , , , and
, , , , , , , , , and
Abstract
:Simple Summary
Abstract
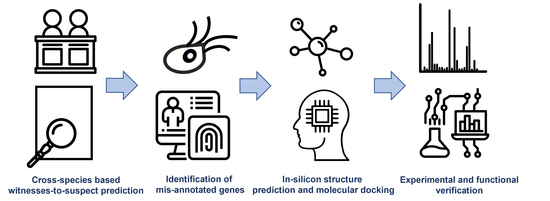
1. Introduction
2. Materials and Methods
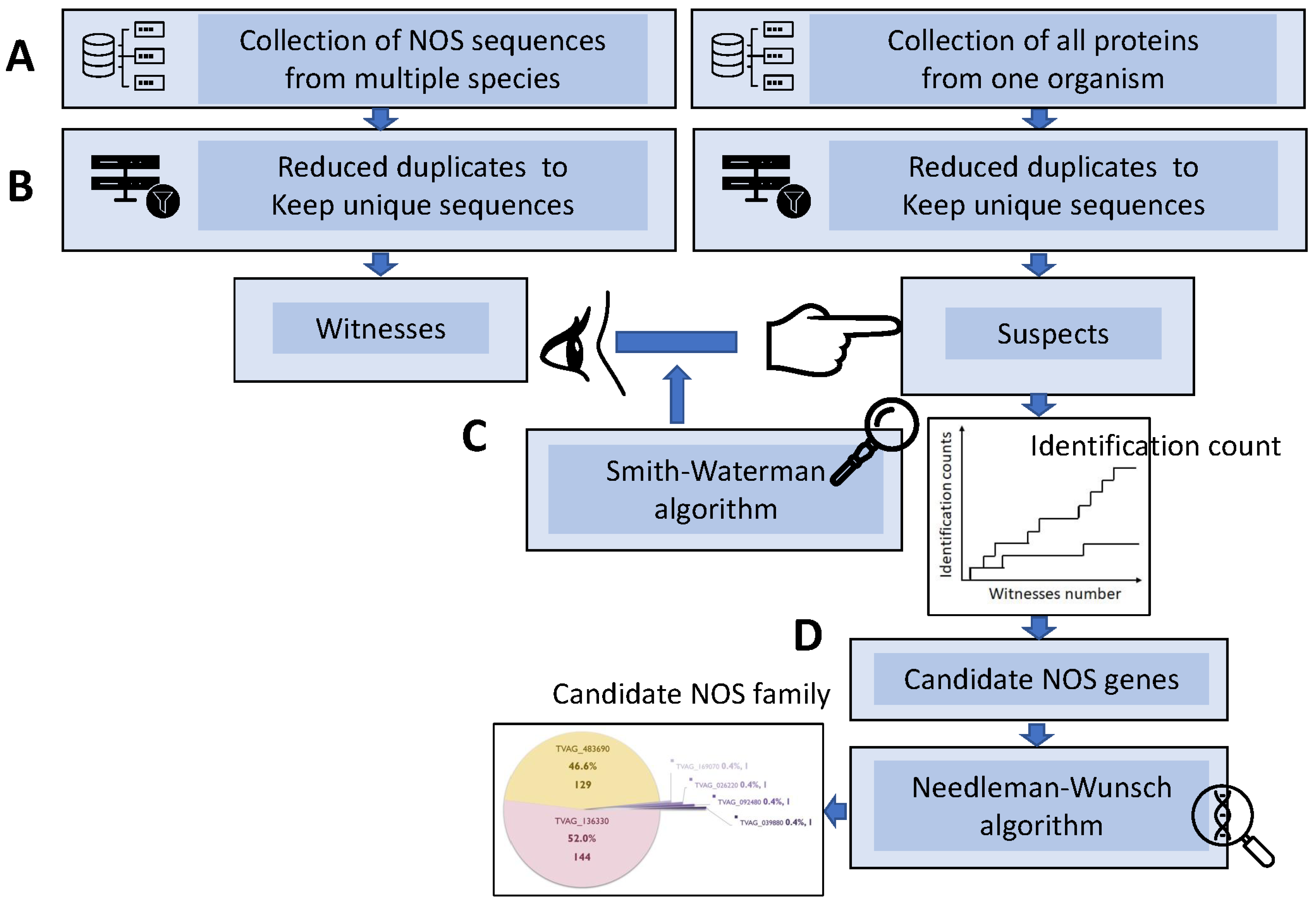
2.1. Sequence Retrieval, Data Reduction, Witness-vs.-Suspect Search, and Hybrid-Alignment Annotation Method
2.2. Structure Prediction and Models of TV
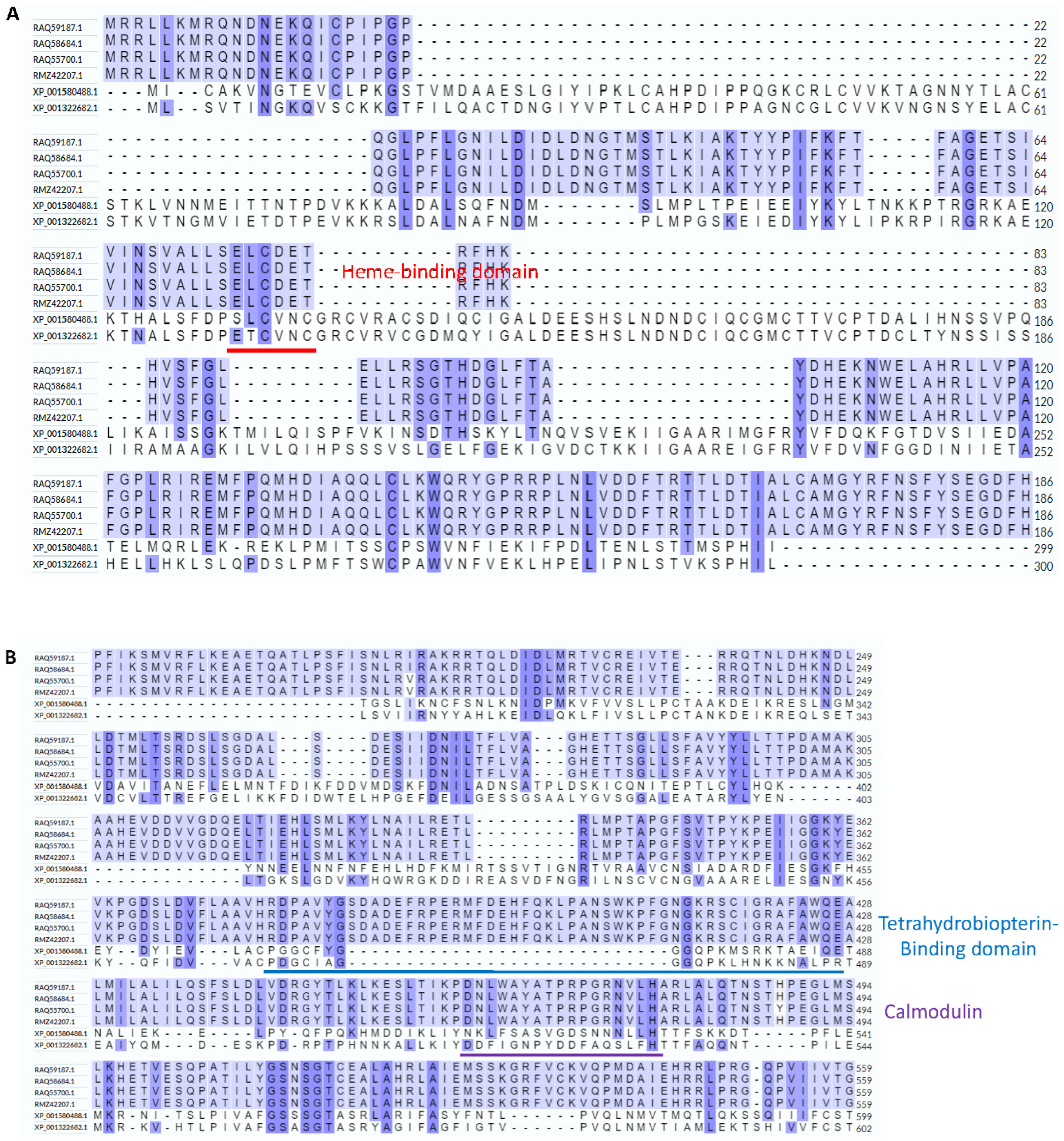
2.3. Predicting Domains Using a Bioinformatics Tool and Manually
2.4. Predicting Calmodulin Binding Sites Using the Calmodulin Target Database and HDOCK Server
2.5. Predicting Cofactor Binding Sites Using AutoDock FR Software
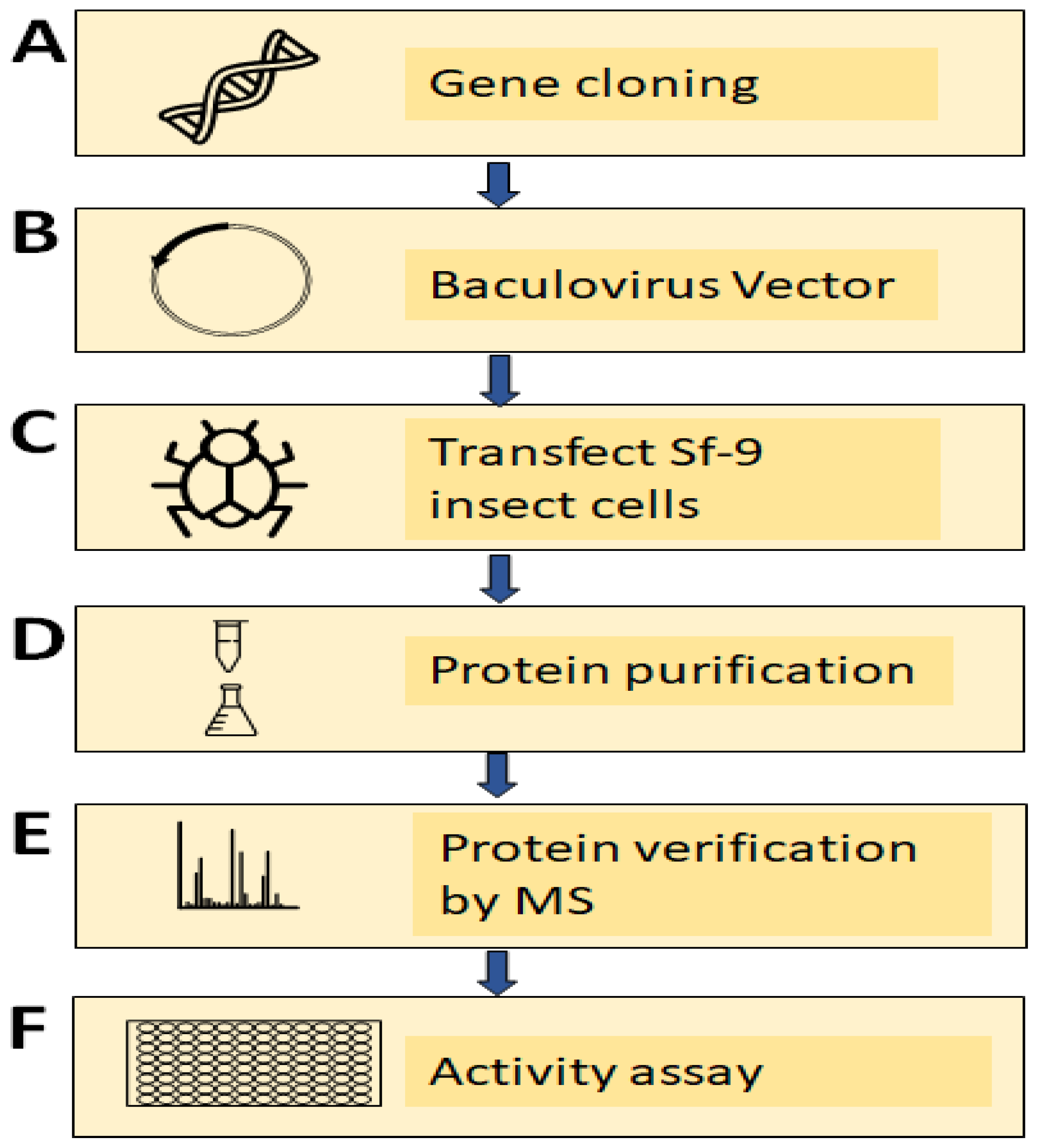
2.6. Cloning of TV Nitric Oxide Synthase (TVAG_136330) Using a PCR-Based Accurate Synthesis Method
2.7. Subcloning of Gene and Expression of Recombinant TV NOS Proteins
2.8. Verification of Recombinant TV NOS Proteins by Mass Spectrometry
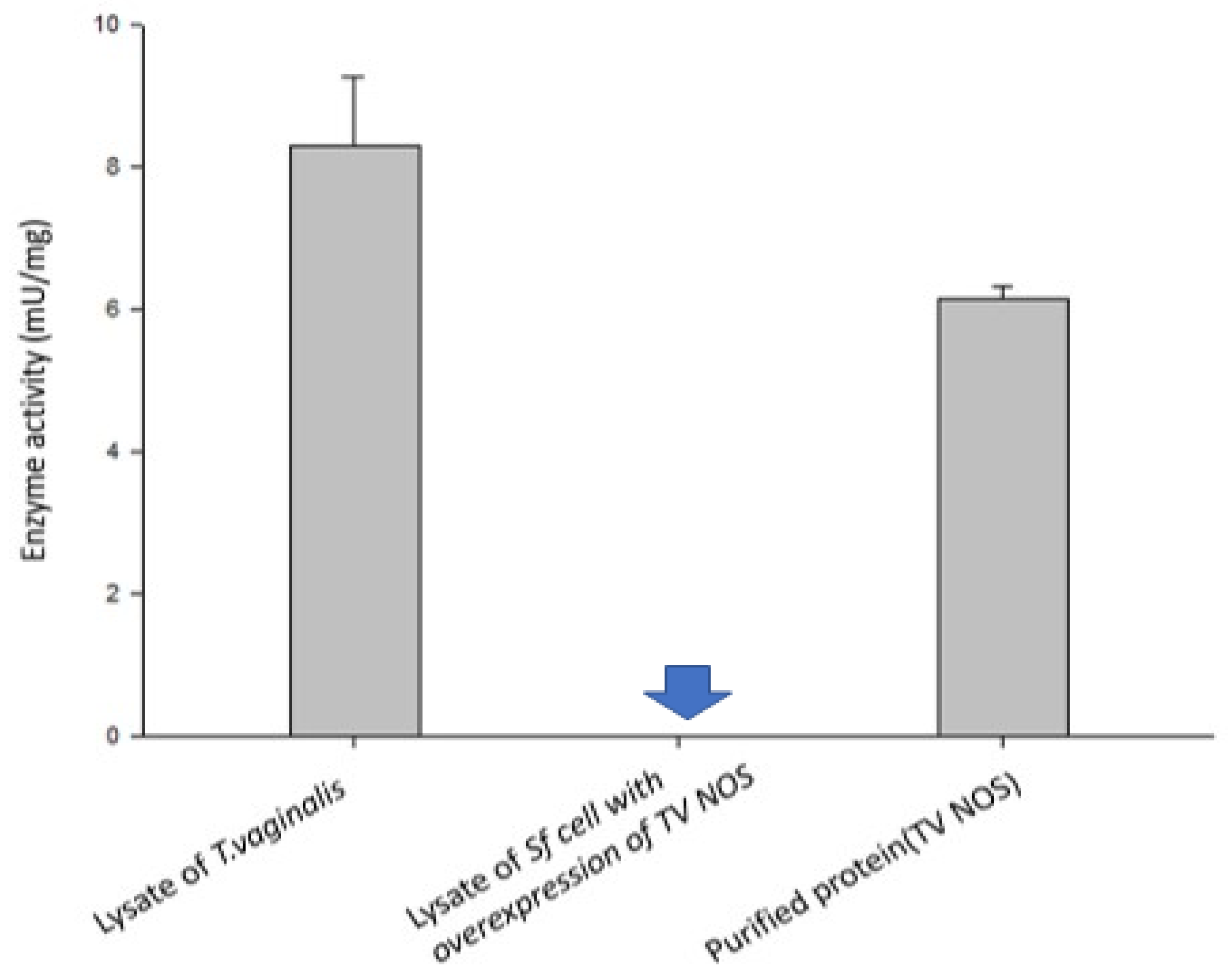
2.9. NOS Activity Assay
3. Results
3.1. Hybrid Annotation Method by Combining the Smith–Waterman Algorithm and the Needleman–Wunsch Algorithm
3.2. Protein Structure Prediction and Simulations of TV NOS and by RoseTTAFold and AlpaFold2
3.3. Molecular Docking of TV NOS with Cofactors and NOS Inhibitor (L-NMMA)
3.4. Prediction of the Protein–Protein Interaction of TV NOS with Calmodulin Protein
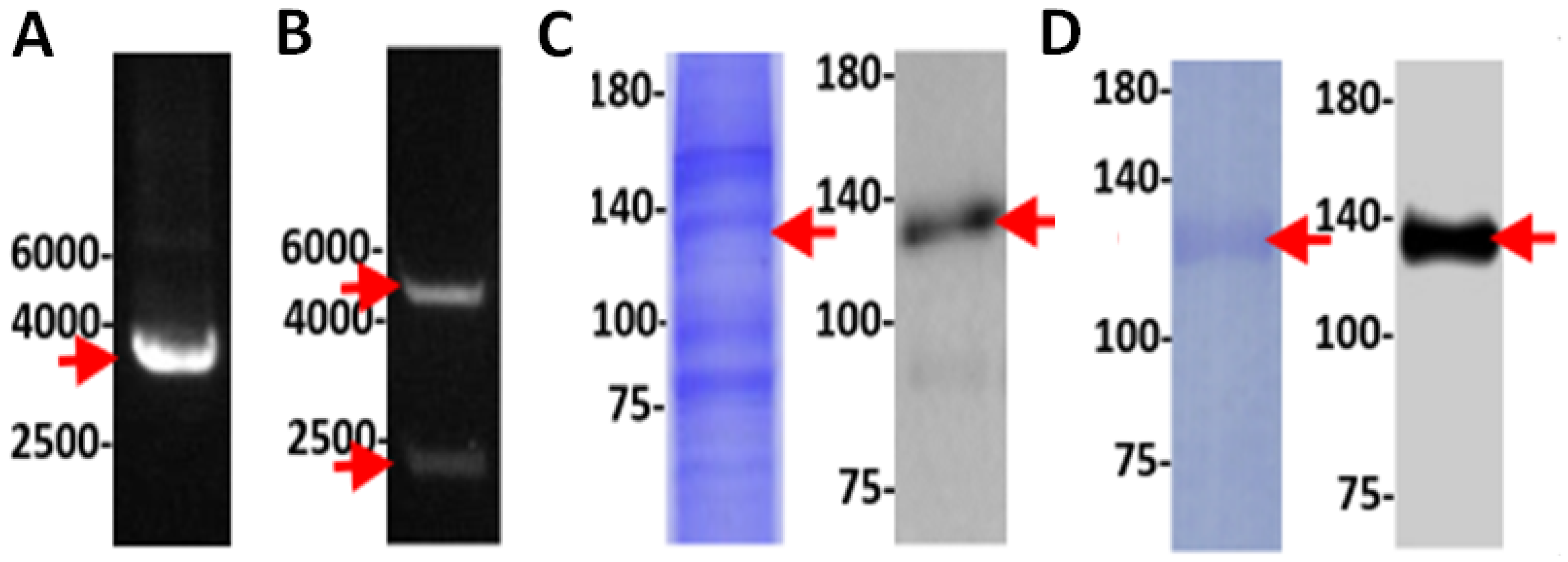
3.5. Experimental Validation of the NOS Gene in TV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TV | Trichomonas vaginalis or T. vaginalis |
| NOS | nitric oxide synthase |
| NGS | next-generation sequencing |
| NOSoxy | NOS oxygenase domain |
| NOSred | NOS reductase domain |
| CaM | calmodulin |
| bNOS | bacterial NOS |
| ADH | arginine dihydrolase |
| ADI | arginine deiminase |
| IPGs | identical protein groups |
| FMN | flavine mononucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| BH4 | tetrahydrobiopterin |
References
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Wetmore, K.M.; Waters, R.J.; Callaghan, M.; Ray, J.; Liu, H.; Kuehl, J.V.; Melnyk, R.A.; Lamson, J.S.; Suh, Y.; et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 2018, 557, 503–509. [Google Scholar] [CrossRef]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H.; et al. Functional annotation of a full-length mouse cDNA collection. Nature 2001, 409, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Promponas, V.J.; Iliopoulos, I.; Ouzounis, C.A. Annotation inconsistencies beyond sequence similarity-based function prediction–phylogeny and genome structure. Stand. Genom. Sci. 2015, 10, 108. [Google Scholar] [CrossRef]
- Harris, K.M. Determining the Role of Polyamine Metabolism in Two Human Pathogenicprotozoa: Tichomonas Vaginalis and Giardia Intestinalis; Cardiff University: Cardiff, UK, 2007. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Foresi, N.; Correa-Aragunde, N.; Parisi, G.; Caló, G.; Salerno, G.; Lamattina, L. Characterization of a nitric oxide synthase from the plant kingdom: No generation from the green alga Ostreococcus tauri is light irradiance and growth phase dependent. Plant Cell 2010, 22, 3816–3830. [Google Scholar] [CrossRef]
- Rafferty, S. Nitric oxide synthases of bacteria and other unicellular organisms. Open Nitric Oxide J. 2011, 3, 25–32. [Google Scholar] [CrossRef]
- Loshchinina, E.A.; Nikitina, V.E. Role of the NO synthase system in response to abiotic stress factors for basidiomycetes Lentinula edodes and Grifola frondosa. Microbiology 2016, 85, 165–171. [Google Scholar] [CrossRef]
- Nishimura, A.; Kawahara, N.; Takagi, H. The flavoprotein Tah18-dependent NO synthesis confers high-temperature stress tolerance on yeast cells. Biochem. Biophys. Res. Commun. 2013, 430, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Gorren, A.C.; Mayer, B. Nitric-oxide synthase: A cytochrome P450 family foster child. Biochim. Biophys. Acta 2007, 1770, 432–445. [Google Scholar] [CrossRef]
- Jeandroz, S.; Wipf, D.; Stuehr, D.J.; Lamattina, L.; Melkonian, M.; Tian, Z.; Zhu, Y.; Carpenter, E.J.; Wong, G.K.S.; Wendehenne, D. Occurrence, structure, and evolution of nitric oxide synthase–Like proteins in the plant kingdom. Sci. Signal. 2016, 9, re2. [Google Scholar] [CrossRef] [PubMed]
- Correa-Aragunde, N.; Foresi, N.; Lamattina, L. Structure diversity of nitric oxide synthases (NOS): The emergence of new forms in photosynthetic organisms. Front. Plant Sci. 2013, 4, 232. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Su, X.S.; Zhang, S.H. Structure of nitric oxide synthase and its catalytic mechanism. Chin. J. Org. Chem. 2005, 25, 342–345. [Google Scholar]
- Andreakis, N.; D’Aniello, S.; Albalat, R.; Patti, F.P.; Garcia-Fernandez, J.; Procaccini, G.; Sordino, P.; Palumbo, A. Evolution of the nitric oxide synthase family in metazoans. Mol. Biol. Evol. 2011, 28, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Golderer, G.; Werner, E.R.; Leitner, S.; Gröbner, P.; Werner-Felmayer, G. Nitric oxide synthase is induced in sporulation of Physarum polycephalum. Genes Dev. 2001, 15, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.K.; Li, H.; Jing, Q.; Kang, S.; Richo, J.; Silverman, R.B.; Poulos, T.L. Structural and biological studies on bacterial nitric oxide synthase inhibitors. Proc. Natl. Acad. Sci. USA 2013, 110, 18127–18131. [Google Scholar] [CrossRef]
- Surdel, M.C.; Dutter, B.F.; Sulikowski, G.A.; Skaar, E.P. Bacterial nitric oxide synthase is required for the Staphylococcus aureus response to heme stress. ACS Infect. Dis. 2016, 2, 572–578. [Google Scholar] [CrossRef]
- Carlton, J.M.; Hirt, R.P.; Silva, J.C.; Delcher, A.L.; Schatz, M.; Zhao, Q.; Wortman, J.R.; Bidwell, S.L.; Alsmark, U.C.M.; Besteiro, S. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 2007, 315, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Goldberg, B.; Biagini, G.A.; Lloyd, D. Trichomonas vaginalis and Giardia intestinalis produce nitric oxide and display NO-synthase activity. J. Eukaryot. Microbiol. 2006, 53 (Suppl. S1), S182–S183. [Google Scholar] [CrossRef]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.; Žárský, V.; Barlow, L.D.; Herman, E.K. A eukaryote without a mitochondrial organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.G.; McArthur, A.G.; Gillin, F.D.; Aley, S.B.; Adam, R.D.; Olsen, G.J.; Best, A.A.; Cande, W.Z.; Chen, F.; Cipriano, M.J.; et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 2007, 317, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Hirt, R.P.; Sherrard, J. Trichomonas vaginalis origins, molecular pathobiology and clinical considerations. Curr. Opin. Infect. Dis. 2015, 28, 72–79. [Google Scholar] [CrossRef]
- Gould, S.B.; Woehle, C.; Kusdian, G.; Landan, G.; Tachezy, J.; Zimorski, V.; Martin, W.F. Deep sequencing of Trichomonas vaginalis during the early infection of vaginal epithelial cells and amoeboid transition. Int. J. Parasitol. 2013, 43, 707–719. [Google Scholar] [CrossRef]
- Cheng, W.H.; Huang, K.Y.; Huang, P.J.; Hsu, J.H.; Fang, Y.K.; Chiu, C.H.; Tang, P. Nitric oxide maintains cell survival of Trichomonas vaginalis upon iron depletion. Parasit. Vectors 2015, 8, 393. [Google Scholar] [CrossRef]
- Franzen, O.; Jerlström-Hultqvist, J.; Castro, E.; Sherwood, E.; Ankarklev, J.; Reiner, D.S.; Palm, D.; Andersson, J.O.; Andersson, B.; Svärd, S.G. Draft genome sequencing of giardia intestinalis assemblage B isolate GS: Is human giardiasis caused by two different species? PLoS Pathog. 2009, 5, e1000560. [Google Scholar] [CrossRef]
- Jerlström-Hultqvist, J.; Franzén, O.; Ankarklev, J.; Xu, F.; Nohýnková, E.; Andersson, J.O.; Svärd, S.G.; Andersson, B. Genome analysis and comparative genomics of a Giardia intestinalis assemblage E isolate. BMC Genom. 2010, 11, 543. [Google Scholar] [CrossRef]
- Liu, H.L.; Chu, C.M. Genome Annotation for Nitric Oxide Synthase of Trichomonas vaginalis by Smith-Waterman Algorithm based on the NCBI Protein Database. Master’s Thesis, National Defense Medical Center, Taipei, Taiwan, 2012. [Google Scholar]
- Yang, Y.T.; Chu, C.M. Using Smith-Waterman Alignment to Annotate Nitric Oxide Synthase in Trichomonas vaginalis. Master’s Thesis, National Defense Medical Center, Taipei, Taiwan, 2017. [Google Scholar]
- Crane, B.R.; Sudhamsu, J.; Patel, B.A. Bacterial nitric oxide synthases. Annu. Rev. Biochem. 2010, 79, 445–470. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Carter, J.V.; Pan, J.; Rai, S.N.; Galandiuk, S. ROC-ing along: Evaluation and interpretation of receiver operating characteristic curves. Surgery 2016, 159, 1638–1645. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score= 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Wu, C.H.; Siva, V.S.; Song, Y.L. An evolutionarily ancient NO synthase (NOS) in shrimp. Fish Shellfish Immunol. 2013, 35, 1483–1500. [Google Scholar] [CrossRef]
- Foresi, N.; Correa-Aragunde, N.; Santolini, J.; Lamattina, L. Analysis of the expression and activity of nitric oxide synthase from marine photosynthetic microorganisms. Methods Mol. Biol. 2016, 1424, 149–162. [Google Scholar]
- Harel, A.; Bromberg, Y.; Falkowski, P.G.; Bhattacharya, D. Evolutionary history of redox metal-binding domains across the tree of life. Proc. Natl. Acad. Sci. USA 2014, 111, 7042–7047. [Google Scholar] [CrossRef]
- Sarkar, T.S.; Biswas, P.; Ghosh, S.K.; Ghosh, S. Nitric oxide production by necrotrophic pathogen Macrophomina phaseolina and the host plant in charcoal rot disease of jute: Complexity of the interplay between necrotroph–host plant interactions. PLoS ONE 2014, 9, e107348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Lawson, R.J.; Buddha, M.R.; Wei, C.C.; Crane, B.R.; Munro, A.W.; Stuehr, D.J. Bacterial flavodoxins support nitric oxide production by Bacillus subtilis nitric-oxide synthase. J. Biol. Chem. 2007, 282, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef]
- Markoš, A.; Miretsky, A.; Müller, M. A glyceraldehyde-3-phosphate dehydrogenase with eubacterial features in the amitochondriate eukaryote, Trichomonas vaginalis. J. Mol. Evol. 1993, 37, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Viscogliosi, E.; Philippe, H.; Baroin, A.; Perasso, R.; Brugerolle, G. Phylogeny of trichomonads based on partial sequences of large subunit rRNA and on cladistic analysis of morphological data. J. Eukaryot. Microbiol. 1993, 40, 411–421. [Google Scholar] [CrossRef]
- Messner, S.; Leitner, S.; Bommassar, C.; Golderer, G.; Gröbner, P.; Werner, E.R.; Werner-Felmayer, G. Physarum nitric oxide synthases: Genomic structures and enzymology of recombinant proteins. Biochem. J. 2009, 418, 691–700. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Gharahdaghi, F.; Weinberg, C.R.; Meagher, D.A.; Imai, B.S.; Mische, S.M. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: A method for the removal of silver ions to enhance sensitivity. Electrophoresis 1999, 20, 601–605. [Google Scholar] [CrossRef]
- Madda, R.; Chen, C.M.; Wang, J.Y.; Chen, C.F.; Chao, K.Y.; Yang, Y.M.; Wu, H.Y.; Chen, W.M.; Wu, P.K. Proteomic profiling and identification of significant markers from high-grade osteosarcoma after cryotherapy and irradiation. Sci. Rep. 2020, 10, 2105. [Google Scholar] [CrossRef]
- Chiang, C.H.; Wu, C.C.; Lee, L.Y.; Li, Y.C.; Liu, H.P.; Hsu, C.W.; Lu, Y.C.; Chang, J.T.; Cheng, A.J. Proteomics analysis reveals involvement of Krt17 in areca nut-induced oral carcinogenesis. J. Proteome Res. 2016, 15, 2981–2997. [Google Scholar] [CrossRef]
- Ke, C.H.; Wang, Y.S.; Chiang, H.C.; Wu, H.Y.; Liu, W.J.; Huang, C.C.; Huang, Y.C.; Lin, C.S. Xenograft cancer vaccines prepared from immunodeficient mice increase tumor antigen diversity and host T cell efficiency against colorectal cancers. Cancer Lett. 2022, 526, 66–75. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | RoseTTAFold (Confidence Score) | AlphaFold2 (pLDDT) | AlphaFold2 TM Score |
|---|---|---|---|
| TV NOS | 0.82 (all models) | 84.05 (model 1) | 0.6576 (model 3) |
| Protein | Docking Score | Ligand rmsd | Receptor–Ligand Interface Residue Pair |
|---|---|---|---|
| TV NOS (receptor) | −197.83 | 83.17 | 513–527 |
| Calmodulin (Ligand) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-C.; Shui, H.-A.; Huang, K.-Y.; Lin, W.-Z.; Chang, H.-Y.; Lee, H.-J.; Lin, Y.-C.; Huang, Y.-S.; Chen, G.-R.; Yang, Y.-T.; et al. Innovative Hybrid-Alignment Annotation Method for Bioinformatics Identification and Functional Verification of a Novel Nitric Oxide Synthase in Trichomonas vaginalis. Biology 2022, 11, 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11081210
Lin H-C, Shui H-A, Huang K-Y, Lin W-Z, Chang H-Y, Lee H-J, Lin Y-C, Huang Y-S, Chen G-R, Yang Y-T, et al. Innovative Hybrid-Alignment Annotation Method for Bioinformatics Identification and Functional Verification of a Novel Nitric Oxide Synthase in Trichomonas vaginalis. Biology. 2022; 11(8):1210. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11081210
Chicago/Turabian StyleLin, Hung-Che, Hao-Ai Shui, Kuo-Yang Huang, Wei-Zhi Lin, Hsin-Yi Chang, Hwei-Jen Lee, Ying-Chih Lin, Yuahn-Sieh Huang, Guan-Ru Chen, Ya-Ting Yang, and et al. 2022. "Innovative Hybrid-Alignment Annotation Method for Bioinformatics Identification and Functional Verification of a Novel Nitric Oxide Synthase in Trichomonas vaginalis" Biology 11, no. 8: 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11081210