In Silico Drug Design for Purinergic GPCRs: Overview on Molecular Dynamics Applied to Adenosine and P2Y Receptors



Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Biomolecules 2020, 10(6), 812; https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060812
Submission received: 23 April 2020
/
Revised: 21 May 2020
/
Accepted: 22 May 2020
/
Published: 26 May 2020
(This article belongs to the Collection In Silico Drug Design for GPCRs: Big Data for Small Molecule Discovery)
Abstract
:Molecular modeling has contributed to drug discovery for purinergic GPCRs, including adenosine receptors (ARs) and P2Y receptors (P2YRs). Experimental structures and homology modeling have proven to be useful in understanding and predicting structure activity relationships (SAR) of agonists and antagonists. This review provides an excursus on molecular dynamics (MD) simulations applied to ARs and P2YRs. The binding modes of newly synthesized A1AR- and A3AR-selective nucleoside derivatives, potentially of use against depression and inflammation, respectively, have been predicted to recapitulate their SAR and the species dependence of A3AR affinity. P2Y12R and P2Y1R crystallographic structures, respectively, have provided a detailed understanding of the recognition of anti-inflammatory P2Y14R antagonists and a large group of allosteric and orthosteric antagonists of P2Y1R, an antithrombotic and neuroprotective target. MD of A2AAR (an anticancer and neuroprotective target), A3AR, and P2Y1R has identified microswitches that are putatively involved in receptor activation. The approach pathways of different ligands toward A2AAR and P2Y1R binding sites have also been explored. A1AR, A2AAR, and A3AR were utilizes to study allosteric phenomena, but locating the binding site of structurally diverse allosteric modulators, such as an A3AR enhancer LUF6000, is challenging. Ligand residence time, a predictor of in vivo efficacy, and the structural role of water were investigated through A2AAR MD simulations. Thus, new MD and other modeling algorithms have contributed to purinergic GPCR drug discovery.
1. Introduction
Molecular modeling has contributed to drug design and discovery for 40 years. Molecular docking and virtual screening techniques were initially employed to prefilter huge chemical libraries to reduce the burden of compounds and resources that are needed for in vitro testing. Structure-based drug design (SBDD) relies on the availability of the target macromolecule structure, as well as the commercial availability and informatic accessibility of now ultra-large libraries of small chemicals [1]. Moreover, the possibility of utilizing virtual libraries enables the investigation of chemical space in regions that have not yet been synthetically explored [2]. Over the last 20 years, hundreds of experimental structures have been solved for G protein-coupled receptors (GPCRs). These structures provide a snapshot of individual conformational states of the system, and they are useful for SBDD campaigns, with proven success in the identification of potential therapeutic compounds [3]. However, GPCRs in membranes are characterized by a high conformational plasticity, and molecular docking techniques starting from a static experimental structure neglect the target’s flexibility, as well as the membrane and solvent environment [4]. Recent software and hardware advances have extended the capabilities of another technique, molecular dynamics (MD), from an academic exercise to a useful drug discovery tool. The use of MD in the study of ligand-target complexes enables the inclusion of protein flexibility, with a better evaluation of the system stability. Furthermore, MD simulations can account for explicit solvent, ions, and membranes (in the case of membrane-bound proteins). Moreover, this might lead to the identification of protein’s druggable binding cavities, to the investigation of drug’s mechanistic pathways, and consequently to the computation of kinetic parameters [5]. In this review, we focus on recent MD studies of purinergic GPCRs, including adenosine receptors (ARs, also known as P1 receptors) and P2Y receptors (P2YRs) [6,7]. Both these receptor groups belong to the rhodopsin-like branch of Family A GPCRs. There are also purinergic ligand-gated P2X receptors (P2XRs) that are activated by ATP [8]. Structures have been determined for some of the seven P2XR subtypes, which act together as functional trimers, and modeling ligand interactions has been performed [9].
ARs are divided into four subtypes, all being activated by adenosine, but coupled to different G proteins. A1 and A3 ARs are coupled to Gi protein, and A2A and A2B ARs are coupled to Gs protein [10] (see Table 1). The four AR subtypes have been the subject of intense drug discovery efforts, leading to the identification of both agonists and antagonists [11,12]. Numerous A2AAR experimental structures have been released since 2008, as summarized in Table 2. In total, 49 structures for A2AAR have been solved and deposited in the Protein Data Bank (PDB) [13]: 40 antagonist-bound X-ray structures [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]; seven agonist-bound intermediate-state X-ray structures [32,33,34,35]; one agonist-bound active G protein-bound X-ray structure [36]; and one agonist-bound active G protein-bound cryogenic electron microscopy (cryo-EM) structure [37]. In addition, two X-ray structures of A1AR (antagonist-bound) have been reported [24,38], together with one agonist-bound active G protein-bound cryo-EM structure [39].
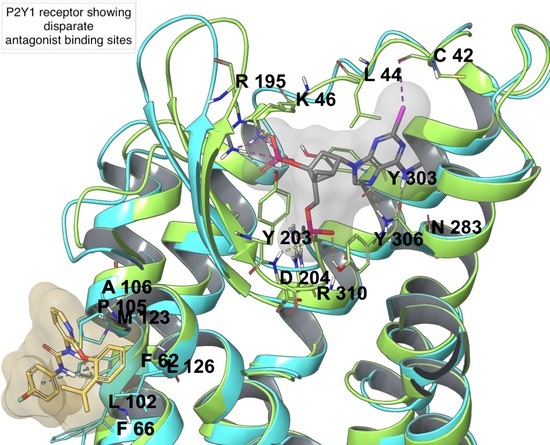
On the P2YR side, there are eight receptor subtypes, with P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11Rs coupled to Gq (and Gi in the case of P2Y2 and P2Y4, and Gs in the case of P2Y11) and belonging to the P2Y1-like subfamily, and P2Y12, P2Y13 and P2Y14Rs coupled to Gi and belonging to the P2Y12-like subfamily. P2YRs are activated by diverse mono- and dinucleotides, as summarized in Table 1, with P2Y1, P2Y12, and P2Y13Rs activated by ADP, P2Y2 and P2Y4 by UTP and ATP (and GTP in the case of P2Y4), P2Y6 and P2Y14Rs by UDP (and UDP-glucose in the case of P2Y14), and P2Y11R by ATP [7]. P2Y1 and P2Y12Rs are the only P2YRs having an X-ray crystal structure deposited in the PDB. Two X-ray structures are available for P2Y1R [40], in complex with two different antagonists: MRS2500, a hydrophilic, nucleotide-like compound, bound to the orthosteric binding site within the transmembrane bundle, and BPTU, which is a hydrophobic diarylurea derivative, bound to an allosteric pocket outside the transmembrane bundle at the phospholipid interface. Two agonist-bound (2MeSADP and 2MeSATP) X-ray structures were solved for the P2Y12R [41], in addition to an antagonist-bound (AZD1283) structure where the conserved disulfide bond connecting TM3 and ECL2 was broken [42].
A2AAR is a cancer target, as antagonists block the local immunosuppressive effects of adenosine. P2Y1R antagonists are also potential antithrombotic and neuroprotective therapies. A1AR- and A3AR-selective agonists are potentially of use for treating depression and inflammation, respectively. A2AAR agonists and P2Y14R antagonists have anti-inflammatory properties.
The experimental structures of purinergic receptors have played a fundamental role in SBDD of agonists and antagonists for these receptors [43] and, lately, MD has gained an important role in this field. MD-based techniques have been applied to ARs and P2YRs to investigate association, dissociation, and activation processes, and, in some cases, with approximate free energy-profiles and binding kinetics (as association and dissociation rate constants). Other valuable applications include the simulation of phenomena such as allostery and exploration of the structural role of water. Before describing examples of MD applied to purinergic receptors, we provide an excursus on some of the main techniques that will be cited throughout this manuscript.
2. Excursus on MD Techniques
MD is a computational technique modeling the evolution of molecular systems in time at an atomistic level by approximating energy as a force field and solving Newton’s equations of motion. MD simulations can be used in combination with other structural techniques to assess the stability of complexes over time, to generate structural ensembles, and to identify putative cryptic pockets or allosteric sites. A timestep on the order of femtoseconds, corresponding to the fastest system motions (bond vibrations), is used; thus, MD is currently not suitable for the simulation of slow molecular processes [5]. The exploration of rare events, like receptor binding pathways and activation processes, requires a long timescale (hundreds of μs to ms), which are not easily achievable with classical MD simulations. Examples of long-timescale classical MD simulations are reported in literature, but they demand a huge computational cost and special resources [44]. To overcome this limitation, several techniques, called enhanced sampling techniques, have been developed in recent decades. These techniques are characterized by the addition of a bias potential to the system energy in order to enhance sampling and allow the system to jump with an increased frequency from one minimum to another within the energy landscape. Various enhanced sampling techniques exist, and they can be either based on collective variables (CVs), or not, where CVs are the reaction coordinates describing the process. Enhanced sampling techniques, after adding a bias potential to the original force-field energy of the system, require a subsequent phase of reweighting in order to recover the target system energy [45].
Metadynamics, Temperature-Accelerated MD (TAMD), Umbrella Sampling, Accelerated MD (aMD), and Gaussian Accelerated MD (GaMD) are some of the enhanced sampling MD techniques that will be considered in this review, as applied to ARs and P2YRs. Metadynamics consists of the decomposition of the system into one or more CVs, and of the application of a history-dependent bias potential that discourages the exploration of already visited states [46,47]. The system free-energy surface can then be reconstructed as a function of CVs while using reweighing approaches. TAMD was inspired by metadynamics, and it relies on dynamical collective variables to which an artificial high temperature is applied [48]. Umbrella sampling adds a harmonic compensating function to increase the sampling on high energy states [49]. aMD adds a boost potential to the true potential energy surface of the system to enable the overcoming of high energy barriers [50]. This method does not require a priori knowledge of the system to define CVs, but it suffers from poor energy reweighting due to the use of a high energetic bias. To overcome this limitation, GaMD was developed, where the bias potential is adaptively added to the potential energy surface following a Gaussian distribution, enabling better reweighting [51,52].
Classical MD simulations combined with adaptive sampling techniques can be employed to explore long time-scale events, by using multiple MD simulations sampling the search space on the basis of the collected data results [53]. Among these methods, Supervised MD (SuMD) was initially developed to simulate recognition of ligands by the A2AAR and consists of the supervision of classical MD simulations through a tabu-like algorithm, but without the introduction of any energetic bias [54,55]. A classical MD simulation is subdivided into different steps, during which the distance between the centers of mass of the ligand and of the receptor’s binding site is monitored. At the end of each step, the simulation cycles to a new step if the ligand has moved closer to the receptor; otherwise, the current step is restarted reassigning initial velocities. A SuMD algorithm modification recently enabled the simulation of ligand unbinding [56]. The algorithm was slightly modified in the criteria to define the success of a SuMD step (i.e., ligand moving away from the binding pocket’s center of mass) and by increasing the length of each SuMD step during the simulation.
3. MD Simulations Applied to Adenosine and P2Y Receptors
3.1. Postprocessing of Molecular Docking Poses
MD can be combined with molecular docking to predict a ligand binding mode at a target site, and to refine the consequent pose by taking into account the solvent contribution and the flexibility provided by all of the system’s degrees of freedom. For instance, associating MD simulations with molecular docking has recently been fruitful in rationalizing the AR-antagonist interactions for novel chemotypes identified through the virtual screening of a small library [57].
A methodological work illustrated the efficiency of a combination of molecular docking and MD simulations in reproducing the crystallographic pose of antagonists ZM241385 (ZMA), T4G, T4E, and caffeine, and of the agonist NECA at the A2AAR orthosteric binding site [58]. The computational pipeline consisted of molecular docking, pose clustering, MD simulations, and evaluation of the complex’s interaction energy fingerprints and interaction energy (electrostatic or hydrophobic) weighted by the root mean square deviation (RMSD).
Since 2008, when the A2AAR became the first AR experimental structure to be solved, it has been employed for numerous molecular modeling and computer aided drug design studies. More recently, two antagonist-bound X-ray structures [24,38] and a cryo-EM agonist-bound [39] structure of A1AR were released, increasing the reliability of computational works. A recent study was conducted to suggest a possible binding mode for the selective A1AR agonist N6-dicyclobutylmethyl-adenosine (MRS7469, Figure 1, hA1AR 2.14 nM, >2000-fold selectivity over the other AR subtypes) [59], while using the cryo-EM adenosine-bound A1AR structure (Figure 2A). A binding mode comparable to that of adenosine in the experimental complex was proposed for MRS7469, with the conserved N2546.55 being involved in a bidentate hydrogen bond with N7 and the exocyclic N6H, with a π-π stacking interaction between the adenine aromatic scaffold and F171ECL2, and with H-bonds between 2′-OH and H2787.43, 3′-OH and T2777.42, 5′-OH and H2516.52. All these interactions were maintained during 30 ns MD simulation, and the two cyclobutyl substituents occupied two hydrophobic clefts positioned among ECL2, TM5, and TM6 in one case and TM6 and TM7 in the other. Moreover, E172ECL2 and K265ECL3, which are separated by approximately 7 Å in the cryo-EM structure, narrowed their separation during the simulation, with a possible role in stabilizing the ligand bound state, as known for the E169ECL2-H264ECL3 salt bridge in the case of A2AAR, constituting a lid that caps the orthosteric binding site in several X-ray structures [19]. Moreover, an involvement of residues E172ECL2 and K265ECL3 in agonist binding was consistent with decreased binding affinity of NECA in the E172ECL2A and K265ECL3A mutant A1AR, with the mutation K265ECL3A being suggested by molecular modeling studies on a A1AR model [60].
A number of recent studies on the A3AR aided in the discovery of many nucleoside agonists and partial agonists, consistent with the observed structure activity relationship (SAR). Two prominent features of these A3AR ligands are the enhancement of affinity and selectivity provided by 2-arylethynyl substitution and ribose ring replacement with a chemically constrained (N)-methanocarba (bicyclo[3.1.0]hexane) ring system [61]. No experimental structure is available for A3AR, so molecular modeling studies are based on homology models built using other ARs structures as templates. In fact, homology modeling had previously proved to be an accurate approximation of the crystallographic structure of A2AAR (apart from ECLs and ICLs) [62], so it is a useful technique when the experimental receptor structure of interest is lacking. This is also valid for A2BAR, which also lacks an experimental structure, and whose molecular modeling studies have been recently summarized by Deb et al. [63]. Different A3AR models have been built in recent years [61], with early examples being based on rhodopsin structures [64], which were then replaced by A3AR based on A2AAR or, more recently, A1AR templates [65,66]. An antagonist-bound A2AAR-based A3AR model, for instance, was used to compare the behavior of C2-substituted adenosine and NECA derivatives though MD simulations. It was suggested that the C2 and 5′ substituents cooperate to maintain the interaction pattern of full or partial agonism that might be associated with an anti or syn glycosidic bond conformation [67].
A well established hypothesis, involving an outward movement of the upper portion of TM2 as compared to its position in A2AAR, was introduced in order to explain the binding mode of highly selective C2-arylethynyl substituted A3AR agonists [68]. The outward position of TM2 would enable an adenosine-like positioning of the agonists, avoiding clashes of the C2-substituent with TM2. The first models that were based on this hypothesis were built using an intermediate state agonist-bound structure of A2AAR, and opsin or β2-adrenergic receptor for TM2 [68]. Recently, a revised nucleoside-bound A3AR model was constructed, based on an intermediate state agonist-bound structure of A2AAR and an inactive antagonist-bound structure of A1AR for TM2, which is outwardly displaced from the TM bundle [69].
Human (h) and mouse (m) A3AR hybrid models that were built in this way were recently applied to rationalize the effective contribution of polar substituents on the N6-2-phenylethyl moiety of 4′-truncated (N)-methanocarba 2-phenylethynyl adenosine partial-agonists [69]. In particular, derivatization at the N6 position with a dopamine-like moiety increased both h and mA3AR binding affinity, with a particular increase in the case of mA3AR with respect to the unsubstituted N6-2-phenylethyl. A comparison between the binding modes of the N6-2-phenylethyl (MRS5776) and hydroxy/methoxylated N6-dopamine-like (MRS7591) derivatives was performed at hA3AR and mA3AR orthosteric sites through a combination of molecular docking and classic MD simulations. The compounds’ adenosine-like scaffold similarly bound the two receptors to the binding mode adenosine or other agonists assumed in the experimental structure of A1AR or A2AAR (Figure 2A,B) [32,33,34,35,36,37,39]: a bidentate H-bond between N2506.55 (N2516.55 at mA3AR) and the ligands’ N7 and exocyclic N6H, a π- π stacking between the adenine aromatic scaffold and F168ECL2 (F169ECL2 (m)), and the H-bonds of hydroxyls 2′ and 3′ with H2727.43 and S2717.42 (respectively H2737.43 (m) and S2727.42 (m)), which were less persistent during MD. Contacts with residues that are involved in agonist binding and/or receptor activation according to site-directed mutagenesis (SDM) studies [70,71,72] were observed during the simulations: L903.32 (L913.32 (m)), T943.36 (T953.36 (m)), M1775.38 (M1785.38 (m)), W2436.48 (W2446.48 (m)), L2466.51 (L2476.51 (m)), I2687.39 (I2697.39 (m)), in addition to the aforementioned residues. Together with this, MRS7591 transiently interacted with residues of ECL2 and ECL3 in both hA3AR and mA3AR simulations, possibly explaining the higher affinity of this compound for the receptor in comparison to MRS5776. The enhanced effect in the case of mA3AR could be rationalized by the generally higher polar character of the extracellular regions of this receptor in comparison to the human homologue.
In the aforementioned study, the 2-arylethynyl moiety pointed toward the outwardly displaced TM2, as expected for 2-substituted nucleosides. However, an alternative binding mode was recently proposed for a series of 2-arylalkynyl-adenine hA3AR antagonists, where the lack of the ribose moiety enabled the compounds to occupy the orthosteric site upside down, with a bidentate H-bond between the key N2506.55 and the adenine N3 and N9 positions, and the 2-arylalkynyl group hosted by a hydrophobic pocket between TM5 and TM6 [73]. Thus, the adenine moiety does not need to persist in the same orientation as in the nucleoside when the ribose or pseudoribose is absent.
Another study was recently conducted using the aforementioned hybrid hA3AR model [69] to compare the bound state of a ribose and a (N)-methanocarba agonist analogue, namely MRS7432 and MRS7334 (Figure 3A) [74]. The (N)-methanocarba ligand had higher affinity for the receptor as compared to the ribose analogue, following the typical SAR for A3AR. During MD simulations, the two ligands behaved similarly in maintaining the typical pattern of interactions described above. The puckering parameters were computed for the two ligands in the bound state during time and they were compared to the behavior of the compounds in a free state. The ribose pucker of MRS7432 experienced a wide range of conformations ranging from (N) to (S) and passing through (E) in the free state, but the conformational space it explored in the bound state appeared to be reduced. The lack of this phenomenon in the case of the constrained (N)-methanocarba MRS7334 suggested that an entropy loss upon binding might explain the lower affinity of ribose-containing agonists as compared to conformationally constrained (N)-methanocarba.
The conformational constraint induced by the ribose ring substitution with a methanocarba was also applied to P2YR agonists. Dramatically, the (N)-methanocarba substitution prevented binding, while the isomeric (S)-methanocarba modification of UDP (MRS2975) increased its P2Y6R affinity. Dinucleotide analogues, particularly dinucleoside triphosphates, are also known to activate the P2Y6R. A series of alkyloxyimino-pyrimidine dinucleotides presenting a (S)-methanocarba or a (N)-methanocarba at either one or both nucleoside moieties showed that the highest potency is reached when one nucleotide (MRS4387) was constrained in the (N) and the other in the (S) conformation [75]. The reference compound, MRS2957, bearing two unconstrained ribose rings, was docked to a P2Y6R homology model that was built using the chemokine receptor 4 (CXCR4) as a template, and the complex was subjected to MD simulation. One uridine (called proximal) was hosted by a pocket between the conserved TM3-ECL2 disulfide bond and TM7 and the other (the distal one) lay above the orthosteric binding site. The proximal uridine showed an (S) pucker for the major part of a 100 ns MD simulation, while the distal uridine spent most simulation time in a (N) pucker, consistently with the experimental data.
Among P2YRs, the availability of crystallographic structures for P2Y1R and P2Y12R (Figure 2C,D) in recent years increased the reliability of molecular modeling studies on this subfamily of receptors. A molecular modeling study on P2Y1R combining docking and MD simulations recapitulated the SAR of a series of 100 nucleotide-like biphosphates that were structurally related to MRS2500 [76]. This study highlighted a steric limit for C2-alkyl substituents that cannot fit the hydrophobic pocket defined by ECL3 and N-term and, thus, are solvent exposed, with a balance provided by H-bond capable moieties. The intolerance of C8 substitutions was justified by the incompatibility with the anti-glycosidic bond present in the experimental MRS2500 conformation (Figure 2C) and maintained during the MD simulation. The agonist 2MeSADP showed a preferred (N) conformation during the MD simulation, which gave rationale to the efficient constraint that was provided by the (N)-methanocarba in MRS2500.
The therapeutic relevance of P2Y14R for inflammatory diseases has raised the interest in antagonist development for this receptor subtype, which lacks an experimental structure. A P2Y14R homology model was built on the basis of an agonist-bound P2Y12R structure, belonging to the same P2Y12-like subfamily [77]. The docking of UDP to the P2Y14R model provided a possible syn- or anti-glycosidic bond conformation, and MD simulation was used to discriminate the two, selecting the anti- as being more likely, in agreement with the pose of 2MeSADP bound to the P2Y12R X-ray structure. The substitution of C1945.43 of P2Y12R with F1915.43 in P2Y14R was hypothesized as a selectivity filter allowing pyrimidine and not purine binding. UDP occupied a pocket that was delimited by TMs 3, 4, 5, 6, and 7, with the pyrimidine base making a π-π interaction with Y1023.33, the ribose ring involved in H-bonds with the side chains of H1845.35 and K176ECL2 and the backbone carbonyl of C943.25. The same model was then refined to be employed for SBDD of P2Y14R antagonists [78,79]. The prototype antagonist used for the refinement was the highly active and selective antagonist PPTN, which was docked with an induced fit procedure and subjected to 10 ns MD simulation, showing modification in the model geometry (Figure 3B). In particular, TM7, as well as ECL1 and ECL2, moved outward from the TM bundle, and the axis of TM2 slightly bent toward TM3. PPTN was then re-docked and the complex stability was assessed during MD simulation; the compound interacted through its carboxylate group with K772.60, Y1023.33, and K2777.35 by means of H-bonds (and ionic interactions in the case of charged residues), and through the piperidine nitrogen with the backbone carbonyl of I170ECL2 (replacing the interaction with G802.63 observed in the docking pose) [78]. The poor solubility and low oral bioavailability of PPTN has necessitated the development of new P2Y14R antagonists, and the same protocol made for docking and MD refinement was used for investigating a bioisosteric substitution of the scaffold’s naphthalene portion. Two alternatives were considered, an alkyne and a triazole substitution, with the latter maximizing the pattern of interactions during MD simulations, in agreement with its higher in vitro affinity for the receptor when compared to the alkyne. While the alkyne derivative lost the H-bond with Y1023.33 and had a higher root mean square fluctuation (RMSF) as compared to the precursor, the triazole derivative had a lower RMSF and it was additionally stabilized by a π-π interaction between the triazole and Y1023.33 and between the 4-(4-trifluoromethyl)-phenyl group and His1845.36, and by a π-cation interaction between the triazole and R2536.55 [78]. In agreement with this, MRS4217, a triazole analogue of PPTN, was considered to be the lead compound for a series of analogues, whose SAR, in agreement with molecular modeling results, highlighted the requirement of the carboxylate and of a hydrophobic substituent preferentially in p- position on the 4-aryl group attached to the triazole [78,79]. Different substituents of the 5-(4-piperidin)-phenyl moiety were analyzed trying to maximize interactions with ECLs: the replacement of the phenyl ring with a thiophene and of piperidine with a propylamine were suggested by the docking results. A comparison of the two analogues with an amide or sulfonamide linker showed a similar behavior in MD simulations, with an H-bond between the carboxylate and Y1023.33, and between the amine and G802.63, a π−π stacking between the 4-(4-trifluoromethyl)-phenyl moiety and H1845.36 and between the triazole and Y1023.33. Two salt bridges between the carboxylate and K772.60 and K2777.35 were present with the sulfonamide compound, but they occurred between the carboxylate and K772.60 and R2536.55 with the amide ligand. The amide compound (MRS4458, the most potent of the series) was more rigid and fitted in a slightly deeper position, granting an additional ionic interaction between the carboxylate and R2536.55. Another compound (MRS4478), with a 5-(4-carboxamide)-phenyl substituent, showed a promising binding affinity to P2Y14R and it behaved similarly to the other two ligands during MD simulations, with, as only the difference, an interaction between the amide and N903.21 [79].
An advanced MD technique can be applied in order to estimate ligand-target binding affinity, starting from ligand-target complexes that originate from either molecular docking or experimental structures. This technique is called Free Energy Perturbation (FEP) and it consists of a statistical mechanics method computing the free energy change related to an alchemical process, like the transformation of a ligand into another, giving the relative binding free energy of the two compounds to the same target [80]. In the field of purinergic receptors, FEP was used to compare a thiazolo [5,4-d]pyrimidine partial agonist having a 7-prolinol substitution to its antagonist analogue lacking the prolinol’s 2-hydroxymethylene moiety. The simulations of the compounds docked to the binding site of an inactive and an active-like A2AAR structures showed that the loss of the 2-hydroxymethylene group was more unfavorable in the active-like state, in agreement with the potency reduction of the antagonist [81]. Prospective FEP+ (a FEP modification [82]) studies predicted a highly potent A2AAR antagonist in the nanomolar range, with a ten-fold increase in binding affinity as compared to the parent compound [83]. A different application of FEP enabled the exploration of the influence of alanine-scanning in agonist (NECA) and antagonist (ZMA) binding to A2AAR, with a good correlation with the experimental data [84]. In a study on N-(4,6-diarylpyridin-2-yl)acetamides as A3AR antagonists, FEP was used to rationalize the reduction in binding affinity, due to a bioisosteric substitution of N1 with CH and to suggest a likely binding mode [85]. Moreover, examples of using FEP in combination with Fragment-Based Lead Discovery (FBLD) are reported, where MD and FEP played an important role in fragments optimization as A2AAR binders [86,87].
3.2. Activation and Deactivation
Many MD studies have been conducted in order to investigate one of the most intriguing mechanistic processes related to GPCRs, activation, and deactivation. A number of common structural motives, called “microswitches”, are known to play a key role in GPCR activation and, among them: the ionic lock between R3.50 of the conserved E/DR3.50Y motif on TM3 and E6.30 on TM6 typical of the inactive state, the “rotamer toggle switch” or “transmission switch” characterized by a different conformation of W6.48 according to the receptor activation state, and the “tyrosine toggle switch” characterized by a conformational rearrangement of Y7.53 of the conserved NPxxY7.53 motif [88].
The formation and maintenance of a salt bridge between R1023.50 and E2286.30, the so-called ionic lock, typical of GPCR inactive states, was observed in different works simulating pseudo-apo states coming from antagonist-bound structures [89,90]. An alternative salt bridge involving E2286.30 and R1053.55, together with an outward movement of the TM5 intracellular portion, was observed in the X-ray structure of A2AAR bound to a triazole-carboximidamide antagonist (Cmpd-1) [21], which was recently verified through adaptive sampling MD simulations and 19F-NMR spectroscopy [91].
Together with the R1023.50-E2286.30 “ionic lock”, the rotameric transition of W6.48 (“toggle switch”), of the conserved CW6.48xP motif, is considered to play a key role in the GPCR activation process, and a conformational rearrangement was observed for W2466.48 in a pseudo-apo A2AAR MD simulations coming from inactive antagonist (ZMA)-bound structures [89]. Moreover, simulations in the order of μs showed that agonist (NECA) binding to A2AAR induced a conformational rearrangement of W2466.48. This movement allowed for water flow through the transmembrane helical (TM) bundle [92], which provoked a rotational switch of residue Y2887.53 (of the conserved NPxxY7.53 motif), an outward movement of TM6 and inward movement of TM7, causing an opening of the receptor’s internal portion, which could enable G protein binding [93]. The concerted movement starting from the toggle switch to the TM6 outward movement upon agonist binding is in agreement with a previous study, where TM6 appeared to assume an active-like, inactive-like, and intermediate conformation in agonist-bound, antagonist-bound, and pseudo-apo simulations, respectively [94]. An adaptive sampling study combined with Markov State Models computation lead to the simulation of the activation energy landscape of an apo A2AAR, which could be approximated by four predominant states. These four states are: (1) inactive antagonist-bound like state with the ionic lock between R1023.50 and E2286.30 and TM6 in proximity to TM3; (2) the apo intermediate-inactive state with disruption of the ionic lock and its own peculiar TM6 conformation; (3) the agonist-competent state, with TM6 moved outward and Y2887.53 pointing toward TM5 preventing G protein binding; and, (4) the active state, with TM6 pointing outward and an overall more open intracellular portion of the receptor. The active state was minimally populated in the explored activation energy landscape, in agreement with the concept of the basal activity of GPCRs [95]. A common character of all simulations of A2AAR activation/inactivation processes is an outward TM6 movement during receptor activation upon agonist binding. This phenomenon was experimentally confirmed by the experimental structures of a fully active A2AAR bound to agonist NECA and an engineered G protein, where the TM6 intracellular portion is 14 Å away from the central axis of the TM bundle, and TM5 and TM7 slightly deviate [36,37]. In another study, the MD simulations showed decreased entropy of the extracellular portion when agonists (adenosine or NECA) bound an intermediate-active or active (G protein-bound or free) receptor rather than the inactive one. In parallel, a flexibility increase in the intracellular portion was observed when agonists bound the intermediate-active or G protein-free active state as compared to the inactive one, but stabilization was observed in the G protein-bound active state. The agonist’s geometric stability was higher, the orthosteric pocket volume was reduced, and the pattern of ligand-receptor interactions stabilized in the intermediate-active and G-protein bound active receptor. All these observations well supported the gradually increased computed agonist binding affinity going from the inactive, the intermediate-active and G-protein bound active receptor [96].
A2AAR has been, for a long time, the test case for many molecular modeling studies as an AR prototype, thanks to the early availability of many X-ray crystal structures. More recently, for the A1AR two inactive antagonist-bound structures [24,38] and a cryo-EM active agonist-bound structure [39] were released, so more reliable molecular modeling studies are now possible. Recently, GaMD investigated the flexibility profile of the active adenosine-bound and inactive PSB36-bound A1AR [97]. In both simulations, conformations of the ECL2 region and the intracellular end of TM6 fluctuated, with higher flexibility of these receptor portions and of the intracellular end of TM5 in the active state.
The A3AR activation mechanism has been recently investigated through the simulation of three constitutively active mutants (CAMs), R108K3.50, R108A3.50, and A229E6.34, and a wild-type structures, modelled on the inactive and active (G protein bound and unbound) X-ray structures of A2AAR. The salt-bridges D1073.49-R1083.50 (with the two residues belonging to the conserved E/DR3.50Y motif on TM3) and E2556.30-R1113.53 characterized the inactive state, while salt-bridges D1073.49-R1113.53 and E2556.30-R2055.66 characterized the active one. Together with this, different conformations of W2436.48 (microswitch) were observed in the active and inactive states, an interaction between a Na+ ion and D582.50 characterized the inactive state, and an increased flow of water molecules characterized the active one [98].
A study on agonist- and antagonist-bound structures of P2Y1R was reported, simulating the receptor bound to BPTU (in its extrahelical binding site), MR2500, and ADP through long classical MD simulations [99]. An ionic-lock between D204ECL2 and R3107.39 was present in both antagonist-bound P2Y1R simulations, while it was broken in the agonist-bound simulation; these residues were confirmed to be essential for activation by SDM studies. ADP was predicted to bind the receptor extracellular portion, similarly to MRS2500, but the binding pocket in the agonist-bound state appeared to be larger than in the antagonist-bound one. The increased pocket volume was due to a continuum of water flux that spanned the TM bundle with a consequent rotameric switch of Y3247.53 (of the conserved NPxxY7.53 motif), and a shift of the cytoplasmic region of TM3, TM6, and TM7 that enlarged the intracellular cavity for G protein binding. The presence of the ionic lock between D204ECL2 and R3107.39 was also confirmed for the BPTU-bound P2Y1R in another study, but interaction between D204ECL2 and K2806.55 was instead observed as a characteristic of 3′,5′-biphosphate ligands [76]. Differently, in the same study, simulations of the 2MeSADP-bound receptor (built in place on the basis of MRS2500) showed a deviation of K2806.55 from the binding site and an interaction with Y2145.35, H1323.33, Y1363.37, in agreement with the reduced ligand affinity or potency in the case of K2806.55A, H1323.33A, and Y1363.37A mutations [100]. The same study confirmed also that the water molecule diffusion within the TM bundle was enhanced in the case of the pseudo-apo or nucleotide agonist-bound form of P2Y1R, while it was reduced in the MRS2500-bound receptor simulation.
A D204ECL2-R3107.39 homologue ionic lock was also observed in the models of P2Y2R and P2Y4R built on the inactive P2Y1R template, with D185ECL2-R2927.39 and D187 ECL2-R2927.39 being present in P2Y2R and P2Y4R, respectively. Agonists’ phosphate groups are predicted to interact with these residues, suggesting an activation mechanism that involves the disruption of the ionic lock, similarly to P2Y1 [101,102,103,104].
Another study on the agonist 2MeSADP-P2Y1R complex suggested a different binding site for the ligand, different from MRS2500, but comparable to the conformation of the same ligand in the X-ray structure of 2MeSADP-P2Y12R and compatible with SDM data [105]. This binding mode was obtained by docking the ligand in an ECL2 deprived inactive P2Y1R and subjecting the complex (after restoring ECL2) to aMD. The ligand was found to participate in π-π stacking and a cation-π interaction with H1323.33 and K2806.55 through the aromatic purine ring, a H-bond with Y2225.43 through the exocyclic amino group, a H-bond with Y1363.37 and T2215.42 through N1, and ionic and polar interactions with R1283.29, R2876.62, R3107.39, K2806.55, and Y3067.35 through the pyrophosphates. Moreover, the agonist-bound receptor underwent an outward movement of TM6 in the intracellular extremity, a S1463.47-Y2375.58 H-bond rupture, with the formation of a Y2375.58-V2626.37 (backbone O) H-bond and rotamer modification of F2696.44, which were not observed in the simulation of the MRS2500-bound inactive receptor in the same work.
3.3. Association Process
In the last years, a paradigm shift has been accompanying drug discovery, with increased attention to kinetic parameters in addition to binding affinities. For this reason, several methods have been developed to compute binding rate constants and study binding pathways [106].
The binding pathway of high affinity antagonist ZMA to the A2AAR was explored using well-tempered metadynamics [107]. The bound state showing lowest energy presented a salt bridge between E169ECL2 and H264ECL3, and its calculated binding free energy compared well to the experimental one. A metastable binding site was found in a vestibule that formed by ECL1 and ECL2 and the tips of TM1, TM2, and TM7, with two of the engaged residues, S672.65 and L2677.32, known to increase the residence time of ZMA without influencing binding affinity if mutated to Ala. A transient low populated state showed the interaction with K153ECL2, which is reported to decrease the dissociation rate of ZMA if mutated into Ala.
SuMD simulations investigated the involvement of the A2AAR ECLs in the initial stages of the recognition of various ligands, where the binding event of ZMA, T4G, and T4E (reaching a bound state comparable to the crystallographic one in approximately 60 ns, 65 ns, and 110 ns of SuMD simulation time, respectively) was anticipated by contacts with residues of ECL2 and ECL3 [54]. In the same way, adenosine (as well as its metabolite inosine) approached A2AAR breaking the ionic interaction between E169ECL2 and H264ECL3 (in agreement with the rupture of this interaction in previously reported pseudo-apo simulations of A2AAR [89]) [108,109]. This binding occurred through two main pathways, one involving in the first stages of recognition ECL2, ECL3 and the tips of TM5 and TM6 (similarly to what observed also for the agonist NECA [55]), and the other just ECL2. In the orthosteric binding pocket adenosine was observed to explore different bound states with the ribose pointing toward either the intracellular or the extracellular region. A bound state that was similar to the crystallographic one was only reached in the G protein-bound receptor simulation, supporting the hypothesis that adenosine binds and stabilizes through conformational selection for the receptor active state. A speculative 2:1 binding stoichiometry of adenosine to A2AAR was also suggested using SuMD simulations, supposing a kinetic rather than a thermodynamic driven event [110].
A recent application of SuMD to a hybrid A2BAR-A2AAR model contributed to the investigation of the role of ECL2 in defining the different binding affinity of A2AAR and A2BAR for the endogenous agonist adenosine [111]. The two AR subtypes have a high sequence identity (59%), with the bigger differences relative to ECL2 (34%, with A2BAR ECL2 longer than that of A2AAR), but A2AAR has high affinity for adenosine (nanomolar), while A2BAR has low affinity (micromolar). The introduction of A2BAR ECL2 in A2AAR reduced the affinity for adenosine by ~20-fold. A hypothetical explanation was provided through a SuMD trajectory showing a metastable binding site at the ECL2, which seemed to be energetically favored in comparison to the orthosteric site.
The recognition of adenosine by A3AR was also simulated with SuMD, observing also in this case a first contact with ECL2 and ECL3, and a final stabilization in the orthosteric binding site in 20 ns, with contacts with the conserved N2506.55, F168ECL2, W2436.48, and I2697.39 [112].
An application of SuMD to the P2Y subfamily was performed while simulating the antagonist ticagrelor’s P2Y12R binding pathway: the ligand was not able to reach the putative orthosteric binding site (supposedly comparable to the crystallographic 2MeSADP one), but it was stuck in metastable binding sites engaged in numerous polar and ionic interactions with residues belonging to ECL2 and the tips of TM5, TM6, and TM7 [113]. Differently from the ARs scenario, this could support the idea of a possible plasticity of the extracellular region of P2Y12R, which should be able to recognize different ligands.
The simulation of the approach of ADP, placed initially about 15 Å from the orthosteric binding site, to P2Y1R was studied with long classical MD simulations [99]. ADP was predicted to bind similarly to the antagonist MRS2500, in the extracellular vestibule, with the purine base undertaking a π-π interaction with Y3037.32, the ribose interacting with the same residue through a H-bond network that is mediated by water, and the pyrophosphate interacting through charge interactions with K411.41, K461.46, R195EL2, and R2876.38, consistent with SDM data [40,114].
The antagonist BPTU’s binding process from the bulk solvent to its extrahelical P2Y1R binding site was simulated through well-tempered metadynamics, and three stages were identified from the Free Energy Surface: the ligand entered the phospholipid bilayer, diffused in the bilayer, and interacted with I118ECL1 and F119ECL1 making contacts with ECL2, and finally reached the final position in the lipid-receptor binding site, with a RMSD < 2 Å from the crystallographic pose, with two single ligand H-bonds with L1022.55 [115].
3.4. Allostery
GPCRs inherently function as allosteric engines, because a conformational change in one location (extracellular) can affect the conformation at a remote location, enabling coupling to intracellular messengers. Different partners induce a population shift in the conformational equilibrium of GPCRs [116]. Unfortunately, structural information on GPCRs bound to allosteric modulators is limited, so the description of allostery from a structural perspective relies especially on MD simulations, which helps to fill a gap in this field.
GaMD recently investigated the binding of positive allosteric modulators (PAMs) to A1AR. The spontaneous binding of 2-amino-3-benzoylthiophenes, PD81723 and VCP171, was simulated in the presence of NECA that was bound to the orthosteric binding site [117]. Both PAMs bound a site in ECL2, consistent with mutagenesis data, and they were observed to stabilize the binding of NECA, especially through the formation of a salt bridge between E172ECL2 and K265ECL3. In fact, NECA explored a larger conformational space and it could also dissociate from the receptor in the absence of the PAM in the ECL2 cleft. The involvement of ECL2 in the binding of allosteric ligands seems promising for addressing the selectivity issue in AR drug discovery, with this receptor region showing low sequence identity in comparison to the TM bundle among the receptors and structural diversity in the experimental A1AR and A2AAR structures. Moreover, E172ECL2 appeared to be relevant in A1AR binding of both PD81723 and VCP171 in a previous study employing a receptor homology model, and that showed a pharmacological reduction of the allosteric ligand affinity for the E172ECL2A mutant receptor [118].
Locating the binding site of structurally diverse allosteric modulators, such as PAM LUF6000, has been challenging in most cases, even when SDM data are available. SuMD was employed to investigate the possible allosteric pathways of LUF6000 to A3AR in presence of adenosine bound to the orthosteric pocket. Two allosteric mechanisms were observed: binding to ECL2 with the induction of conformational changes able to stabilize adenosine deeper in the orthosteric pocket or capping the orthosteric pocket to form a ternary complex with adenosine and the receptor [112].
Among the well-known allosteric modulators of GPCRs, sodium is widely reported with its crystallographic conserved allosteric binding site among GPCRs [119]. An MD study was performed to compare the influence of the ion on an inactive and intermediate-active conformation of A2AAR in the presence of antagonist ZMA or the agonists UK432097 and NECA [120]. In the crystallographic ZMA-bound inactive structure, the ion was coordinated by D522.50, S913.39, and water molecules, mediating interactions with W2466.48, N2807.45, T883.36, and S2817.46. The ionic interaction with D522.50 was monitored and remained stable in all the ZMA-bound simulations, but two alternate states were observed for sodium interacting either with S913.39 or N2807.45. In the presence of sodium and/or the antagonist ZMA, the conformation of W2466.48 remained unaltered, while in the pseudo-apo receptor without sodium, a rotameric transition was observed for that residue (the already mentioned “toggle switch”), which seemed connected with a rotation of N2807.45. From the simulations of the intermediate-active state of A2AAR, bound to an agonist (UK432097 or NECA) or in the pseudo-apo form, binding of sodium and agonists were observed to be mutually exclusive: the ion was unstable, since it escaped from its binding site in some simulations or, in others, stayed in place, but a conformational change of TM7 moving apart from TM3, resembling the inactive state, occurred. Thus, the MD simulations were in agreement with experimental data highlighting the reduction of NECA and enhancement of ZMA binding in the presence of sodium, with a stabilization of the receptor inactive state. A study analyzed the effect of mutations of residues of the first and second sodium coordination shells, in particular, of D52A2.50, S91A3.39, W246A6.48, N280A7.45, and N284A7.49, by means of in vitro radioligand displacement studies and MD simulations [121]. In the case of the D52A2.50 mutant, sodium moved from its site to a pocket that formed by E131.39 and H2787.43, giving a possible explanation to the enhancement of NECA binding at high concentration of NaCl for this mutant receptor. Since sodium can bind to a different pocket, the binding of the ion and the agonist are not exclusive. S91A3.39, W246A6.48, N280A7.45, and N284A7.49 did not affect the ion mobility, but the interactions with the three coordinating residues were lowered during the simulations, and the mutations S91A3.39, W246A6.48, and N280A7.45 had a modest (or none in the case of N284A7.49) in vitro effect on NECA binding. SuMD simulations were performed in order to investigate the binding pathway of sodium from bulk solvent toward its allosteric pocket [122]. The ion reached the binding site of the pseudo-apo A2AAR coming either from an antagonist (ZMA)-bound structure or from an agonist (adenosine)-bound one, but, in the second case, a TM domain rearrangement was required, with TM2 and TM3 increasing their distance and TM7 moving outward. A contribution of W2466.48 was observed in mediating the final transition of the ion to the allosteric site during the binding pathway. Furthermore, SuMD enabled the investigation of adenosine recognition from the sodium-free intermediate-active A2AAR, but the simulation was unfruitful in most replicates of the sodium-bound inactive receptor.
In addition to the well characterized allosteric effect of sodium, a combination of 19F-NMR experiments and MD simulations showed that a high concentration of calcium and magnesium ions acts as A2AAR positive allosteric modulators. MD simulations in large excess of the ions highlighted the formation of stable cation-bridged interactions (E151ECL2-E161ECL2, E151ECL2-E169ECL2-D170ECL2, and D261ECL3-D170ECL2) with a consequent compaction of the receptor’s extracellular portion, which was allosterically propagated to an opening of the intracellular G protein-binding cavity. This would facilitate a receptor allosteric activation, further enhanced by the cations’ interaction with E2286.30 disfavoring the ionic lock [123].
Lipid allostery on A2AAR contributes to an active-like conformation, as shown using unbiased, long (µsec) MD simulations [124,125]. In addition, receptor-cholesterol interactions have been explored with MD simulations [126,127,128], including the observation of cholesterol entering the receptor, going beyond its solely allosteric effect [129].
3.5. Dissociation Process
In recent years, the paradigm of evaluating pharmacological activity in terms of thermodynamic binding parameters has been accompanied by a perspective on ligand residence time [130]. A longer residence time, i.e., slower dissociation rate, is thought to be predictive of in vivo efficacy. Thus, MD simulations recently investigated this phenomenon.
A method, called Smoothened Potential MD, has been proposed to estimate the relative koff ranking within a series of congeneric compounds with respect to a reference. Scaled potentials are applied in order to enhance the transition probability between different energy minima in MD simulations, without the need of CVs. This method was applied to a series of triazine-based antagonists in complex with the A2AAR, which were correctly ranked with a correlation coefficient of 0.95 [131].
In the same year, the aMetaD method was developed combining adiabatic-bias MD and well-tempered metadynamics to rapidly simulate dissociation events [132]. aMetaD evaluates the maximum energy (TS energy) bias that is needed to move a ligand from the starting energy basin to the other, giving a residence time score (RTscore). RTscore was able to discriminate the residence time difference between A2AAR-bound compounds 4e and 4a.
A recent dissociation study focusing on A2AAR developed a method employing ensemble-based steered MD (SMD) to determine the residence time on the basis of a different water-ligand interaction energy (ΔELW) between unbound and bound ligand states [133]. The calculated change in water-ligand interaction energy correlated well (R2 = 0.79) with the experimental residence time for 17 A2AAR ligands. While antagonist ZMA was extracted from the receptor, thirteen residues made intermediate contacts with the ligand during the dissociation pathway. Eight of these residues had been mutated into Ala in a previous study, and seven of them had kinetic and/or thermodynamic effects: I662.64A, S672.65A, K153ECL2A increased residence time, S156ECL2A and Q157ECL2A decreased the binding affinity, T2566.58A increased binding affinity and decreased residence time, and Y2717.36A prevented binding [134].
Other studies explored the dissociation process with a mechanistic perspective. Some GaMD simulations of NECA bound to the A1AR orthosteric site showed ligand dissociation in the absence of a PAM bound to ECL2, as anticipated in the section on allosteric modulation [117]. The dissociation process involved a pathway through ECL2 and ECL3.
In TAMD simulations accelerating the center of mass of ZMA out of A2AAR, contacts between the ligand and 16 residues in the upper receptor region were observed, especially in ECL2 and tip of helices TM2, TM6, and TM7 [134]. Twelve of those residues were mutated into Ala and, among them, E169ECL2A and Y2717.36A disrupted ligand binding, while two residue clusters affected the dissociation kinetics, with E169ECL2A, T2566.58A, and H264ECL3A increasing the dissociation rate, and I662.63A, S672.64A, and L2677.32A lowering it. These two clusters are topologically different; thus, a speculative interpretation of the binding process as multistep was provided. The first triad interacted with ZMA together with a structural water molecule in the initial system (like in the crystallographic structure with PDB code 4EIY [18]), and in the MD simulation the dissociation was preceded by the rupture of the interaction between E169ECL2 and H264ECL3 and by the loss of the triad-ligand interaction. An alternative binding intermediate during dissociation involved the ligand interaction with the second cluster, in a fashion that is similar to a different A2AAR-ZMA crystallographic structure, in which the E169ECL2-H264ECL3 bridge is broken (PDB code: 3PWH [15]).
The disruption of the salt bridge between E169ECL2 and H264ECL3 was also observed during the unbinding process of ZMA and LUF5452 from A2AAR through parallel tempering metadynamics simulations and, similarly, a bias had to be added to break the interaction between E172ECL2 and K265ECL3 to simulate the compounds’ dissociation from A1AR [135].
The influence of the E169ECL2-H264ECL3 salt bridge on the dissociation rate was also studied in order to rationalize the different A2AAR residence time of ZMA and four related ligands, whose X-ray structures showed a stabilizing interaction with H264ECL3 for compounds with longer residence time [19]. Metadynamics simulations were used to estimate the energy that is required to break the E169ECL2-H264ECL3 ionic bond, which was higher in the case of compounds with longer residence time.
Recently, the SuMD algorithm has been reversed and slightly modified to simulate ligand dissociation from its target [56]. The dissociation events of ZMA, adenosine, and NECA from A2AAR and adenosine from A1AR were simulated.
3.6. Water Contribution in Ligand-Binding
Water molecules have an important contribution in SBDD, together with the role they can play in modulating protein structure and function. In terms of contribution to ligand binding, in general, the binding site desolvation is one of the principal components of binding free energy, since water molecules are often entropically unfavored (limited degrees of freedom). However, there may or may not be an enthalpy compensation stabilizing the water molecule in the protein pocket, according to the protein environment. Water molecules can be classified as “happy” and “unhappy”, or respectively “cold” and “hot”, according to their stability on the protein surface as compared to the bulk solvent. The displacement of “happy” water molecules by a ligand is unfavored, with these molecules playing a structural role and mediating protein-ligand interactions; conversely, the displacement of “unhappy” water molecules, generally residing in hydrophobic environments, is favored, with consequent implications in SBDD [136,137].
Several computational methods have been developed for characterizing hydration sites on the protein surface. Among them, a method, called WaterMap, calculates the entropic and enthalpic components of water molecules through a statistical thermodynamic analysis of water clusters (hydration sites) obtained from MD simulations [138]. This tool was applied to characterize the hydration sites within the A2AAR binding site and analyze the SAR of a series of triazolylpurines that could not be explained by ligand-protein interactions, steric or desolvation effects. Small aliphatic substituents at the purine C2 position decreased the compound potency by occupying a region predicted to host stable water molecules. Longer aliphatic substituents increased the potency, because they extended to a region containing unstable waters, with a consequent compensation and favorable predicted binding free energy. The computed binding energies associated with water displacement correlated well with the experimental data [139].
Moreover, the identification and characterization of hydration sites is not merely an indicator for replacement, but it can be exploited to study possible water-mediated ligand–protein interactions. In fact, the inclusion of some explicit water molecules identified though WaterMap in a virtual screening applied to A2AAR increased the enrichment of active compounds over decoys [140].
An analysis of the water hotspots in the binding site of the crystallographic compounds T4E and T4G, together with a series of 5,6-diaryl-1,2,4-triazines having different residence time, was performed with a pipeline, including WaterMap [17,141]. A low residence time characterized compounds with a benzene ring at position 6. In these cases, a hydration site with “unhappy” water molecules was observed between TM2, TM3, and TM7 and stabilized by H2787.43. The benzene ring at position 6 pointed toward the hotspot, and this could explain the low residence time of these molecules. Compounds with a p-hydroxy-benzene ring at position 6 had just one water molecule in the hotspot, but with no “unhappy” character and stabilized by an interaction with the hydroxy group, which could explain their increased residence time (Figure 4). The H-bond network was also stabilized by compounds with a pyridine at position 6, being characterized by an increased ligand residence time.
A similar rationalization was provided while analyzing the water network in the A2AAR with two other tools, employing the fluid dynamics properties of water (RMSF < 1.4Å during 200 ps) to identify hydration hot spots in the protein binding site and subsequently computing the water residence time (occupancy) in each hotspot. A first version projected the residence time in a two-dimensional (2D)-grid, called Water Fluid Dynamics (WFD) map [142]. The WFD of a ZMA-bound A2AAR identified high occupancy hotspots in proximity of Y91.35, E131.39, and H2787.43 mediating interactions with the ligand triazolotriazine core, and in proximity of N2536.55 and E169ECL2 mediating interactions with the compound exocyclic nitrogen, in good agreement with crystallographic water molecules. The same water network was found in presence of NECA and overlapped with the one obtained from a pseudo-apo simulation. A similar algorithm, AquaMMapS, was developed to make three-dimensional (3D) maps of hydration sites on the basis of the fluid dynamics properties of water [143]. Simulations and analysis of ZMA-bound and pseudo-apo trajectories gave results similar to those that were obtained by WFD. The binding pocket region at the ligand-TM1 interface was populated by stationary water molecules also during a SuMD trajectory of ZMA approaching to the receptor.
Another work introduced an alternative method to investigate water hot spots on protein surfaces from MD simulations [144]. In this protocol, most persistent waters on the protein surface are identified and then SMD is used to force hydration site desolvation, with the aim to identify regions that are easily depleted of water molecules, which are potentially the most advantageous to be filled by the ligand. This method was validated using A2AAR and the aforementioned triazolotriazine test set and showed good agreement with experimental data.
In a work cited in the previous paragraph, we have described the use of atomistic ensemble-based steered molecular dynamics to study the dissociation of 17 ligands from A2AAR [133]. In that study, a change in the water-ligand interaction energy (ΔELW) between unbound and bound ligand states was computed during the simulations and correlated well with the compounds’ residence time, but not with their binding affinity. The rationale is that ligands with high ΔELW are badly hydrated in the bound state, so their hydrophilic interactions are buried, and this kind of interaction are known to increase the ligand residence time [145]. The derivatization of NECA with an arylalkyl extension at position C2 (CGS21680), and with an arylalkyl substituent at N6 in addition to an extended moiety at C2 (UK432,097) increased the residence time. Each of these substituents points toward the extracellular vestibule and they can prevent the entrance of water into the binding pocket.
A method combining SuMD, metadynamics, and suMetaD, enables the investigation of ligand binding and dissociation and retrieval of the transition state conformations of the two processes [146]. This algorithm was applied to A2AAR and six ligands, three xanthines (XAC, DPCPX, KW3902), and three triazolotriazines (ZMA, Z48, Z80), whose transition state thermodynamics appeared to be connected to the characteristics of water molecules solvating the binding pocket and the compounds. The transition state free energy was similar for all the compounds, but different enthalpic and entropic contributions were observed. In the case of binding, a transition state with ligand in the binding pocket was associated to a high enthalpic barrier (i.e., ZMA and Z80), due to the displacement of stable water molecules; while a transition state with ligand located in proximity of the vestibule was associated with a higher entropic barrier (i.e., XAC, KW3902, DPCPX, Z48), due to the water entrapped in the orthosteric site. Similarly, in the case of dissociation, transition states with high enthalpic barriers corresponded to a ligand in the orthosteric pocket (i.e., ZMA), while transition states with higher entropic barriers corresponded to all ligands in proximity to the vestibule.
4. Conclusions
The enhancement of computing power provided by new hardware and software technologies has been making MD simulations a helpful tool in molecular modeling and SBDD. The inclusion of protein flexibility and explicit solvent provides two advantages over molecular docking. Their inclusion enables the assessment of complex stability over time and can identify alternative binding mode(s) through induced fit phenomena, together with the interaction with structurally relevant water molecules. Moreover, the analysis of water molecules in explicit-solvent MD simulations might suggest chemical modification of ligands maximizing the replacement of thermodynamically unfavored water molecules and minimizing the replacement of favored waters.
New algorithms enhancing the classical MD simulations have enabled the exploration of long-timescale phenomena, like receptor activation, recognition, and dissociation pathways, filling the gap in experimental structural information necessary for investigating these processes at an atomistic level. This has implications in the drug discovery field, offering, for example, the possibility to rationally develop allosteric modulators or ligands focusing on optimized kinetics together with thermodynamic parameters.
Author Contributions
Wrote and corrected the manuscript: V.S., K.A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIDDK Intramural Research grant number ZIADK31126.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.-P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Chakraborty, S.; Warnick, E.; Crane, S.; Gao, Z.-G.; O’Connor, R.; Jacobson, K.A.; Carlsson, J. Structure-Based Screening of Uncharted Chemical Space for Atypical Adenosine Receptor Agonists. ACS Chem. Biol. 2016, 11, 2763–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lazim, R.; Macalino, S.J.Y.; Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 2019, 55, 147–153. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Delicado, E.G.; Gachet, C.; Kennedy, C.; von Kügelgen, I.; Li, B.; Miras-Portugal, M.T.; Novak, I.; Schöneberg, T.; Perez-Sen, R.; et al. Update of P2Y receptor pharmacology: IUPHAR Review 27. Br. J. Pharmacol. 2020, 177, 2413–2433. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signalling: Therapeutic developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, A.; Dayl, S.; Schmid, R. Homology modeling of P2X receptors. Methods Mol. Biol. 2020, 2041, 65–75. [Google Scholar]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.-G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on adenosine receptors as a potential targeted therapy in human diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- Batyuk, A.; Galli, L.; Ishchenko, A.; Han, G.W.; Gati, C.; Popov, P.A.; Lee, M.-Y.; Stauch, B.; White, T.A.; Barty, A.; et al. Native phasing of X-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2016, 2, e1600292. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Bachhawat, P.; Chu, M.L.-H.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, I.; Polovinkin, V.; Kovalev, K.; Gushchin, I.; Shevtsov, M.; Shevchenko, V.; Mishin, A.; Alekseev, A.; Rodriguez-Valera, F.; Borshchevskiy, V.; et al. Fast iodide-SAD phasing for high-throughput membrane protein structure determination. Sci. Adv. 2017, 3, e1602952. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Broecker, J.; Morizumi, T.; Ou, W.-L.; Klingel, V.; Kuo, A.; Kissick, D.J.; Ishchenko, A.; Lee, M.-Y.; Xu, S.; Makarov, O.; et al. High-throughput in situ X-ray screening of and data collection from protein crystals at room temperature and under cryogenic conditions. Nat. Protoc. 2018, 13, 260–292. [Google Scholar] [CrossRef]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef] [Green Version]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Doré, A.S. Towards high throughput GPCR crystallography: In Meso soaking of Adenosine A2A Receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Martin-Garcia, J.M.; Zhu, L.; Mendez, D.; Lee, M.-Y.; Chun, E.; Li, C.; Hu, H.; Subramanian, G.; Kissick, D.; Ogata, C.; et al. High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 2019, 6, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, Y.; Tono, K.; Tanaka, T.; Yamanaka, Y.; Nakane, T.; Mori, C.; Terakado Kimura, K.; Fujiwara, T.; Sugahara, M.; Tanaka, R.; et al. High-viscosity sample-injection device for serial femtosecond crystallography at atmospheric pressure. J. Appl. Crystallogr. 2019, 52, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.-G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebon, G.; Edwards, P.C.; Leslie, A.G.W.; Tate, C.G. Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol. Pharmacol. 2015, 87, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- García-Nafría, J.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7, 35946. [Google Scholar] [CrossRef]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [Green Version]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.-L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.-A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, Z.-G.; Zhang, K.; Kiselev, E.; Crane, S.; Wang, J.; Paoletta, S.; Yi, C.; Ma, L.; Zhang, W.; et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 2015, 520, 317–321. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, K.; Gao, Z.-G.; Paoletta, S.; Zhang, D.; Han, G.W.; Li, T.; Ma, L.; Zhang, W.; Müller, C.E.; et al. Agonist-bound structure of the human P2Y12 receptor. Nature 2014, 509, 119–122. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, J.; Gao, Z.-G.; Zhang, D.; Zhu, L.; Han, G.W.; Moss, S.M.; Paoletta, S.; Kiselev, E.; Lu, W.; et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 2014, 509, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Ciancetta, A.; Jacobson, K.A. Breakthrough in GPCR Crystallography and Its Impact on Computer-Aided Drug Design. Methods Mol. Biol. 2018, 1705, 45–72. [Google Scholar]
- Velgy, N.; Hedger, G.; Biggin, P.C. GPCRs: What Can We Learn from Molecular Dynamics Simulations? Methods Mol. Biol. 2018, 1705, 133–158. [Google Scholar]
- Shen, T.; Hamelberg, D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008, 129, 034103. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Bhattarai, A.; Miao, Y. Gaussian accelerated molecular dynamics for elucidation of drug pathways. Expert Opin. Drug Discov. 2018, 13, 1055–1065. [Google Scholar] [CrossRef]
- Bowman, G.R.; Ensign, D.L.; Pande, V.S. Enhanced modeling via network theory: Adaptive sampling of Markov state models. J. Chem. Theory Comput. 2010, 6, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef]
- Deganutti, G.; Moro, S.; Reynolds, C.A. A Supervised Molecular Dynamics Approach to Unbiased Ligand-Protein Unbinding. J. Chem. Inf. Model. 2020, 60, 1804–1817. [Google Scholar] [CrossRef]
- Lagarias, P.; Vrontaki, E.; Lambrinidis, G.; Stamatis, D.; Convertino, M.; Ortore, G.; Mavromoustakos, T.; Klotz, K.-N.; Kolocouris, A. Discovery of Novel Adenosine Receptor Antagonists through a Combined Structure- and Ligand-Based Approach Followed by Molecular Dynamics Investigation of Ligand Binding Mode. J. Chem. Inf. Model. 2018, 58, 794–815. [Google Scholar] [CrossRef]
- Sabbadin, D.; Ciancetta, A.; Moro, S. Bridging molecular docking to membrane molecular dynamics to investigate GPCR-ligand recognition: The human A₂A adenosine receptor as a key study. J. Chem. Inf. Model. 2014, 54, 169–183. [Google Scholar] [CrossRef]
- Tosh, D.K.; Rao, H.; Bitant, A.; Salmaso, V.; Mannes, P.; Lieberman, D.I.; Vaughan, K.L.; Mattison, J.A.; Rothwell, A.C.; Auchampach, J.A.; et al. Design and in Vivo Characterization of A1 Adenosine Receptor Agonists in the Native Ribose and Conformationally Constrained (N)-Methanocarba Series. J. Med. Chem. 2019, 62, 1502–1522. [Google Scholar] [CrossRef]
- Nguyen, A.T.N.; Baltos, J.-A.; Thomas, T.; Nguyen, T.D.; Muñoz, L.L.; Gregory, K.J.; White, P.J.; Sexton, P.M.; Christopoulos, A.; May, L.T. Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol. Pharmacol. 2016, 90, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Ciancetta, A.; Jacobson, K.A. Structural probing and molecular modeling of the A₃ adenosine receptor: A focus on agonist binding. Molecules 2017, 22, 449. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.A.; Barak, D.; Jacobson, K.A. Evaluation of homology modeling of G-protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J. Med. Chem. 2009, 52, 3284–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, P.K.; Chandrasekaran, B.; Mailavaram, R.; Tekade, R.K.; Jaber, A.M.Y. Molecular modeling approaches for the discovery of adenosine A2B receptor antagonists: Current status and future perspectives. Drug Discov. Today 2019, 24, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Gao, Z.-G.; Jeong, L.S.; Jacobson, K.A. Docking studies of agonists and antagonists suggest an activation pathway of the A3 adenosine receptor. J. Mol. Graph. Model. 2006, 25, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Floris, M.; Sabbadin, D.; Medda, R.; Bulfone, A.; Moro, S. Adenosiland: Walking through adenosine receptors landscape. Eur. J. Med. Chem. 2012, 58, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, E.; Moro, S. A comparison in the use of the crystallographic structure of the human A1 or the A2A adenosine receptors as a template for the construction of a homology model of the A3 subtype. Appl. Sci. 2019, 9, 821. [Google Scholar] [CrossRef] [Green Version]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Kachler, S.; Falgner, N.; Marucci, G.; Thomas, A.; Cristalli, G.; Volpini, R.; Klotz, K.-N. Different efficacy of adenosine and NECA derivatives at the human A3 adenosine receptor: Insight into the receptor activation switch. Biochem. Pharmacol. 2014, 87, 321–331. [Google Scholar] [CrossRef]
- Tosh, D.K.; Deflorian, F.; Phan, K.; Gao, Z.-G.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012, 55, 4847–4860. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Salmaso, V.; Rao, H.; Bitant, A.; Fisher, C.L.; Lieberman, D.I.; Vorbrüggen, H.; Reitman, M.L.; Gavrilova, O.; Gao, Z.-G.; et al. Truncated (N)-Methanocarba Nucleosides as Partial Agonists at Mouse and Human A3 Adenosine Receptors: Affinity Enhancement by N6-(2-Phenylethyl) Substitution. J. Med. Chem. 2020, 63, 4334–4348. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Chen, A.; Barak, D.; Kim, S.-K.; Müller, C.E.; Jacobson, K.A. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J. Biol. Chem. 2002, 277, 19056–19063. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, L.A.; Kellam, B.; Briddon, S.J.; Hill, S.J. Effect of a toggle switch mutation in TM6 of the human adenosine A₃ receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br. J. Pharmacol. 2014, 171, 3827–3844. [Google Scholar] [CrossRef] [PubMed]
- Stamatis, D.; Lagarias, P.; Barkan, K.; Vrontaki, E.; Ladds, G.; Kolocouris, A. Structural Characterization of Agonist Binding to an A3 Adenosine Receptor through Biomolecular Simulations and Mutagenesis Experiments. J. Med. Chem. 2019, 62, 8831–8846. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mannes, P.; Jung, Y.-H.; Ciancetta, A.; Bitant, A.; Lieberman, D.I.; Khaznadar, S.; Auchampach, J.A.; Gao, Z.-G.; Jacobson, K.A. Structure activity relationship of 2-arylalkynyl-adenine derivatives as human A3 adenosine receptor antagonists. Medchemcomm 2018, 9, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Salmaso, V.; Rao, H.; Campbell, R.; Bitant, A.; Gao, Z.-G.; Auchampach, J.A.; Jacobson, K.A. Direct Comparison of (N)-Methanocarba and Ribose-Containing 2-Arylalkynyladenosine Derivatives as A3 Receptor Agonists. ACS Med. Chem. Lett. 2020, in press. [Google Scholar] [CrossRef]
- Toti, K.S.; Jain, S.; Ciancetta, A.; Balasubramanian, R.; Chakraborty, S.; Surujdin, R.; Shi, Z.-D.; Jacobson, K.A. Pyrimidine Nucleotides Containing a (S)-Methanocarba Ring as P2Y6 Receptor Agonists. Medchemcomm 2017, 8, 1897–1908. [Google Scholar] [CrossRef]
- Ciancetta, A.; O’Connor, R.D.; Paoletta, S.; Jacobson, K.A. Demystifying P2Y1 Receptor Ligand Recognition through Docking and Molecular Dynamics Analyses. J. Chem. Inf. Model. 2017, 57, 3104–3123. [Google Scholar] [CrossRef]
- Trujillo, K.; Paoletta, S.; Kiselev, E.; Jacobson, K.A. Molecular modeling of the human P2Y14 receptor: A template for structure-based design of selective agonist ligands. Bioorg. Med. Chem. 2015, 23, 4056–4064. [Google Scholar] [CrossRef] [Green Version]
- Junker, A.; Balasubramanian, R.; Ciancetta, A.; Uliassi, E.; Kiselev, E.; Martiriggiano, C.; Trujillo, K.; Mtchedlidze, G.; Birdwell, L.; Brown, K.A.; et al. Structure-Based Design of 3-(4-Aryl-1H-1,2,3-triazol-1-yl)-Biphenyl Derivatives as P2Y14 Receptor Antagonists. J. Med. Chem. 2016, 59, 6149–6168. [Google Scholar] [CrossRef]
- Yu, J.; Ciancetta, A.; Dudas, S.; Duca, S.; Lottermoser, J.; Jacobson, K.A. Structure-Guided Modification of Heterocyclic Antagonists of the P2Y14 Receptor. J. Med. Chem. 2018, 61, 4860–4882. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420. [Google Scholar] [CrossRef]
- Jespers, W.; Oliveira, A.; Prieto-Díaz, R.; Majellaro, M.; Åqvist, J.; Sotelo, E.; Gutiérrez-de-Terán, H. Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors. Molecules 2017, 22, 1945. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, R.A. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Gutiérrez-de-Terán, H.; Åqvist, J. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, J.; Jespers, W.; Yaziji, V.; Mallo, A.; Majellaro, M.; Caamaño, O.; Loza, M.I.; Cadavid, M.I.; Brea, J.; Åqvist, J.; et al. Effect of Nitrogen Atom Substitution in A3 Adenosine Receptor Binding: N-(4,6-Diarylpyridin-2-yl)acetamides as Potent and Selective Antagonists. J. Med. Chem. 2017, 60, 7502–7511. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; IJzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Matricon, P.; Ranganathan, A.; Warnick, E.; Gao, Z.-G.; Rudling, A.; Lambertucci, C.; Marucci, G.; Ezzati, A.; Jaiteh, M.; Dal Ben, D.; et al. Fragment optimization for GPCRs by molecular dynamics free energy calculations: Probing druggable subpockets of the A2A adenosine receptor binding site. Sci. Rep. 2017, 7, 6398. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs--theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Piñeiro, Á.; Gutiérrez-de-Terán, H. Molecular dynamics simulations reveal insights into key structural elements of adenosine receptors. Biochemistry 2011, 50, 4194–4208. [Google Scholar] [CrossRef]
- Caliman, A.D.; Swift, S.E.; Wang, Y.; Miao, Y.; McCammon, J.A. Investigation of the conformational dynamics of the apo A2A adenosine receptor. Protein Sci. 2015, 24, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Landin, E.J.B.; Lovera, S.; de Fabritiis, G.; Kelm, S.; Mercier, J.; McMillan, D.; Sessions, R.B.; Taylor, R.J.; Sands, Z.A.; Joedicke, L.; et al. The Aminotriazole Antagonist Cmpd-1 Stabilises a Distinct Inactive State of the Adenosine 2A Receptor. Angew. Chem. Int. Ed. Engl. 2019, 58, 9399–9403. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Filipek, S.; Palczewski, K.; Vogel, H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat. Commun. 2014, 5, 4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. Engl. 2015, 54, 556–559. [Google Scholar] [PubMed]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-dependent activation and deactivation of the human adenosine A2A receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovera, S.; Cuzzolin, A.; Kelm, S.; De Fabritiis, G.; Sands, Z.A. Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci. Rep. 2019, 9, 14199. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Nivedha, A.K.; Tate, C.G.; Vaidehi, N. Dynamic role of the G protein in stabilizing the active state of the adenosine A2A receptor. Structure 2019, 27, 703–712.e3. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, A.; Wang, J.; Miao, Y. G-Protein-Coupled Receptor-Membrane Interactions Depend on the Receptor Activation State. J. Comput. Chem. 2020, 41, 460–471. [Google Scholar] [CrossRef]
- Ciancetta, A.; Rubio, P.; Lieberman, D.I.; Jacobson, K.A. A3 adenosine receptor activation mechanisms: Molecular dynamics analysis of inactive, active, and fully active states. J. Comput. Aided Mol. Des. 2019, 33, 983–996. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Vogel, H.; Filipek, S.; Stevens, R.C.; Palczewski, K. The molecular mechanism of P2Y1 receptor activation. Angew. Chem. Int. Ed. Engl. 2016, 55, 10331–10335. [Google Scholar] [CrossRef]
- Jiang, Q.; Guo, D.; Lee, B.X.; Van Rhee, A.M.; Kim, Y.C.; Nicholas, R.A.; Schachter, J.B.; Harden, T.K.; Jacobson, K.A. A mutational analysis of residues essential for ligand recognition at the human P2Y1 receptor. Mol. Pharmacol. 1997, 52, 499–507. [Google Scholar] [CrossRef]
- Neumann, A.; Müller, C.E.; Namasivayam, V. P2Y1-like nucleotide receptors—Structures, molecular modeling, mutagenesis, and oligomerization. In WIREs Computational Molecular Science; Wiley Periodicals, Inc.: Hoboken, NJ, USA, 2020. [Google Scholar] [CrossRef] [Green Version]
- Rafehi, M.; Neumann, A.; Baqi, Y.; Malik, E.M.; Wiese, M.; Namasivayam, V.; Müller, C.E. Molecular Recognition of Agonists and Antagonists by the Nucleotide-Activated G Protein-Coupled P2Y2 Receptor. J. Med. Chem. 2017, 60, 8425–8440. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, M.; Malik, E.M.; Neumann, A.; Abdelrahman, A.; Hanck, T.; Namasivayam, V.; Müller, C.E.; Baqi, Y. Development of Potent and Selective Antagonists for the UTP-Activated P2Y4 Receptor. J. Med. Chem. 2017, 60, 3020–3038. [Google Scholar] [CrossRef] [PubMed]
- Attah, I.Y.; Neumann, A.; Al-Hroub, H.; Rafehi, M.; Baqi, Y.; Namasivayam, V.; Müller, C.E. Ligand binding and activation of UTP-activated G protein-coupled P2Y2 and P2Y4 receptors elucidated by mutagenesis, pharmacological and computational studies. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129501. [Google Scholar] [CrossRef]
- Li, Y.; Yin, C.; Liu, P.; Li, D.; Lin, J. Identification of a Different Agonist-Binding Site and Activation Mechanism of the Human P2Y1 Receptor. Sci. Rep. 2017, 7, 13764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, N.J.; Ganotra, G.K.; Kokh, D.B.; Sadiq, S.K.; Wade, R.C. New approaches for computing ligand-receptor binding kinetics. Curr. Opin. Struct. Biol. 2018, 49, 1–10. [Google Scholar] [CrossRef]
- Cao, R.; Giorgetti, A.; Bauer, A.; Neumaier, B.; Rossetti, G.; Carloni, P. Role of extracellular loops and membrane lipids for ligand recognition in the neuronal adenosine receptor type 2A: An enhanced sampling simulation study. Molecules 2018, 23, 2616. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. Medchemcomm 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Deganutti, G.; Welihinda, A.; Moro, S. Comparison of the Human A2A Adenosine Receptor Recognition by Adenosine and Inosine: New Insight from Supervised Molecular Dynamics Simulations. ChemMedChem 2017, 12, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deganutti, G.; Salmaso, V.; Moro, S. Could Adenosine Recognize its Receptors with a Stoichiometry Other than 1:1? Mol. Inform. 2018, 37, e1800009. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Müller, C.E. A2A and A2B adenosine receptors: The extracellular loop 2 determines high (A2A) or low affinity (A2B) for adenosine. Biochem. Pharmacol. 2020, 172, 113718. [Google Scholar] [CrossRef]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Paoletta, S.; Sabbadin, D.; von Kügelgen, I.; Hinz, S.; Katritch, V.; Hoffmann, K.; Abdelrahman, A.; Straßburger, J.; Baqi, Y.; Zhao, Q.; et al. Modeling ligand recognition at the P2Y12 receptor in light of X-ray structural information. J. Comput. Aided Mol. Des. 2015, 29, 737–756. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Hoffmann, C.; Jacobson, K.A. Role of the extracellular loops of G protein-coupled receptors in ligand recognition: A molecular modeling study of the human P2Y1 receptor. Biochemistry 1999, 38, 3498–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Raniolo, S.; Limongelli, V.; Xu, Y. The Molecular Mechanism Underlying Ligand Binding to the Membrane-Embedded Site of a G-Protein-Coupled Receptor. J. Chem. Theory Comput. 2018, 14, 2761–2770. [Google Scholar] [CrossRef]
- Lamim Ribeiro, J.M.; Filizola, M. Allostery in G protein-coupled receptors investigated by molecular dynamics simulations. Curr. Opin. Struct. Biol. 2019, 55, 121–128. [Google Scholar] [CrossRef]
- Miao, Y.; Bhattarai, A.; Nguyen, A.T.N.; Christopoulos, A.; May, L.T. Structural basis for binding of allosteric drug leads in the adenosine A1 receptor. Sci. Rep. 2018, 8, 16836. [Google Scholar] [CrossRef]
- Nguyen, A.T.N.; Vecchio, E.A.; Thomas, T.; Nguyen, T.D.; Aurelio, L.; Scammells, P.J.; White, P.J.; Sexton, P.M.; Gregory, K.J.; May, L.T.; et al. Role of the second extracellular loop of the adenosine A1 receptor on allosteric modulator binding, signaling, and cooperativity. Mol. Pharmacol. 2016, 90, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-de-Terán, H.; Massink, A.; Rodríguez, D.; Liu, W.; Han, G.W.; Joseph, J.S.; Katritch, I.; Heitman, L.H.; Xia, L.; Ijzerman, A.P.; et al. The role of a sodium ion binding site in the allosteric modulation of the A2A adenosine G protein-coupled receptor. Structure 2013, 21, 2175–2185. [Google Scholar] [CrossRef] [Green Version]
- Massink, A.; Gutiérrez-de-Terán, H.; Lenselink, E.B.; Ortiz Zacarías, N.V.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; IJzerman, A.P. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, M.; Bolcato, G.; Deganutti, G.; Sturlese, M.; Moro, S. Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules 2019, 24, 2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomès, R.; García, A.E.; Ernst, O.P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, A.; Dalton, J.A.R.; Giraldo, J. Insights into adenosine A2A receptor activation through cooperative modulation of agonist and allosteric lipid interactions. PLoS Comput. Biol. 2020, 16, e1007818. [Google Scholar] [CrossRef]
- Song, W.; Yen, H.-Y.; Robinson, C.V.; Sansom, M.S.P. State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 2019, 27, 392–403.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Lyman, E. Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc. 2012, 134, 16512–16515. [Google Scholar] [CrossRef] [Green Version]
- Lyman, E.; Higgs, C.; Kim, B.; Lupyan, D.; Shelley, J.C.; Farid, R.; Voth, G.A. A role for a specific cholesterol interaction in stabilizing the Apo configuration of the human A2A adenosine receptor. Structure 2009, 17, 1660–1668. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Patel, R.; Lyman, E. Ligand-dependent cholesterol interactions with the human A2A adenosine receptor. Chem. Phys. Lipids 2013, 169, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Martí-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 14505. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef]
- Mollica, L.; Decherchi, S.; Zia, S.R.; Gaspari, R.; Cavalli, A.; Rocchia, W. Kinetics of protein-ligand unbinding via smoothed potential molecular dynamics simulations. Sci. Rep. 2015, 5, 11539. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, A.; Deflorian, F.; Weiss, D.R.; Mason, J.S. Decoding the Role of Water Dynamics in Ligand-Protein Unbinding: CRF1R as a Test Case. J. Chem. Inf. Model. 2015, 55, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Potterton, A.; Husseini, F.S.; Southey, M.W.Y.; Bodkin, M.J.; Heifetz, A.; Coveney, P.V.; Townsend-Nicholson, A. Ensemble-Based Steered Molecular Dynamics Predicts Relative Residence Time of A2A Receptor Binders. J. Chem. Theory Comput. 2019, 15, 3316–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Pan, A.C.; Dror, R.O.; Mocking, T.; Liu, R.; Heitman, L.H.; Shaw, D.E.; IJzerman, A.P. Molecular Basis of Ligand Dissociation from the Adenosine A2A Receptor. Mol. Pharmacol. 2016, 89, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Mattedi, G.; Deflorian, F.; Mason, J.S.; de Graaf, C.; Gervasio, F.L. Understanding ligand binding selectivity in a prototypical GPCR family. J. Chem. Inf. Model. 2019, 59, 2830–2836. [Google Scholar] [CrossRef]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Ahmed, M.H.; Bayden, A.S.; Cozzini, P.; Mozzarelli, A.; Kellogg, G.E. The roles of water in the protein matrix: A largely untapped resource for drug discovery. J. Med. Chem. 2017, 60, 6781–6827. [Google Scholar] [CrossRef]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, C.; Beuming, T.; Sherman, W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Lenselink, E.B.; Beuming, T.; Sherman, W.; van Vlijmen, H.W.T.; IJzerman, A.P. Selecting an optimal number of binding site waters to improve virtual screening enrichments against the adenosine A2A receptor. J. Chem. Inf. Model. 2014, 54, 1737–1746. [Google Scholar] [CrossRef]
- Bortolato, A.; Tehan, B.G.; Bodnarchuk, M.S.; Essex, J.W.; Mason, J.S. Water network perturbation in ligand binding: Adenosine A2A antagonists as a case study. J. Chem. Inf. Model. 2013, 53, 1700–1713. [Google Scholar] [CrossRef]
- Sabbadin, D.; Ciancetta, A.; Moro, S. Perturbation of fluid dynamics properties of water molecules during G protein-coupled receptor-ligand recognition: The human A2A adenosine receptor as a key study. J. Chem. Inf. Model. 2014, 54, 2846–2855. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Deganutti, G.; Salmaso, V.; Sturlese, M.; Moro, S. AquaMMapS: An Alternative Tool to Monitor the Role of Water Molecules During Protein-Ligand Association. ChemMedChem 2018, 13, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Zia, S.R.; Gaspari, R.; Decherchi, S.; Rocchia, W. Probing Hydration Patterns in Class-A GPCRs via Biased MD: The A2A Receptor. J. Chem. Theory Comput. 2016, 12, 6049–6061. [Google Scholar] [CrossRef]
- Schmidtke, P.; Luque, F.J.; Murray, J.B.; Barril, X. Shielded hydrogen bonds as structural determinants of binding kinetics: Application in drug design. J. Am. Chem. Soc. 2011, 133, 18903–18910. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Zhukov, A.; Deflorian, F.; Federico, S.; Spalluto, G.; Cooke, R.M.; Moro, S.; Mason, J.S.; Bortolato, A. Impact of protein-ligand solvation and desolvation on transition state thermodynamic properties of adenosine A2A ligand binding kinetics. Silico Pharmacol. 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Structures of ARs and P2YRs (both orthosteric and allosteric) ligands discussed.

Figure 2.
(A) cryogenic electron microscopy (cryo-EM) structure of the active-state A1AR (light green) bound to adenosine (gray) [39]. (B) X-ray structure of the intermediate-state A2AAR (light green) bound to adenosine (gray) [33]. (C) Superposition of the X-ray structures of MRS2500-bound and BPTU-bound P2Y1Rs [40]. MRS2500 is depicted by grays sticks and the corresponding receptor in light green, while BPTU is reported in orange sticks and the relative receptor in cyan. (D) Superposition of the X-ray structures of 2MeSADP-bound and AZD1283-bound P2Y12Rs [41,42]. 2MeSADP is depicted by gray sticks and the P2Y12R in light green, while AZD1283 is reported in orange sticks and the receptor in cyan.
Figure 2.
(A) cryogenic electron microscopy (cryo-EM) structure of the active-state A1AR (light green) bound to adenosine (gray) [39]. (B) X-ray structure of the intermediate-state A2AAR (light green) bound to adenosine (gray) [33]. (C) Superposition of the X-ray structures of MRS2500-bound and BPTU-bound P2Y1Rs [40]. MRS2500 is depicted by grays sticks and the corresponding receptor in light green, while BPTU is reported in orange sticks and the relative receptor in cyan. (D) Superposition of the X-ray structures of 2MeSADP-bound and AZD1283-bound P2Y12Rs [41,42]. 2MeSADP is depicted by gray sticks and the P2Y12R in light green, while AZD1283 is reported in orange sticks and the receptor in cyan.

Figure 3.
(A) Docking pose of MRS7334 (gray) [74] at an A3AR model (light green) [69]. (B) Docking pose of PPTN (gray) at a P2Y14R model (light green) [78].

Figure 4.
Superposition of the X-ray structures of A2A AR bound to T4E (receptor in light green and ligand in gray; PDB ID: 3UZC [17]) and T4G (receptor in cyan and ligand in orange; PDB ID: 3UZA [17]). The area of “unhappy” water molecules favorably displaced by the hydroxyl moiety of T4E is highlighted by a red circle.
Figure 4.
Superposition of the X-ray structures of A2A AR bound to T4E (receptor in light green and ligand in gray; PDB ID: 3UZC [17]) and T4G (receptor in cyan and ligand in orange; PDB ID: 3UZA [17]). The area of “unhappy” water molecules favorably displaced by the hydroxyl moiety of T4E is highlighted by a red circle.


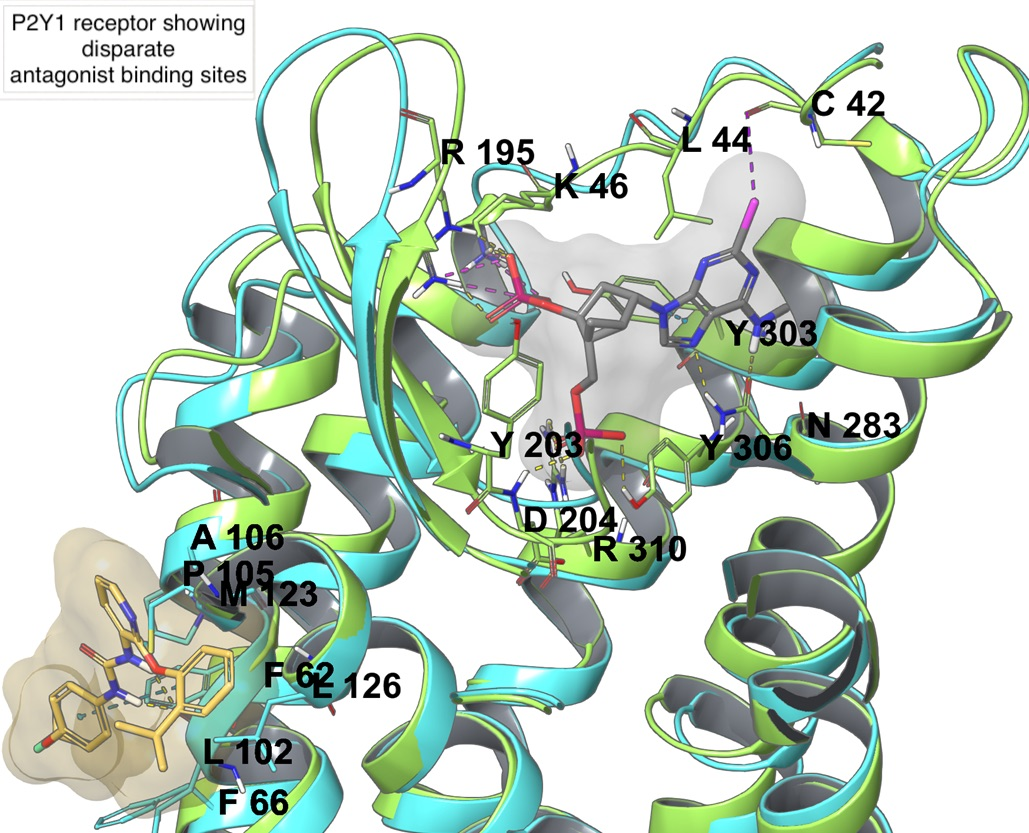{kind=link}
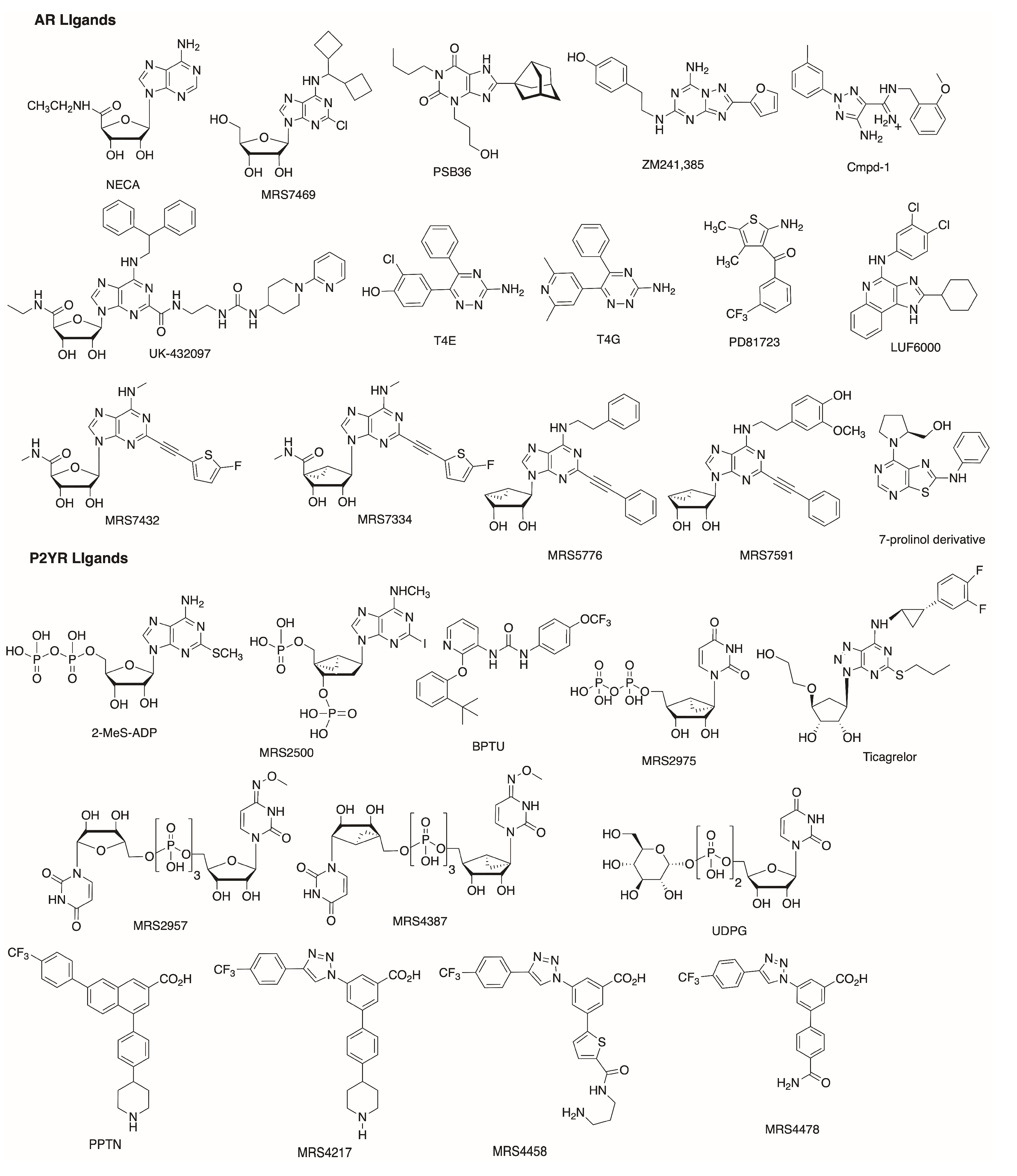{kind=link}
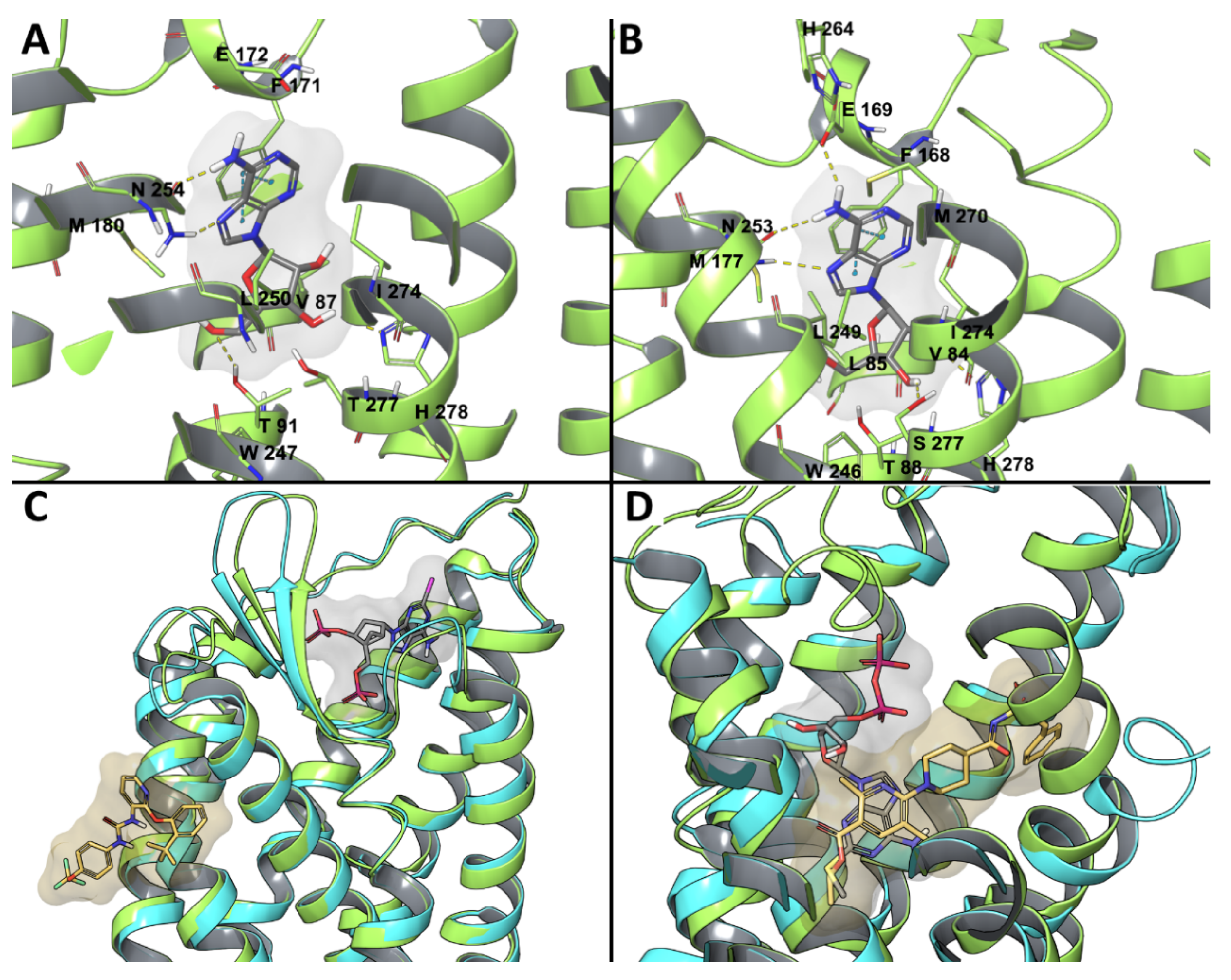{kind=link}
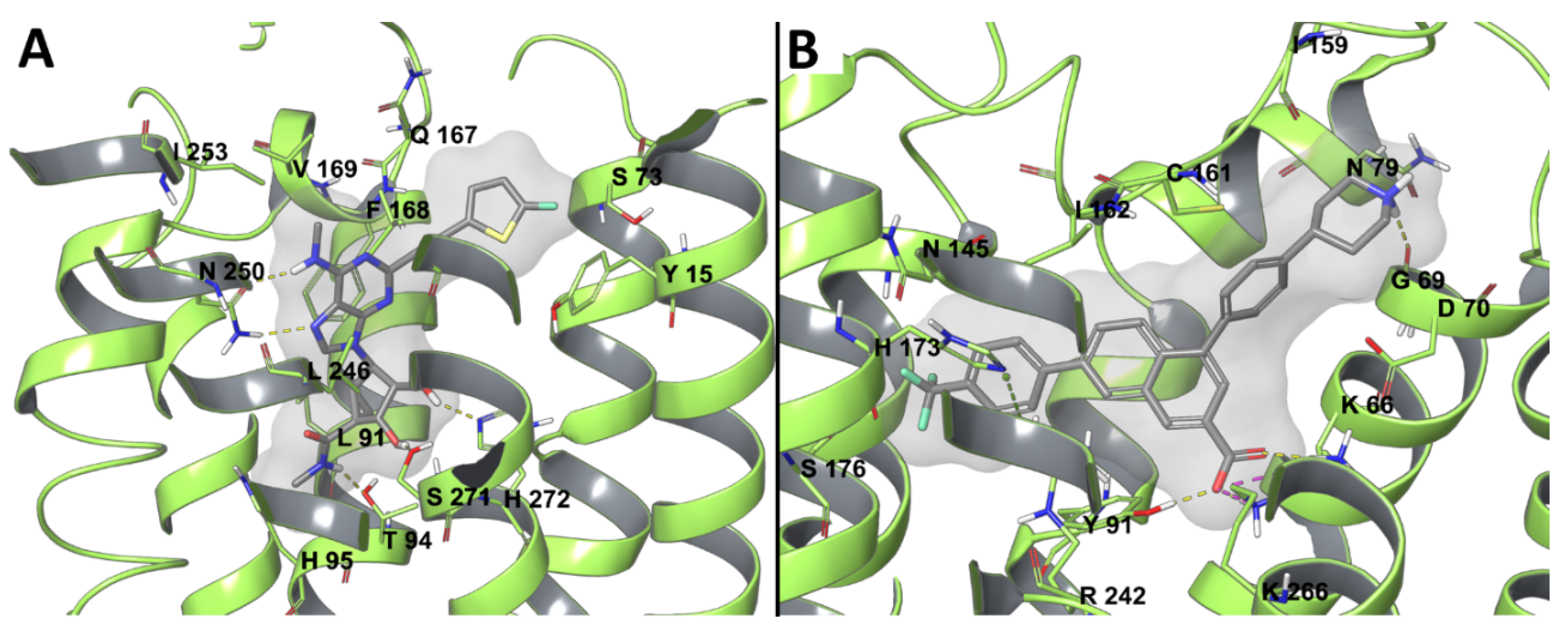{kind=link}
{kind=link}
Table 1.
Subtypes of adenosine receptors (ARs) and P2YRs and their characteristics.
| Receptor Class | Receptor Subtype | G Protein Types | Endogenous Agonists | |
|---|---|---|---|---|
| Adenosine Receptors (P1 Receptors) | A1 | Gi, Go | Adenosine | |
| A2A | Gs, Golf | Adenosine | ||
| A2B | Gs, Gq | Adenosine | ||
| A3 | Gi | Adenosine, Inosine | ||
| P2Y Receptors | P2Y1-like | P2Y1 | Gq | ADP |
| P2Y2 | Gq, Gi | UTP, ATP | ||
| P2Y4 | Gq, Gi | UTP, ATP, GTP | ||
| P2Y6 | Gq | UDP | ||
| P2Y11 | Gq, Gs | ATP | ||
| P2Y12-like | P2Y12 | Gi | ADP | |
| P2Y13 | Gi | ADP | ||
| P2Y14 | Gi | UDP-glucose, UDP, other UDP-sugars | ||
Table 2.
Reported AR and P2YR experimental structures (in the Protein Data Bank (PDB)) and their characteristics.
Table 2.
Reported AR and P2YR experimental structures (in the Protein Data Bank (PDB)) and their characteristics.
| Receptor | Activation State | Ligand | Technique | PDB ID [Reference] |
|---|---|---|---|---|
| Adenosine Receptors (ARs) | ||||
| A1AR | Inactive | PSB36 | X-ray | 5N2S [24] |
| DU172 (covalent) | X-ray | 5UEN [38] | ||
| Active (Gi) | Adenosine | Cryo-EM | 6D9H [39] | |
| A2AAR | Inactive | ZM241384 | X-ray | 3EML [14], 3PWH [15], 3VG9 [16], 3VGA [16], 5UVI [22], 5JTB [23], 6AQF [27], 6MH8 [29] |
| ZM241384, Na+ allosteric mod. | X-ray | 4EIY [18], 5IU4 [19], 5K2A [20], 5K2B [20], 5K2C [20], 5K2D [20], 5NLX [25], 5NM2 [25], 5NM4 [25], 5VRA [26], 5OLG [28], 6JZH [30], 6PS7 [31] | ||
| 12b, Na+ allosteric mod. | X-ray | 5IUA [19] | ||
| 12x, Na+ allosteric mod. | X-ray | 5IUB [19] | ||
| 12c, Na+ allosteric mod. | X-ray | 5IU7 [19] | ||
| 12f, Na+ allosteric mod. | X-ray | 5IU8 [19] | ||
| XAC | X-ray | 3REY [15] | ||
| Caffeine | X-ray | 3RFM [15] | ||
| Caffeine, Na+ allosteric mod. | X-ray | 5MZP [24] | ||
| Teophylline, Na+ allosteric mod. | X-ray | 5MZJ [24] | ||
| PSB36, Na+ allosteric mod. | X-ray | 5N2R [24] | ||
| T4G | X-ray | 3UZA [17] | ||
| T4E | X-ray | 3UZC [17] | ||
| T4E, Na+ allosteric mod. | X-ray | 5OLZ [28], 5OM1 [28], 5OM4 [28] | ||
| Tozadenant, Na+ allosteric mod. | X-ray | 5OLO [28] | ||
| Cmpd-1 | X-ray | 5UIG [21] | ||
| LUAA47070, Na+ allosteric mod. | X-ray | 5OLV [28] | ||
| Vipadenant, Na+ allosteric mod. | X-ray | 5OLH [28] | ||
| AZD4635, Na+ allosteric mod. | X-ray | 6GT3 | ||
| Intermediate | UK-432097 | X-ray | 3QAK [32], 5WF5 [35], 5WF6 [35] | |
| Adenosine | X-ray | 2YDO [33] | ||
| NECA | X-ray | 2YDV [33] | ||
| CGS21680 | X-ray | 4UG2 [34], 4UHR [34] | ||
| Active (bound to mini-Gs) | NECA | X-ray | 5G53 [36] | |
| NECA | Cryo-EM | 6GDG [37] | ||
| P2Y Receptors (P2YRs) | ||||
| P2Y1R | Inactive | MRS2500 | X-ray | 4XNV [40] |
| BPTU | X-ray | 4XNW [40] | ||
| P2Y12R | Inactive | AZD1283 | X-ray | 4NTJ [42] |
| Intermediate | 2MeSADP | X-ray | 4PXZ [41] | |
| 2MeSATP | X-ray | 4PY0 [41] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Salmaso, V.; Jacobson, K.A. In Silico Drug Design for Purinergic GPCRs: Overview on Molecular Dynamics Applied to Adenosine and P2Y Receptors. Biomolecules 2020, 10, 812. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060812
AMA Style
Salmaso V, Jacobson KA. In Silico Drug Design for Purinergic GPCRs: Overview on Molecular Dynamics Applied to Adenosine and P2Y Receptors. Biomolecules. 2020; 10(6):812. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060812
Chicago/Turabian StyleSalmaso, Veronica, and Kenneth A. Jacobson. 2020. "In Silico Drug Design for Purinergic GPCRs: Overview on Molecular Dynamics Applied to Adenosine and P2Y Receptors" Biomolecules 10, no. 6: 812. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060812
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.