Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics
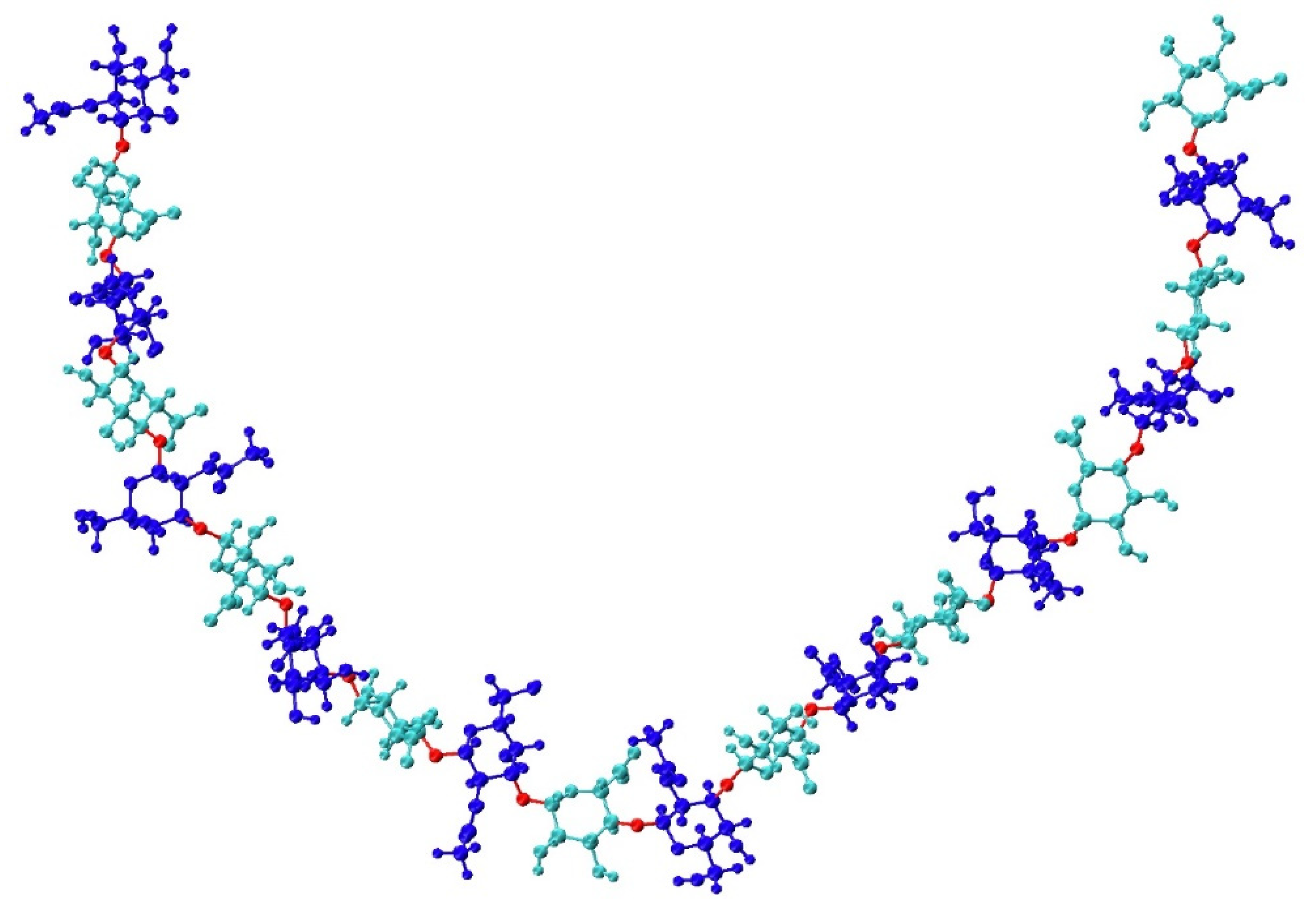
2.1.1. System Construction
2.1.2. Energy Minimization and Heating
2.1.3. Production Simulations
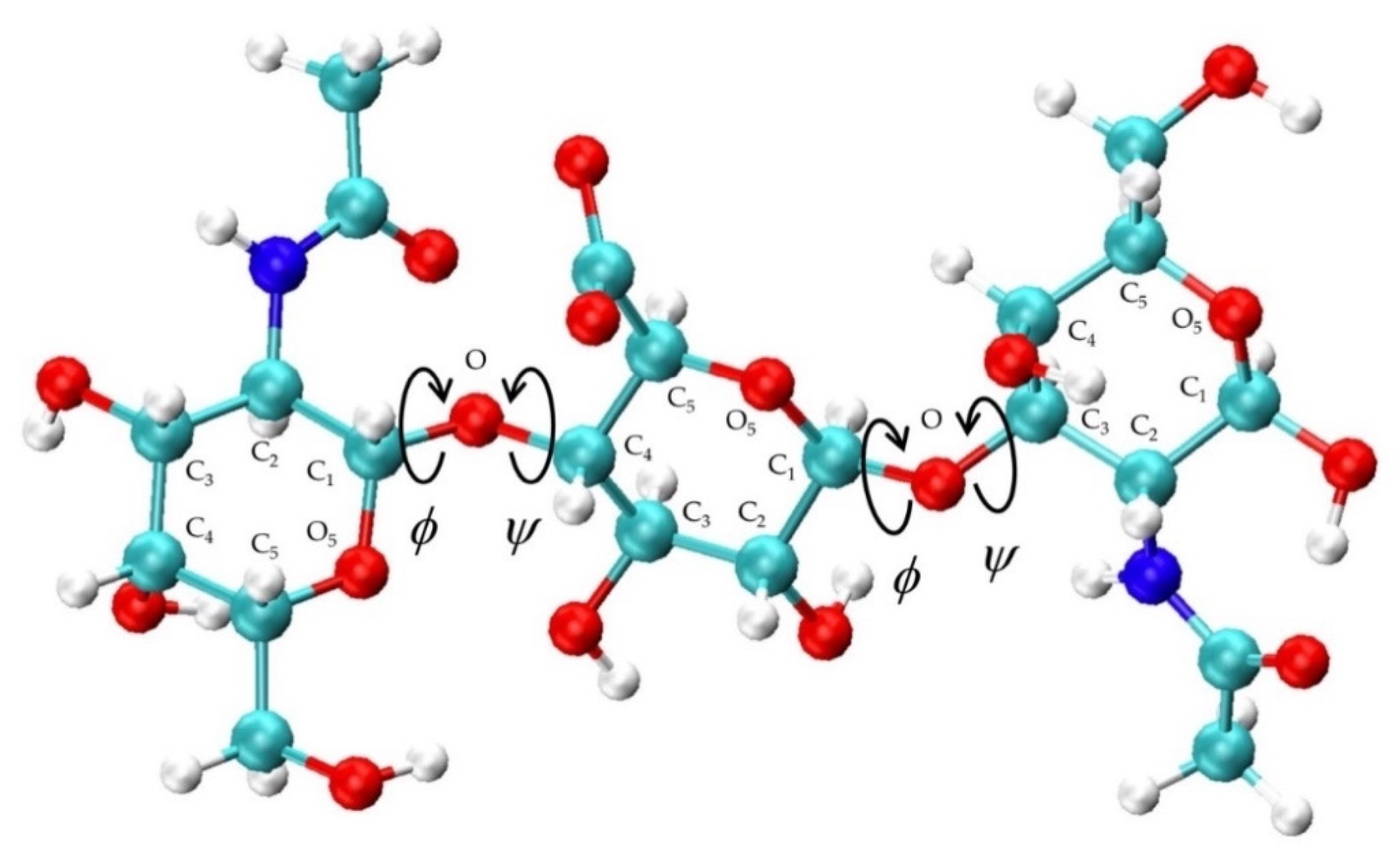
2.1.4. Conformational Analysis
2.2. Construction Algorithm to Generate GAG Conformational Ensembles
- In each constructed polymer conformation, each glycosidic linkage and monosaccharide ring is treated independently, and conformational parameters are randomly selected from the database containing the corresponding linkage or ring conformations from the 20-mer MD trajectories;
- Two CHARMM stream files are written, one to define the sequence and linkages in the polymer and another to perform the following procedure for each frame: (1) All internal geometry conformation values selected by the algorithm are assigned and used to construct atomic coordinates. (2) End-to-end distance (i.e., distance between C1 of the reducing end and C4 of the non-reducing end) and radius of gyration are calculated. (3) A 100-step steepest descent (SD) potential energy minimization followed by a 100-step conjugate gradient minimization, each with intramolecular restraints, is performed to relieve bonded strain and steric clashes. The Lennard–Jones potential () is calculated on an atom–atom pair () basis using an energy switching function, as implemented in CHARMM with ron = 7.5 Å and roff = 8.5 Å [69]. As there is no solvent and thus no solvent screening of electrostatic interactions, electrostatics are excluded from energy calculations to prevent the non-physical intramolecular association of charged and polar groups. All glycosidic linkage and endocyclic ring dihedral angles, along with a dihedral angle in each GlcA carboxylate group (C4-C5-C6-O61) and GalNAc N-acetyl group (C-N-C2-C3), are restrained to their starting values (i.e., those randomly selected from the database) during minimization so as not to change the conformations observed in simulation. Dihedral restraint energy () is calculated by comparing each restrained dihedral angle’s database value () to its value () in the current frame of minimization with a force constant () of 100.0 kcal/mol/radian/radian (Equation (1))
- To ensure conformational ensembles do not contain non-physical conformations, a bond potential energy () cutoff is applied. This cutoff is the sum of a polymer-length-specific cutoff and a constant independent of polymer length. The length-specific component of the cutoff is the bond potential energy after energy minimization, performed using the same restraints and minimization protocol used for each frame of the constructed ensemble (outlined above), of the polymer constructed in a fully-extended conformation (i.e., with the same glycosidic linkage ϕ and ψ angles as the starting conformation for MD simulations). The constant is added as a buffer to account for slight variations in the energies of other extended conformations. As linkage and ring conformations are treated independently and selected at random, it is possible to have a bond piercing another monosaccharide ring that may not be corrected by minimization. To estimate the ring-piercing bond strain energy for each exocyclic bond not participating in a glycosidic linkage, a system containing two non-bonded monosaccharides (i.e., GlcA and GalNAc, GlcA and GlcA, or GalNAc and GalNAc) was constructed such that an exocyclic bond of one monosaccharide pierces the ring of the other. To estimate the bond strain energy for each bond participating in a glycosidic linkage, a system containing one disaccharide unit (i.e., GlcAβ1-3GalNAc or GalNAcβ1-4GlcA) and a single monosaccharide (i.e., GlcA or GalNAc) was constructed such that a linkage bond in the disaccharide pierces the ring of the single monosaccharide. Systems containing interlocking rings (i.e., GlcA-GalNAc, GlcA-GlcA, and GalNAc-GalNAc) were also constructed to estimate the bond strain energy of the bonds piercing the opposite ring. The same energy minimization protocol used in the algorithm was performed on this conformation, as well as a conformation in which the non-bonded saccharide units are 20 Å apart, and the post-minimization lengths of the bond piercing the ring in the initial conformation were compared. The pierced bond length (), the non-pierced bond length (), and the equilibrium bond length () and corresponding force-field bond-stretching constant () from the CHARMM parameter file were used to estimate a lower bound on the energy () resulting from the bond distortion (Equation (2)).Of all conformations that still had a bond piercing a ring after minimization, the smallest = 132.3 kcal/mol. Of the conformations in which ring piercing was corrected during minimization, the maximum < 1 kcal/mol. Thus, a buffer of 100 kcal/mol is added to the post-minimization bond potential of the initial extended conformation for any given polymer length. If the post-minimization bond potential of a given frame is beyond this cutoff, the frame is excluded from the ensemble.
3. Results and Discussion
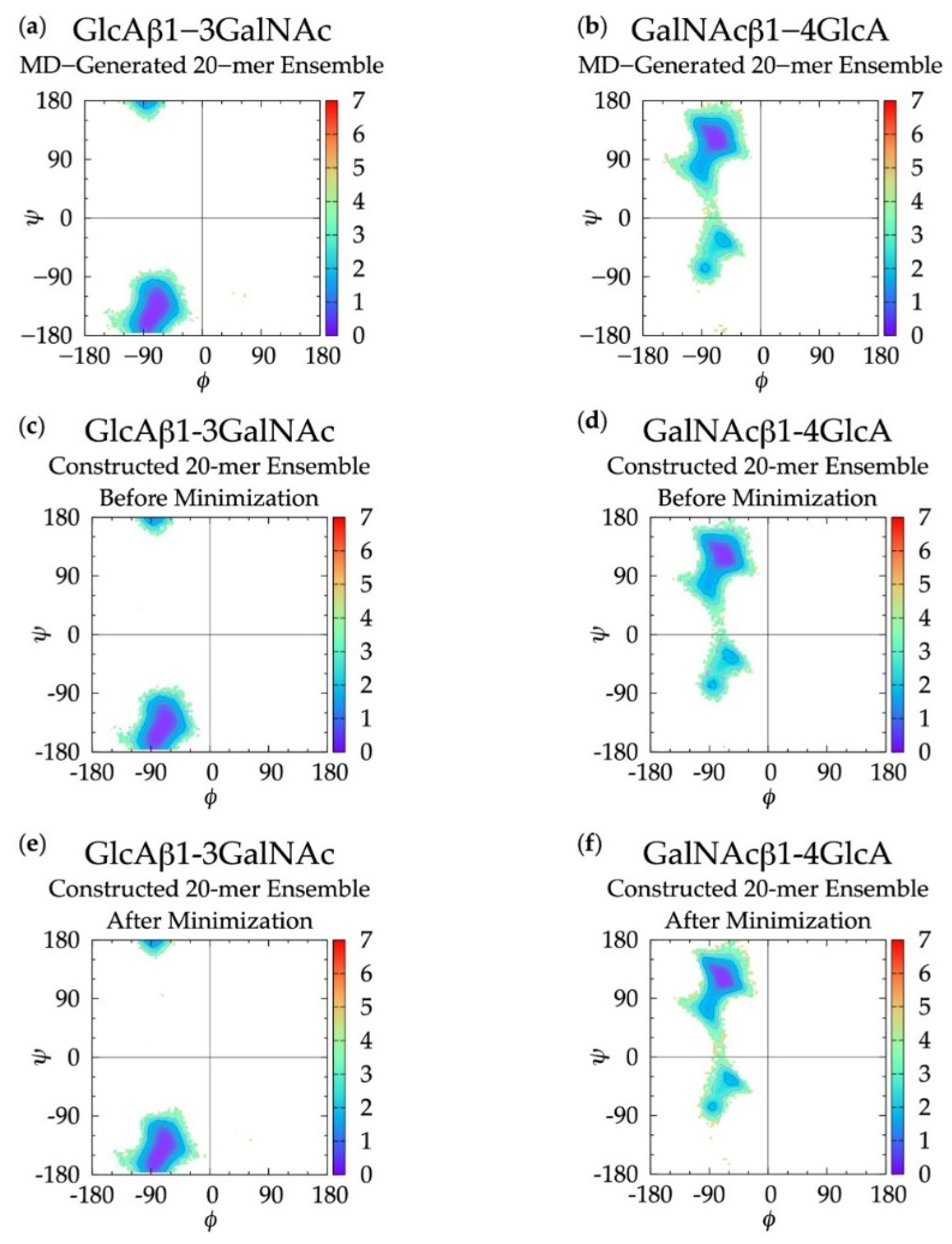
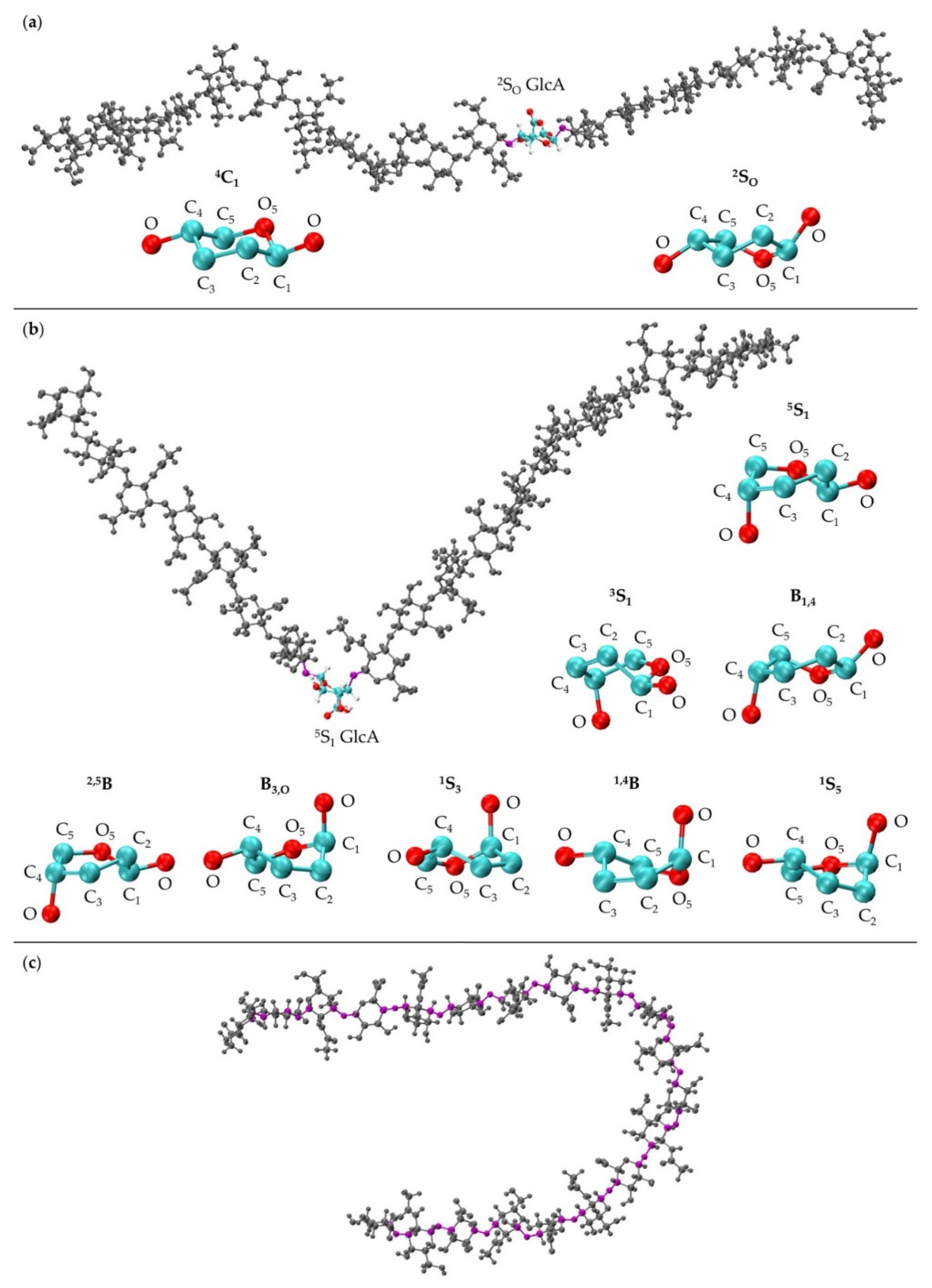
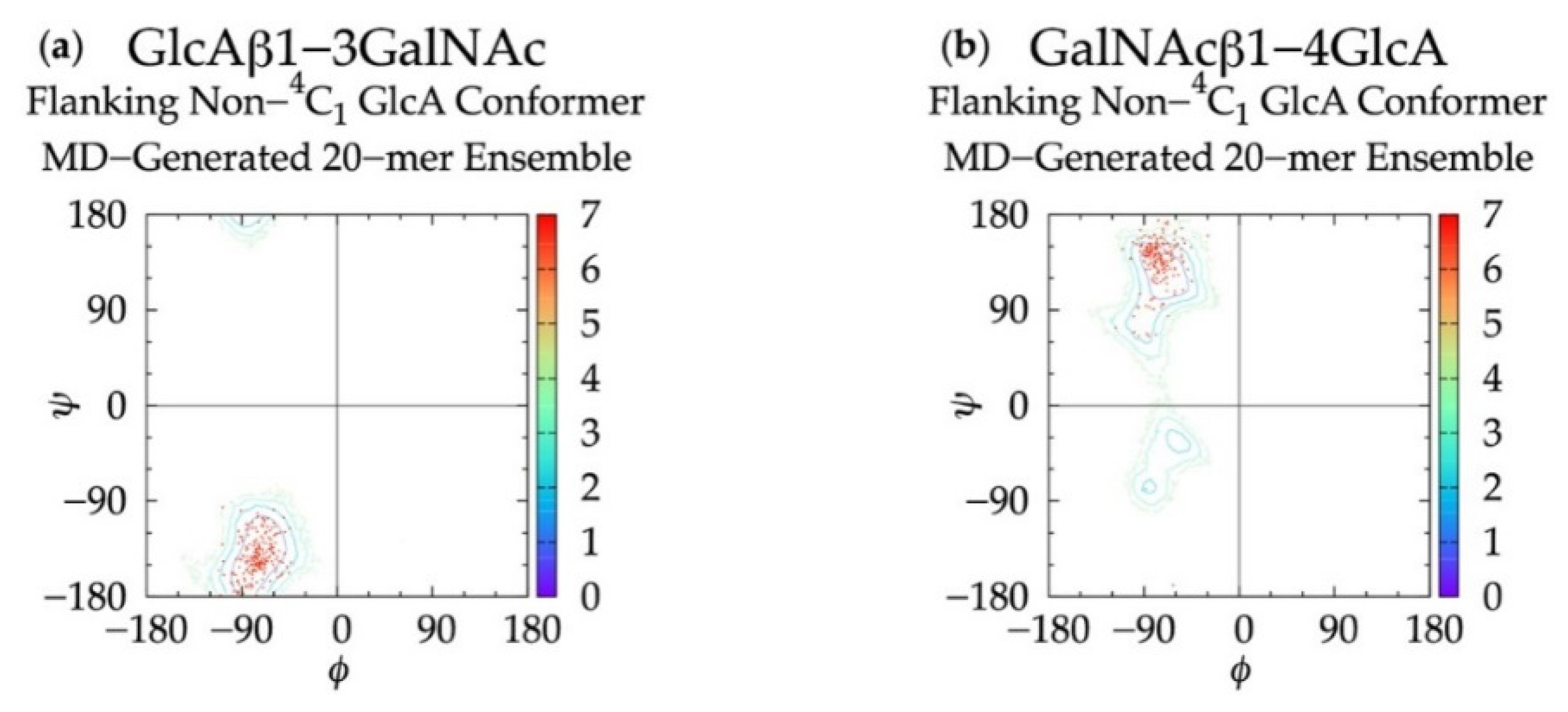
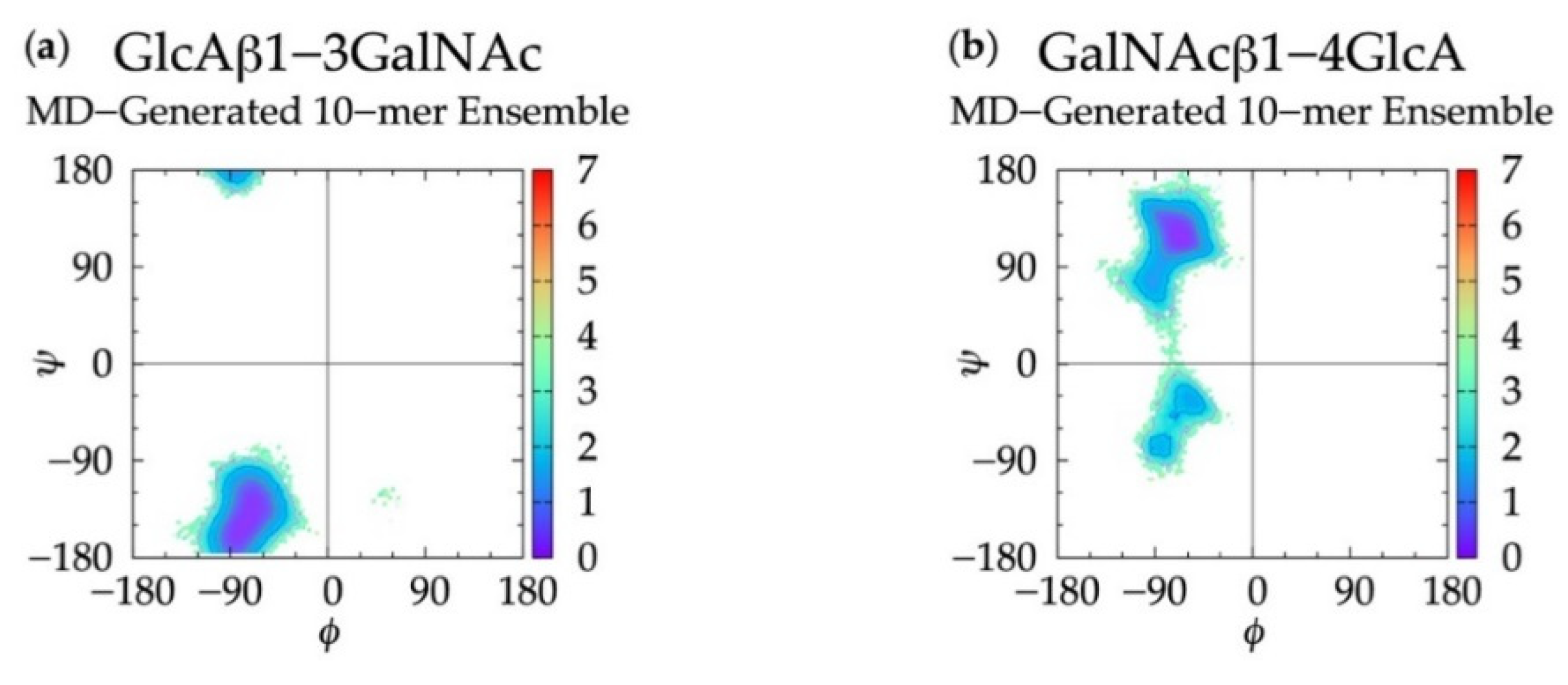
3.1. Glycosidic Linkage Geometries
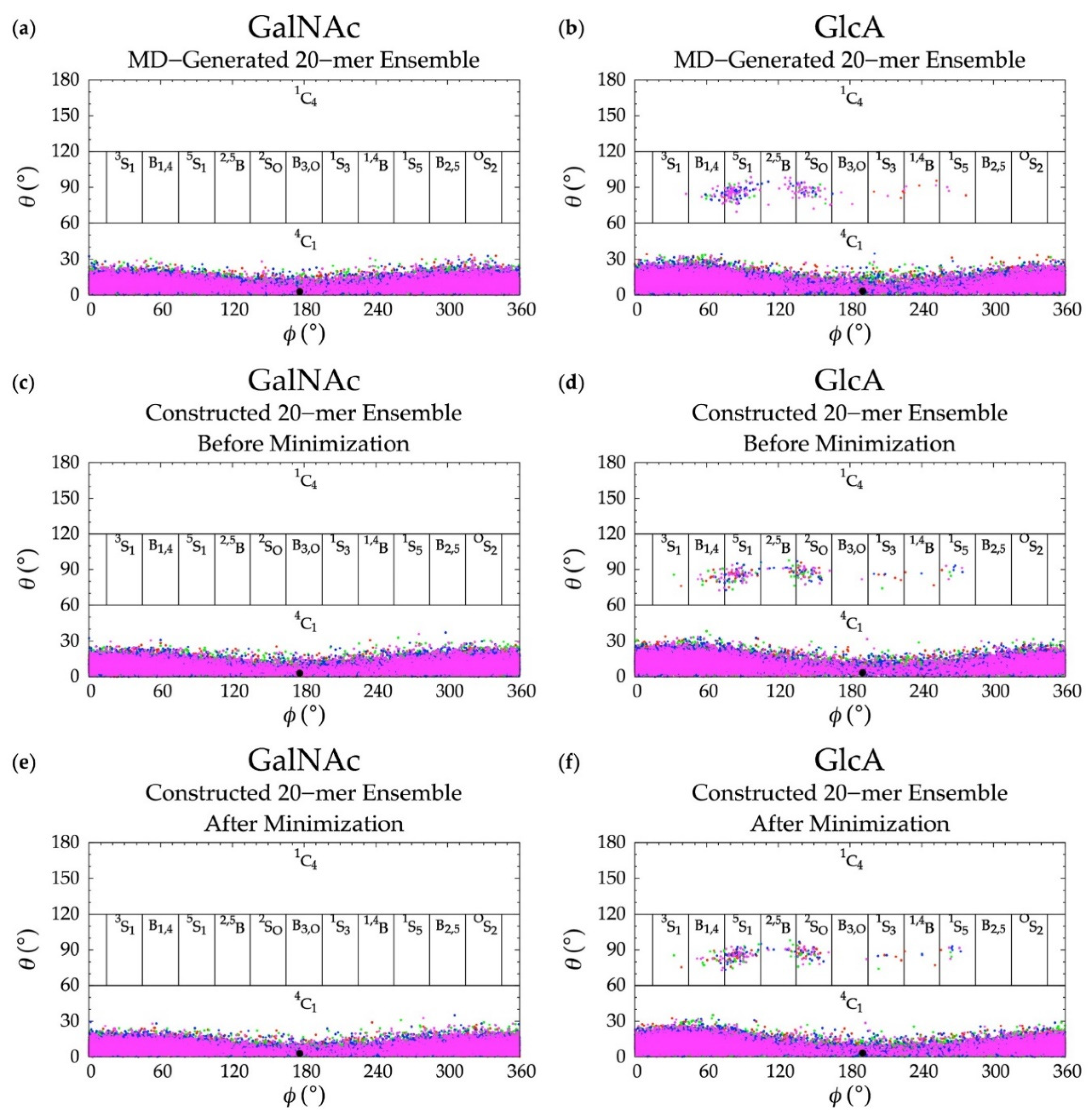
3.2. GlcA Ring Pucker Effects
3.3. Treating Glycosidic Linkage and Ring Pucker Geometries as Independent Variables
3.4. Handling Non-physical Constructed Conformations
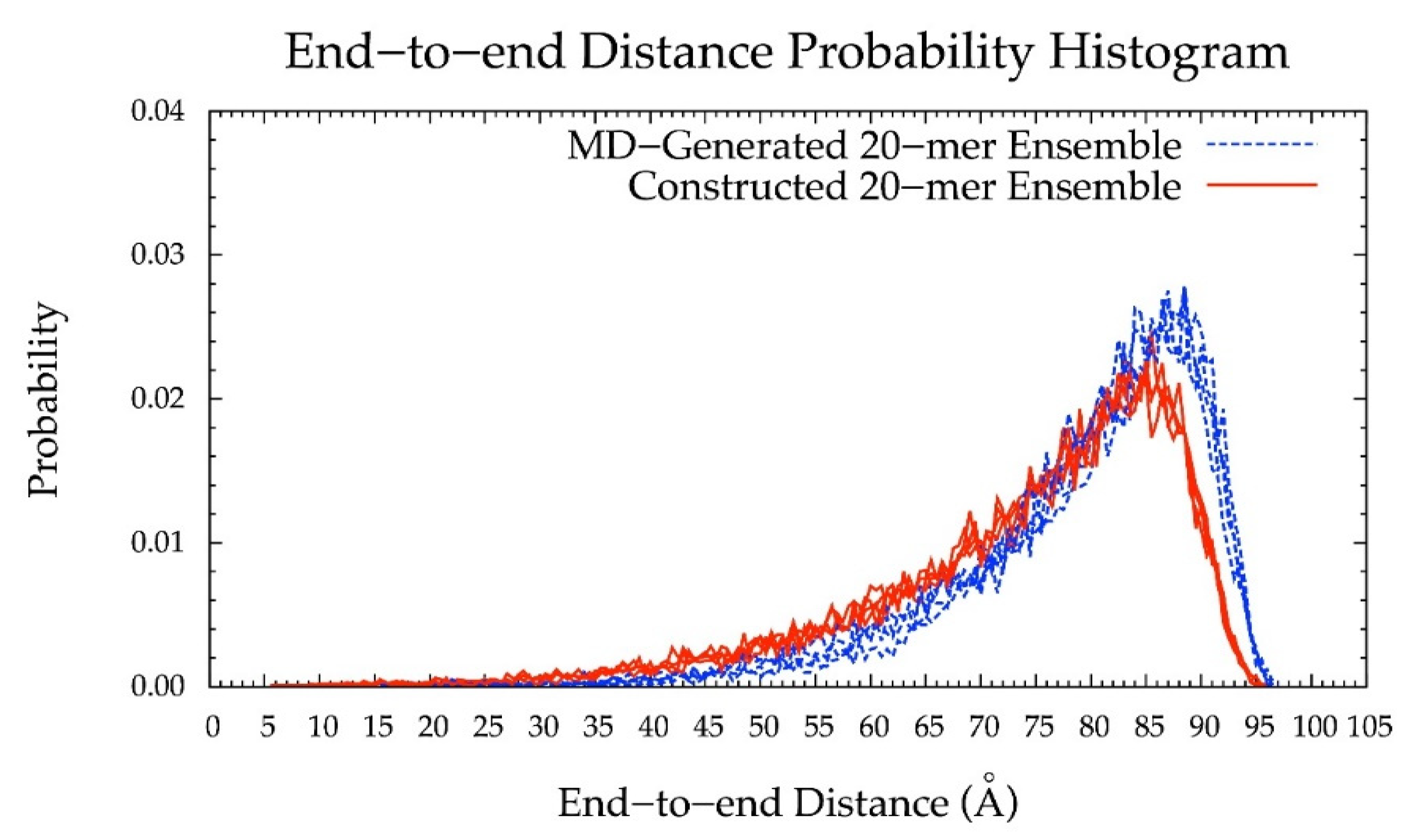
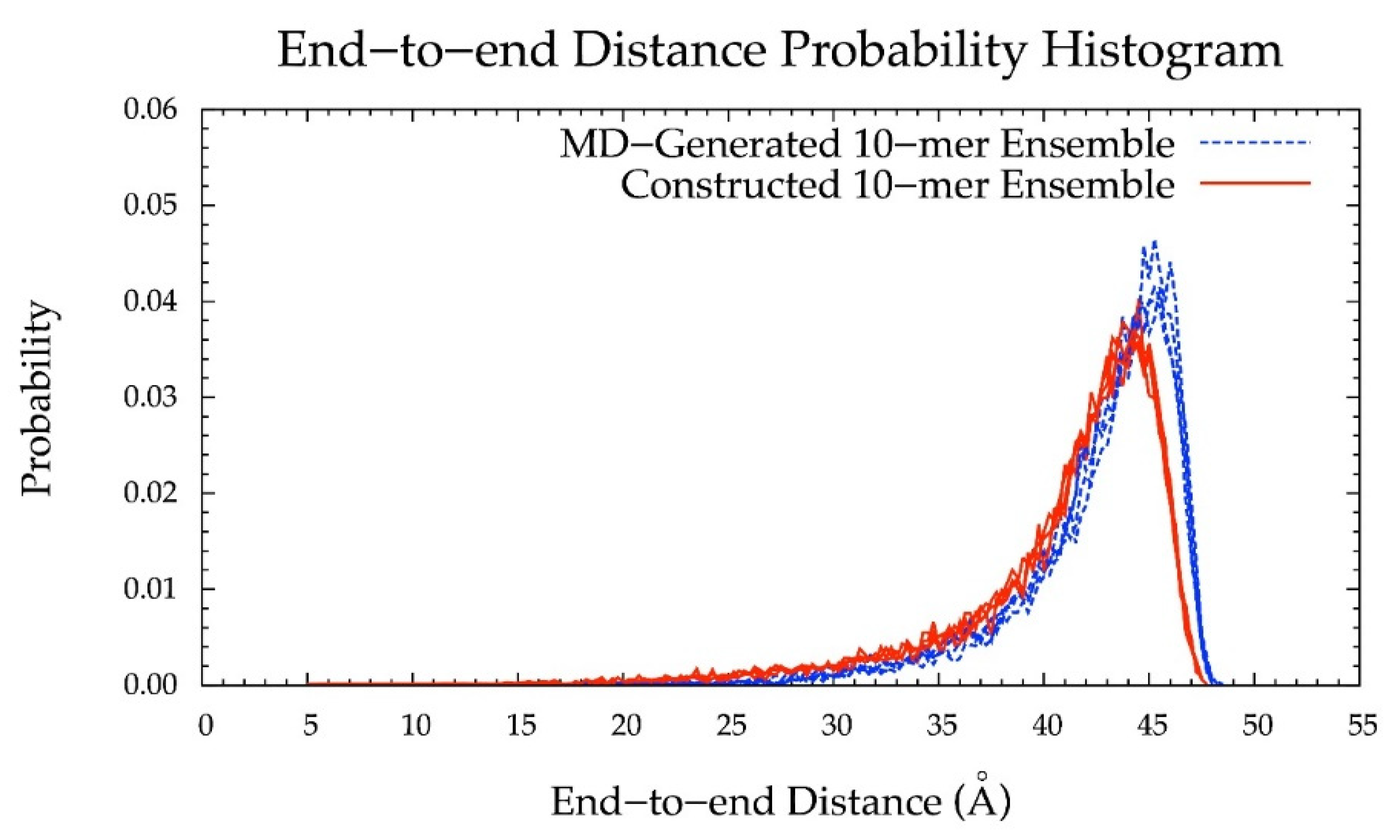
3.5. Internal Validation on 10-mers
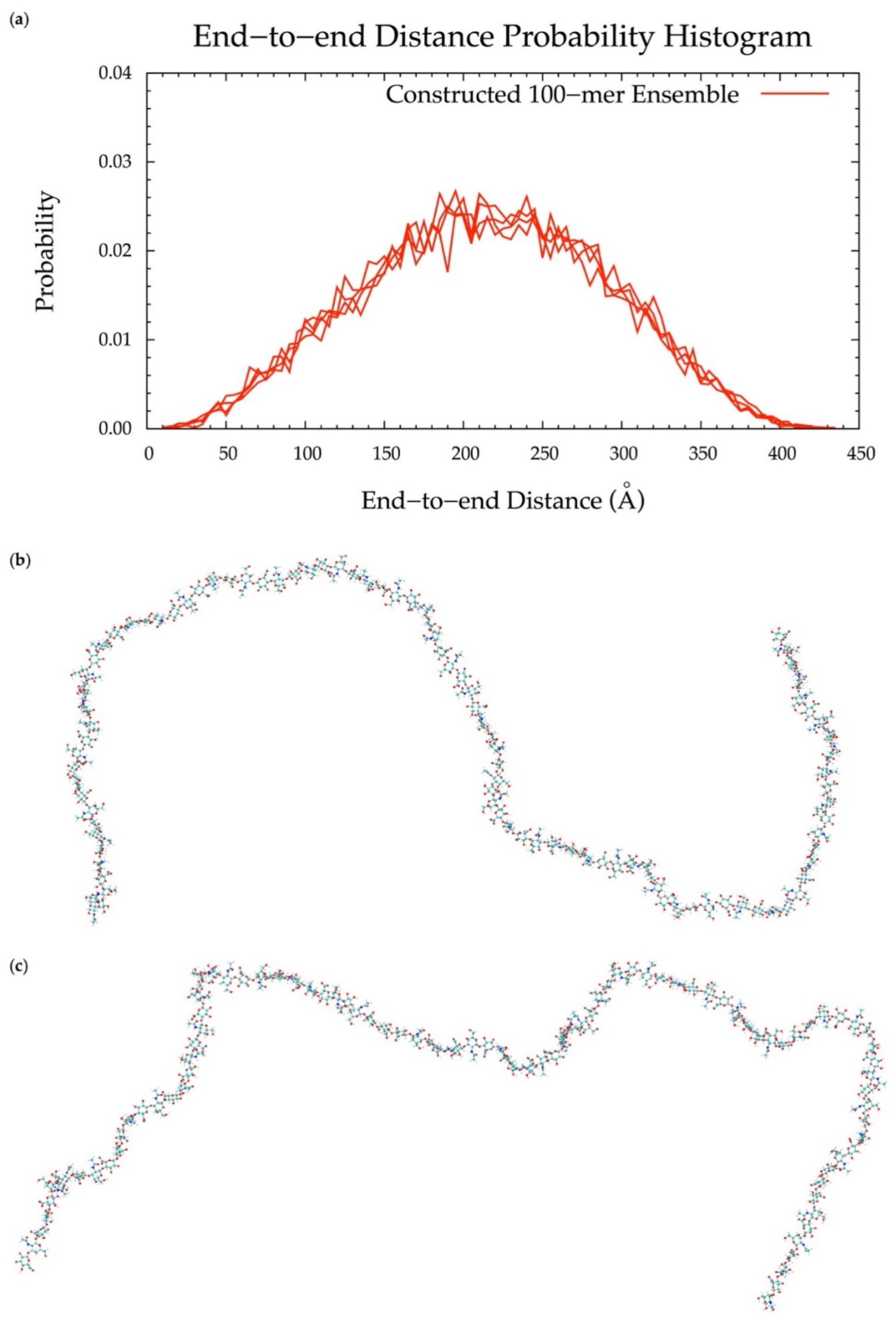
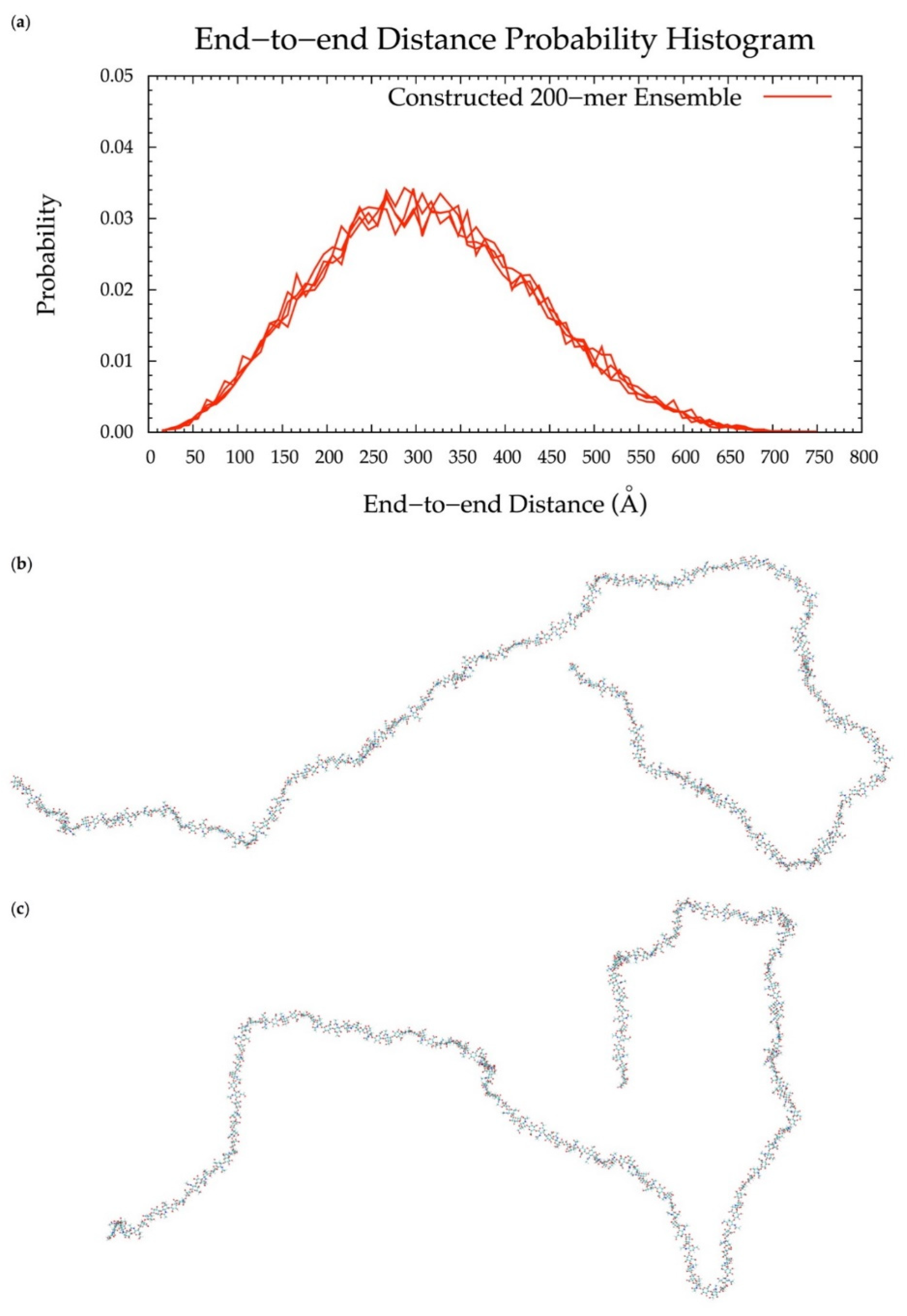
3.6. Application to Longer Chondroitin Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Djerbal, L.; Lortat-Jacob, H.; Kwok, J. Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj. J. 2017, 34, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, A.D.; Selleck, S.B. The elusive functions of proteoglycans: In vivo veritas. J. Cell Biol. 2000, 148, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsten-Williams, K.; Chu, C.L.; Fannon, M.; Buczek-Thomas, J.A.; Nugent, M.A. Control of growth factor networks by heparan sulfate proteoglycans. Ann. Biomed. Eng. 2008, 36, 2134–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhavanandan, V.P.; Davidson, E.A. Mucopolysaccharides associated with nuclei of cultured mammalian cells. Proc. Natl. Acad. Sci. USA 1975, 72, 2032–2036. [Google Scholar] [CrossRef] [Green Version]
- Fedarko, N.S.; Conrad, H.E. A unique heparan sulfate in the nuclei of hepatocytes: Structural changes with the growth state of the cells. J. Cell Biol. 1986, 102, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, M.; Fedarko, N.S.; Conrad, H.E. Transport of heparan sulfate into the nuclei of hepatocytes. J. Biol. Chem. 1986, 261, 13575–13580. [Google Scholar]
- Tumova, S.; Hatch, B.A.; Law, D.J.; Bame, K.J. Basic fibroblast growth factor does not prevent heparan sulphate proteoglycan catabolism in intact cells, but it alters the distribution of the glycosaminoglycan degradation products. Biochem. J. 1999, 337 Pt 3, 471–481. [Google Scholar] [CrossRef]
- Mizumoto, S.; Fongmoon, D.; Sugahara, K. Interaction of chondroitin sulfate and dermatan sulfate from various biological sources with heparin-binding growth factors and cytokines. Glycoconj. J. 2012, 30, 619–632. [Google Scholar] [CrossRef]
- Sugahara, K.; Mikami, T.; Uyama, T.; Mizuguchi, S.; Nomura, K.; Kitagawa, H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. [Google Scholar] [CrossRef]
- Sugahara, K.; Mikami, T. Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 2007, 17, 536–545. [Google Scholar] [CrossRef]
- Kawashima, H.; Atarashi, K.; Hirose, M.; Hirose, J.; Yamada, S.; Sugahara, K.; Miyasaka, M. Oversulfated chondroitin/dermatan sulfates containing GlcAβ1/IdoAα1-3GalNAc(4,6-O-disulfate) interact with L- and P-selectin and chemokines. J. Biol. Chem. 2002, 277, 12921–12930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjellén, L.; Lindahl, U. Specificity of glycosaminoglycan-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Prydz, K.; Pejler, G. Intracellular proteoglycans. Biochem. J. 2004, 379, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, A.; Couchman, J.R. Regulation of cytoskeletal organization by syndecan transmembrane proteoglycans. Matrix Biol. 2003, 22, 25–33. [Google Scholar] [CrossRef]
- Stallcup, W.B.; Dahlin, K.; Healy, P. Interaction of the NG2 chondroitin sulfate proteoglycan with type VI collagen. J. Cell Biol. 1990, 111, 3177–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Susante, J.L.C.; Pieper, J.; Buma, P.; van Kuppevelt, T.H.; van Beuningen, H.; van Der Kraan, P.M.; Veerkamp, J.H.; van den Berg, W.B.; Veth, R.P.H. Linkage of chondroitin-sulfate to type I collagen scaffolds stimulates the bioactivity of seeded chondrocytes in vitro. Biomaterials 2001, 22, 2359–2369. [Google Scholar] [CrossRef]
- Streit, A.; Nolte, C.; Rásony, T.; Schachner, M. Interaction of astrochondrin with extracellular matrix components and its involvement in astrocyte process formation and cerebellar granule cell migration. J. Cell Biol. 1993, 120, 799–814. [Google Scholar] [CrossRef]
- Lukas, M.; Susanne, S.; De Santis Rebecca, D.S.; Hajo, H.; Janine, H.; Lena, H.; Klaus, B.; Gernot, M.; Tobias, S. Peptide 19–2.5 inhibits heparan sulfate-triggered inflammation in murine cardiomyocytes stimulated with human sepsis serum. PLoS ONE 2015, 10, e0127584. [Google Scholar]
- Schmidt, E.P.; Overdier, K.H.; Sun, X.; Lin, L.; Liu, X.; Yang, Y.; Ammons, L.A.; Hiller, T.D.; Suflita, M.A.; Yu, Y. Urinary glycosaminoglycans predict outcomes in septic shock and acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2016, 194, 439–449. [Google Scholar] [CrossRef]
- Fawcett, J.W. The extracellular matrix in plasticity and regeneration after CNS injury and neurodegenerative disease. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 218, pp. 213–226. [Google Scholar]
- Yang, S.; Hilton, S.; Alves, J.N.; Saksida, L.M.; Bussey, T.; Matthews, R.T.; Kitagawa, H.; Spillantini, M.G.; Kwok, J.C.; Fawcett, J.W. Antibody recognizing 4-sulfated chondroitin sulfate proteoglycans restores memory in tauopathy-induced neurodegeneration. Neurobiol. Aging 2017, 59, 197–209. [Google Scholar] [CrossRef]
- Galtrey, C.M.; Fawcett, J.W. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res. Rev. 2007, 54, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Hatamochi, A.; Honda, T.; Yamaguchi, T.; Okamoto, N.; Miyake, N.; Yamada, S.; Sugahara, K. Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 2017, 50, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosho, T. CHST14/D4ST1 deficiency: New form of Ehlers–Danlos syndrome. Pediatr. Int. 2016, 58, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, J. Fell-Muir Lecture: Heparan sulphate and the art of cell regulation: A polymer chain conducts the protein orchestra. Int. J. Exp. Pathol. 2015, 96, 203–231. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Petitou, M.; Provasoli, M.; Sinay, P. Conformational flexibility: A new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem. Sci. 1988, 13, 221–225. [Google Scholar] [CrossRef]
- Mulloy, B.; Forster, M.J.; Jones, C.; Drake, A.F.; Johnson, E.A.; Davies, D.B. The effect of variation of substitution on the solution conformation of heparin: A spectroscopic and molecular modelling study. Carbohydr. Res. 1994, 255, 1–26. [Google Scholar] [CrossRef]
- Zamparo, O.; Comper, W.D. The hydrodynamic frictional coefficient of polysaccharides: The role of the glycosidic linkage. Carbohydr. Res. 1991, 212, 193–200. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Gehrcke, J.-P.; Pisabarro, M.T. Flexibility and explicit solvent in molecular-dynamics-based docking of protein–glycosaminoglycan systems. J. Chem. Inf. Model. 2014, 54, 582–592. [Google Scholar] [CrossRef]
- Shriver, Z.; Raguram, S.; Sasisekharan, R. Glycomics: A pathway to a class of new and improved therapeutics. Nat. Rev. Drug Discov. 2004, 3, 863–873. [Google Scholar] [CrossRef]
- Trottein, F.; Schaffer, L.; Ivanov, S.; Paget, C.; Vendeville, C.; Cazet, A.; Groux-Degroote, S.; Lee, S.; Krzewinski-Recchi, M.-A.; Faveeuw, C.; et al. Glycosyltransferase and sulfotransferase gene expression profiles in human monocytes, dendritic cells and macrophages. Glycoconj. J. 2009, 26, 1259–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, H.; Kinoshita-Toyoda, A.; Selleck, S.B. Structural Analysis of Glycosaminoglycans in Drosophila and Caenorhabditis elegans and Demonstration That tout-velu, a Drosophila Gene Related to EXT Tumor Suppressors, Affects Heparan Sulfate in Vivo. J. Biol. Chem. 2000, 275, 2269–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.O.; Gunay, N.S.; Toida, T.; Kuberan, B.; Yu, G.; Kim, Y.S.; Linhardt, R.J. Preparation and structural determination of dermatan sulfate-derived oligosaccharides. Glycobiology 2000, 10, 1033–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaia, J.; Costello, C.E. Compositional Analysis of Glycosaminoglycans by Electrospray Mass Spectrometry. Anal. Chem. 2001, 73, 233–239. [Google Scholar] [CrossRef]
- Shao, C.; Zhang, F.; Kemp, M.M.; Linhardt, R.J.; Waisman, D.M.; Head, J.F.; Seaton, B.A. Crystallographic analysis of calcium-dependent heparin binding to annexin A2. J. Biol. Chem. 2006, 281, 31689–31695. [Google Scholar] [CrossRef] [Green Version]
- Capila, I.; Hernáiz, M.A.J.; Mo, Y.D.; Mealy, T.R.; Campos, B.; Dedman, J.R.; Linhardt, R.J.; Seaton, B.A. Annexin V–Heparin Oligosaccharide Complex Suggests Heparan Sulfate–Mediated Assembly on Cell Surfaces. Structure 2001, 9, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Dementiev, A.; Petitou, M.; Herbert, J.-M.; Gettins, P.G.W. The ternary complex of antithrombin–anhydrothrombin–heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol. 2004, 11, 863–867. [Google Scholar] [CrossRef]
- Li, W.; Johnson, D.J.D.; Esmon, C.T.; Huntington, J.A. Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol. 2004, 11, 857–862. [Google Scholar] [CrossRef]
- Li, Z.; Kienetz, M.; Cherney, M.M.; James, M.N.G.; Brömme, D. The crystal and molecular structures of a cathepsin K: Chondroitin sulfate complex. J. Mol. Biol. 2008, 383, 78–91. [Google Scholar] [CrossRef]
- Imberty, A.; Lortat-Jacob, H.; Pérez, S. Structural view of glycosaminoglycan–protein interactions. Carbohydr. Res. 2007, 342, 430–439. [Google Scholar] [CrossRef]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Torri, G.; Casu, B.; Gatti, G.; Jacquinet, J.C.; Sinay, P.; Petitou, M.; Choay, J. Evidence for conformational equilibrium of the sulfated L-iduronate residue in heparin and in synthetic heparin mono- and oligo-saccharides: NMR and force-field studies. J. Am. Chem. Soc. 1986, 108, 6773–6778. [Google Scholar] [CrossRef]
- Bossennec, V.; Petitou, M.; Perly, B. 1H-n.m.r. investigation of naturally occurring and chemically oversulphated dermatan sulphates. Identification of minor monosaccharide residues. Biochem. J. 1990, 267, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Casu, B.; Torri, G.; Bossennec, V.; Perly, B.; Sinaÿ, P.; Petitou, M.; Choay, J. Conformer populations of l-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 1990, 195, 157–167. [Google Scholar] [CrossRef]
- Yamada, S.; Yamane, Y.; Sakamoto, K.; Tsuda, H.; Sugahara, K. Structural determination of sulfated tetrasaccharides and hexasaccharides containing a rare disaccharide sequence, -3GalNAc(4,6-disulfate)beta1-4IdoAalpha1-, isolated from porcine intestinal dermatan sulfate. Eur. J. Biochem. 1998, 258, 775–783. [Google Scholar] [CrossRef]
- Almond, A.; Sheehan, J.K. Glycosaminoglycan conformation: Do aqueous molecular dynamics simulations agree with x-ray fiber diffraction? Glycobiology 2000, 10, 329–338. [Google Scholar] [CrossRef]
- Silipo, A.; Zhang, Z.; Cañada, F.J.; Molinaro, A.; Linhardt, R.J.; Jiménez-Barbero, J. Conformational analysis of a dermatan sulfate-derived tetrasaccharide by NMR, molecular modeling, and residual dipolar couplings. ChemBioChem 2008, 9, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Sattelle, B.M.; Shakeri, J.; Cliff, M.J.; Almond, A. Proteoglycans and their heterogeneous glycosaminoglycans at the atomic scale. Biomacromolecules 2015, 16, 951–961. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Can current force fields reproduce ring puckering in 2-O-sulfo-α-l-iduronic acid? A molecular dynamics simulation study. Carbohydr. Res. 2010, 345, 689–695. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Hansen, S.U.; Gardiner, J.; Almond, A. Free energy landscapes of iduronic acid and related monosaccharides. J. Am. Chem. Soc. 2010, 132, 13132–13134. [Google Scholar] [CrossRef]
- Balogh, G.; Gyongyosi, T.S.; Timári, I.; Herczeg, M.; Borbás, A.; Fehér, K.; Kovér, K.E. Comparison of carbohydrate force fields using Gaussian Accelerated Molecular Dynamics simulations and development of force field parameters for heparin-analogue pentasaccharides. J. Chem. Inf. Model. 2019, 59, 4855–4867. [Google Scholar] [CrossRef]
- Ng, C.; Nandha Premnath, P.; Guvench, O. Rigidity and Flexibility in the Tetrasaccharide Linker of Proteoglycans from Atomic-Resolution Molecular Simulation. J. Comput. Chem. 2017, 38, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O. Revealing the Mechanisms of Protein Disorder and N-Glycosylation in CD44-Hyaluronan Binding Using Molecular Simulation. Front. Immunol. 2015, 6, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faller, C.E.; Guvench, O. Sulfation and cation effects on the conformational properties of the glycan backbone of chondroitin sulfate disaccharides. J. Phys. Chem. B 2015, 119, 6063–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favreau, A.J.; Faller, C.E.; Guvench, O. CD44 receptor unfolding enhances binding by freeing basic amino acids to contact carbohydrate ligand. Biophys. J. 2013, 105, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., 2nd; Mackerell, A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Brannigan, G.; Lin, L.C.; Brown, F.L. Implicit solvent simulation models for biomembranes. Eur. Biophys. J. 2006, 35, 104–124. [Google Scholar] [CrossRef]
- Srinivas, G.; Cheng, X.; Smith, J.C. A Solvent-Free Coarse Grain Model for Crystalline and Amorphous Cellulose Fibrils. J. Chem. Theory Comput. 2011, 7, 2539–2548. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; de Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 225–248. [Google Scholar] [CrossRef]
- Guvench, O.; Greene, S.N.; Kamath, G.; Brady, J.W.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D., Jr. Additive Empirical Force Field for Hexopyranose Monosaccharides. J. Comput. Chem. 2008, 29, 2543–2564. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O.; Hatcher, E.R.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallajosyula, S.S.; Guvench, O.; Hatcher, E.; Mackerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J. Chem. Theory Comput. 2012, 8, 759–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Montgomery, D.; Xue, X.; Foley, B.L.; Woods, R.J. GAG Builder: A web-tool for modeling 3D structures of glycosaminoglycans. Glycobiology 2019, 29, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Engelsen, S.B.; Hansen, P.I.; Perez, S. POLYS 2.0: An open source software package for building three-dimensional structures of polysaccharides. Biopolymers 2014, 101, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Kuttel, M.M.; Ståhle, J.; Widmalm, G. CarbBuilder: Software for building molecular models of complex oligo-and polysaccharide structures. J. Comput. Chem. 2016, 37, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Clerc, O.; Deniaud, M.; Vallet, S.D.; Naba, A.; Rivet, A.; Perez, S.; Thierry-Mieg, N.; Ricard-Blum, S. MatrixDB: Integration of new data with a focus on glycosaminoglycan interactions. Nucleic Acids Res. 2018, 47, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Clerc, O.; Mariethoz, J.; Rivet, A.; Lisacek, F.; Pérez, S.; Ricard-Blum, S. A pipeline to translate glycosaminoglycan sequences into 3D models. Application to the exploration of glycosaminoglycan conformational space. Glycobiology 2018, 29, 36–44. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Brooks, B.; Brooks, C.L., III; Nilsson, L.; Roux, B.; Won, Y.; Karplus, M. CHARMM: The Energy Function and Its Parameterization with an Overview of the Program. In Encyclopedia of Computational Chemistry; von Ragué Schleyer, P., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., Schreiner, P.R., Eds.; John Wiley & Sons: Chichester, UK, 1998; Volume 1, pp. 271–277. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Durell, S.R.; Brooks, B.R.; Ben-Naim, A. Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 1994, 98, 2198–2202. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hestenes, M.R.; Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Nat. Bur. Stand. 1952, 49, 409–436. [Google Scholar] [CrossRef]
- Fletcher, R.; Reeves, C.M. Function minimization by conjugate gradients. Comput. J. 1964, 7, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.C. Rattle: A “Velocity” Version of the Shake Algorithm for Molecular Dynamics Calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Jones, J.E.; Chapman, S. On the Determination of Molecular Fields. -II. From the Equation of State of a Gas. Proc. R. Soc. Lond. A-Contain. 1924, 106, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Allen, M.; Tildesley, D. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta 2015, 1850, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Kubo, R.; Toda, M.; Hashitsume, N. Statistical Physics II: Nonequilibrium Statistical Mechanics. In Springer Series in Solid-State Sciences, 2nd ed.; Cardona, M., Fulde, P., von Klitzing, K., Queisser, H.-J., Eds.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1991; Volume 31. [Google Scholar]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef]
- Eastman, P.; Pande, V. OpenMM: A Hardware-Independent Framework for Molecular Simulations. Comput. Sci. Eng. 2010, 12, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastman, P.; Pande, V.S. Constant Constraint Matrix Approximation: A Robust, Parallelizable Constraint Method for Molecular Simulations. J. Chem. Theory Comput. 2010, 6, 434–437. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2012, 9, 461–469. [Google Scholar] [CrossRef]
- Eastman, P.; Pande, V.S. Efficient Nonbonded Interactions for Molecular Dynamics on a Graphics Processing Unit. J. Comput. Chem. 2010, 31, 1268–1272. [Google Scholar] [CrossRef] [Green Version]
- Kiss, P.T.; Sega, M.; Baranyai, A. Efficient Handling of Gaussian Charge Distributions: An Application to Polarizable Molecular Models. J. Chem. Theory Comput. 2014, 10, 5513–5519. [Google Scholar] [CrossRef]
- Gingrich, T.R.; Wilson, M. On the Ewald summation of Gaussian charges for the simulation of metallic surfaces. Chem. Phys. Lett. 2010, 500, 178–183. [Google Scholar] [CrossRef]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Shakeri, J.; Roberts, I.S.; Almond, A. A 3D-structural model of unsulfated chondroitin from high-field NMR: 4-sulfation has little effect on backbone conformation. Carbohydr. Res. 2010, 345, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Casu, B.; Torri, G.; Vercellotti, J.R. Resolution-enhanced 1H-nmr spectra of dermatan sulfate and chondroitin sulfates: Conformation of the uronic acid residues. Carbohydr. Res. 1979, 68, C3–C7. [Google Scholar] [CrossRef]
- Alibay, I.; Bryce, R.A. Ring puckering landscapes of glycosaminoglycan-related monosaccharides from molecular dynamics simulations. J. Chem. Inf. Model. 2019, 59, 4729–4741. [Google Scholar] [CrossRef]
- Nyerges, B.; Kovacs, A. Density functional study of the conformational space of 4C1 D-glucuronic acid. J. Phys. Chem. A 2005, 109, 892–897. [Google Scholar] [CrossRef]
- Scott, J.E. Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J. 1992, 6, 2639–2645. [Google Scholar] [CrossRef]
- Van Boeckel, C.; Van Aelst, S.; Wagenaars, G.; Mellema, J.R.; Paulsen, H.; Peters, T.; Pollex, A.; Sinnwell, V. Conformational analysis of synthetic heparin-like oligosaccharides containing α-L-idopyranosyluronic acid. Recl. Trav. Chim. Pays-Bas 1987, 106, 19–29. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Jacquinet, J.-C.; Krishna, N.R. NMR and molecular modeling studies on two glycopeptides from the carbohydrate-protein linkage region of connective tissue proteoglycans. Glycobiology 1999, 9, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Choe, B.Y.; Ekborg, G.C.; Roden, L.; Harvey, S.C.; Krishna, N.R. High-resolution NMR and molecular modeling studies on complex carbohydrates: Characterization of O-β-D-Gal-(1→3)-O-β-D-Gal-(1→4)-O-β-D-Xyl-(1→ 0)-L-Ser, a carbohydrate-protein linkage region fragment from connective tissue proteoglycans. J. Am. Chem. Soc. 1991, 113, 3743–3749. [Google Scholar] [CrossRef]
- Krishna, N.; Choe, B.; Prabhakaran, M.; Ekborg, G.C.; Rodén, L.; Harvey, S. Nuclear magnetic resonance and molecular modeling studies on O-β-D-galactopyranosyl-(1→4)-O-β-D-xylopyranosyl-(1→0)-L-serine, a carbohydrate-protein linkage region fragment from connective tissue proteoglycans. J. Biol. Chem. 1990, 265, 18256–18262. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MD-Generated 20-mer Ensemble | Constructed 20-mer Ensemble (Before Energy Minimization) | Constructed 20-mer Ensemble (After Energy Minimization) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GlcAβ1-3 GalNAc | GalNAcβ1-4 GlcA | GlcAβ1-3 GalNAc | GalNAcβ1-4 GlcA | GlcAβ1-3 GalNAc | GalNAcβ1-4 GlcA | |||||||
| Min | ϕ, ψ | ΔG | ϕ, ψ | ΔG | ϕ, ψ | ΔG | ϕ, ψ | ΔG | ϕ, ψ | ΔG | ϕ, ψ | ΔG |
| I | −81.25°, −153.75° | 0.00 | −66.25°, 116.25° | 0.00 | −76.25°, −148.75° | 0.00 | −66.25°, 116.25° | 0.00 | −81.25°, −153.75° | 0.00 | −68.75°, 121.25° | 0.00 |
| II | −58.75°, −33.75° | 1.57 | −61.25°, −33.75° | 1.54 | −58.75°, −33.75° | 1.57 | ||||||
| II’ | −86.25°, −73.75° | 1.80 | −86.25°, −73.75° | 1.71 | −86.25°, −78.75° | 1.73 | ||||||
| 20-mer Ensembles | 10-mer Ensembles | |||||
|---|---|---|---|---|---|---|
| MD-Generated d (Å) | Constructed d (Å) | % Difference | MD-Generated d (Å) | Constructed d (Å) | % Difference | |
| Run 1 | 88.5 | 83.0 | 45.25 | 44.50 | ||
| Run 2 | 88.5 | 85.5 | 45.25 | 43.50 | ||
| Run 3 | 86.0 | 85.0 | 45.50 | 44.50 | ||
| Run 4 | 86.5 | 85.0 | 45.25 | 44.25 | ||
| All 2 | 88.5 | 85.0 | 4.03% | 45.25 | 44.50 | 1.671% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitmore, E.K.; Vesenka, G.; Sihler, H.; Guvench, O. Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data. Biomolecules 2020, 10, 537. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040537
Whitmore EK, Vesenka G, Sihler H, Guvench O. Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data. Biomolecules. 2020; 10(4):537. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040537
Chicago/Turabian StyleWhitmore, Elizabeth K., Gabriel Vesenka, Hanna Sihler, and Olgun Guvench. 2020. "Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data" Biomolecules 10, no. 4: 537. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040537