MYB Superfamily in Brassica napus: Evidence for Hormone-Mediated Expression Profiles, Large Expansion, and Functions in Root Hair Development

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval of MYB Genes
2.2. Sequence and Phylogenetic Analyses
2.3. Chromosomal Locations and Collinearity of MYB Genes in B. napus
2.4. Spatial and Hormone-Induced Expression of B. napus MYB Genes
2.5. Cloning of B. napus WER-Homologous Genes
2.6. Plasmid Construction and Transformation
2.7. Growth and Phenotypic Evaluation of Transgenic Plants
3. Results and Discussion
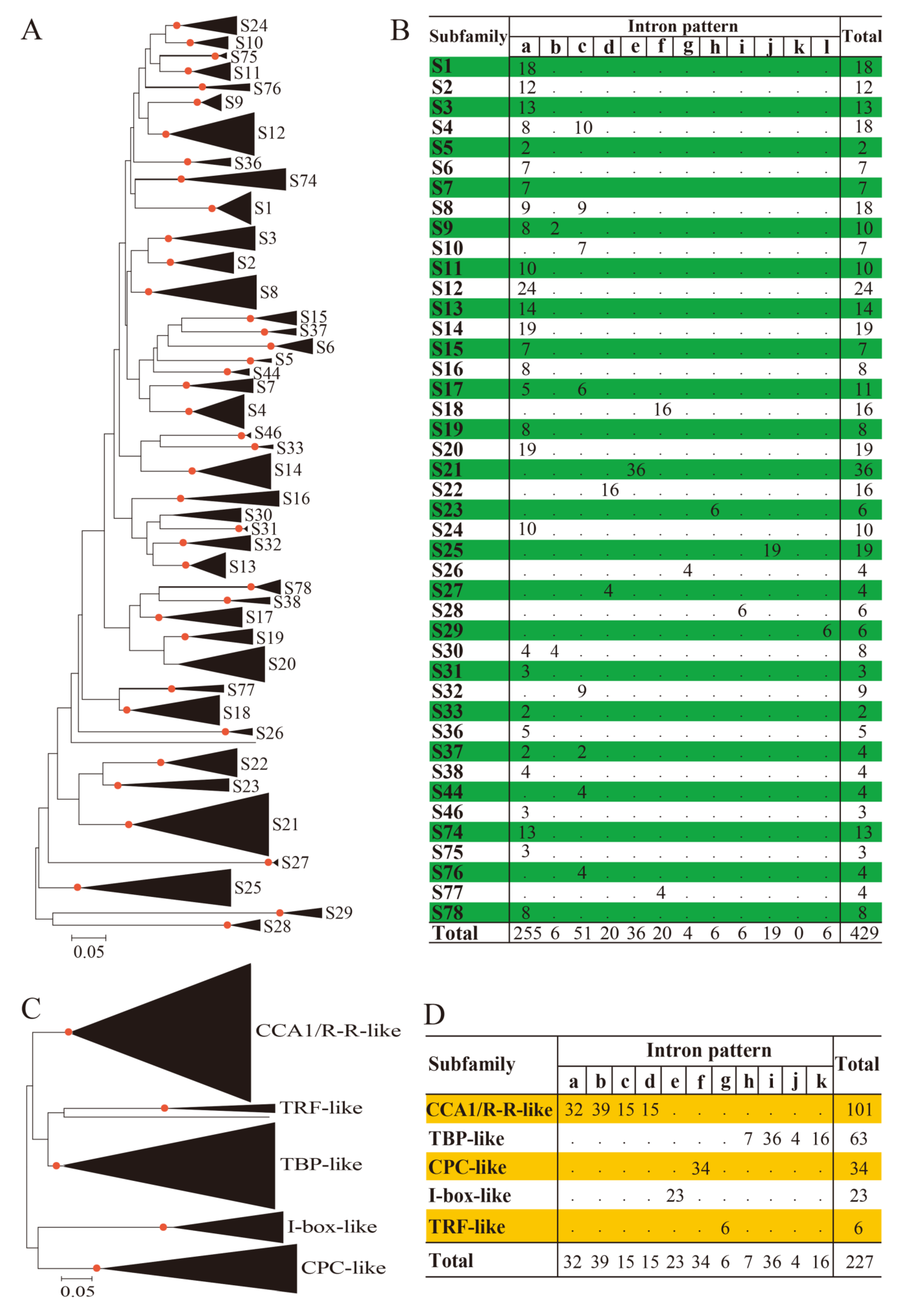
3.1. Identification and Phylogenetic Analysis of BnMYB Proteins
3.2. Diversification of 2R-MYB Genes in Higher Plants
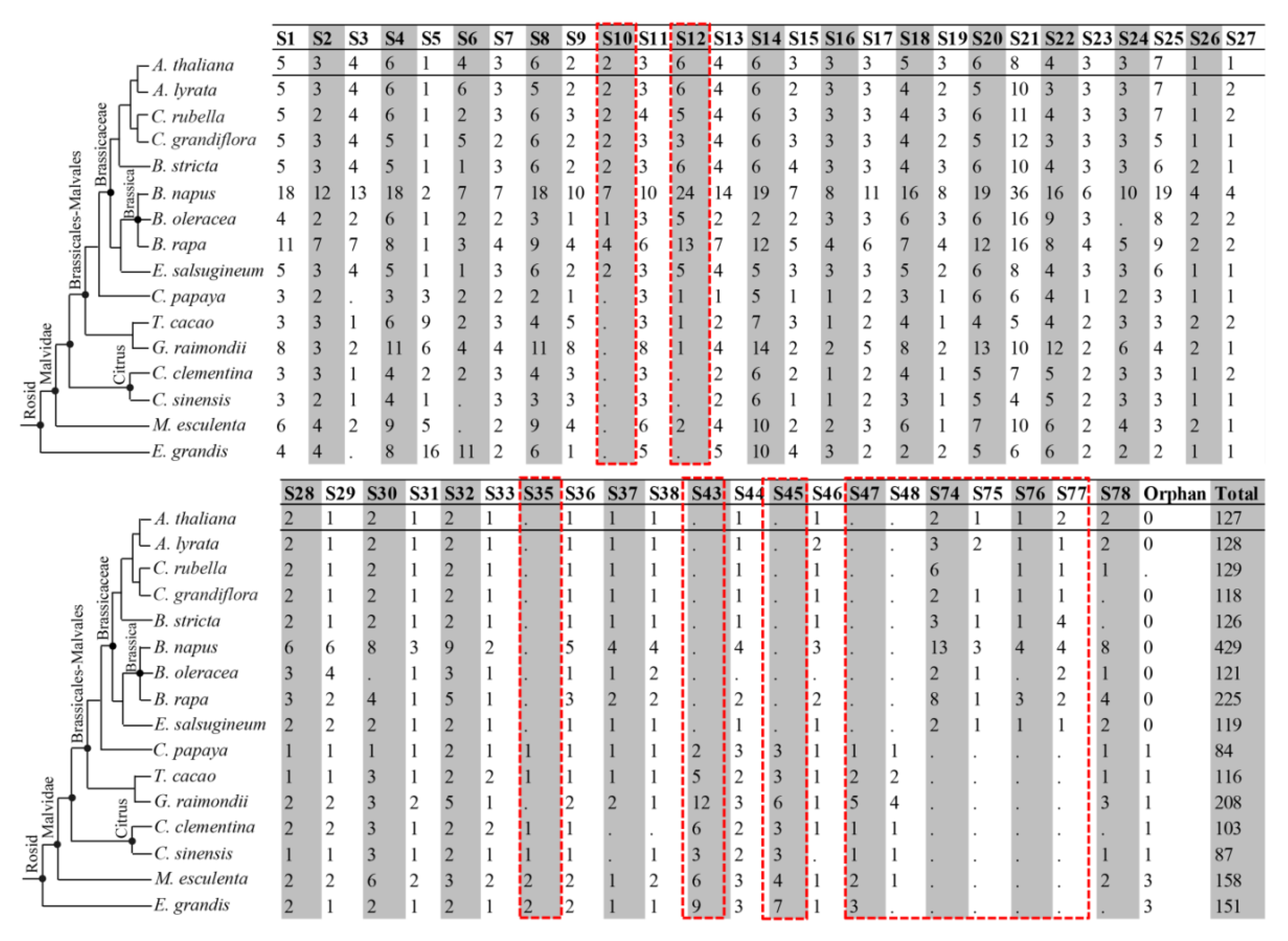
3.3. Chromosomal Distribution and Expansion of the MYB Members in B. napus
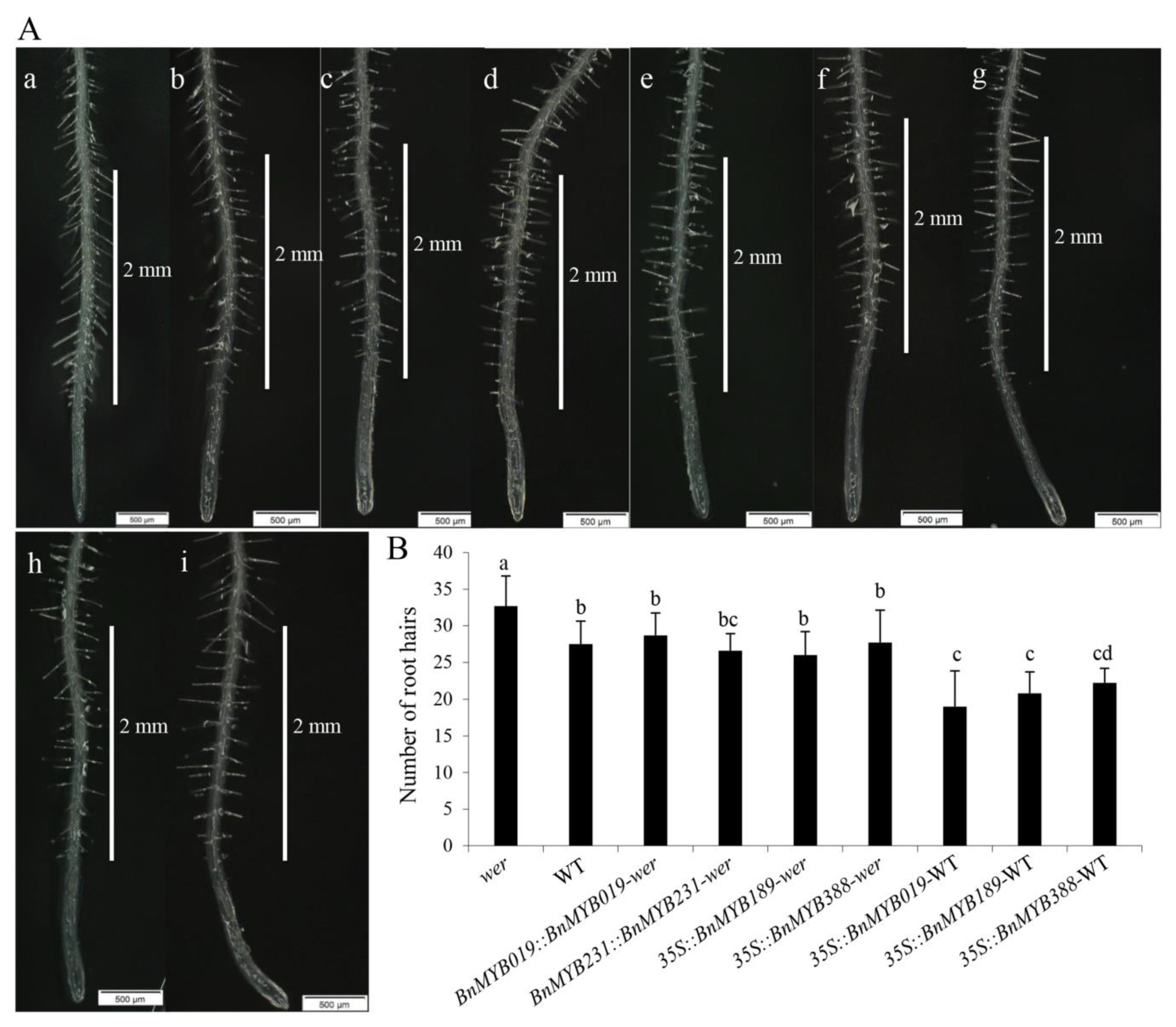
3.4. Conserved Functions of B. napus WER Homologs in Root Hair Development in A. thaliana
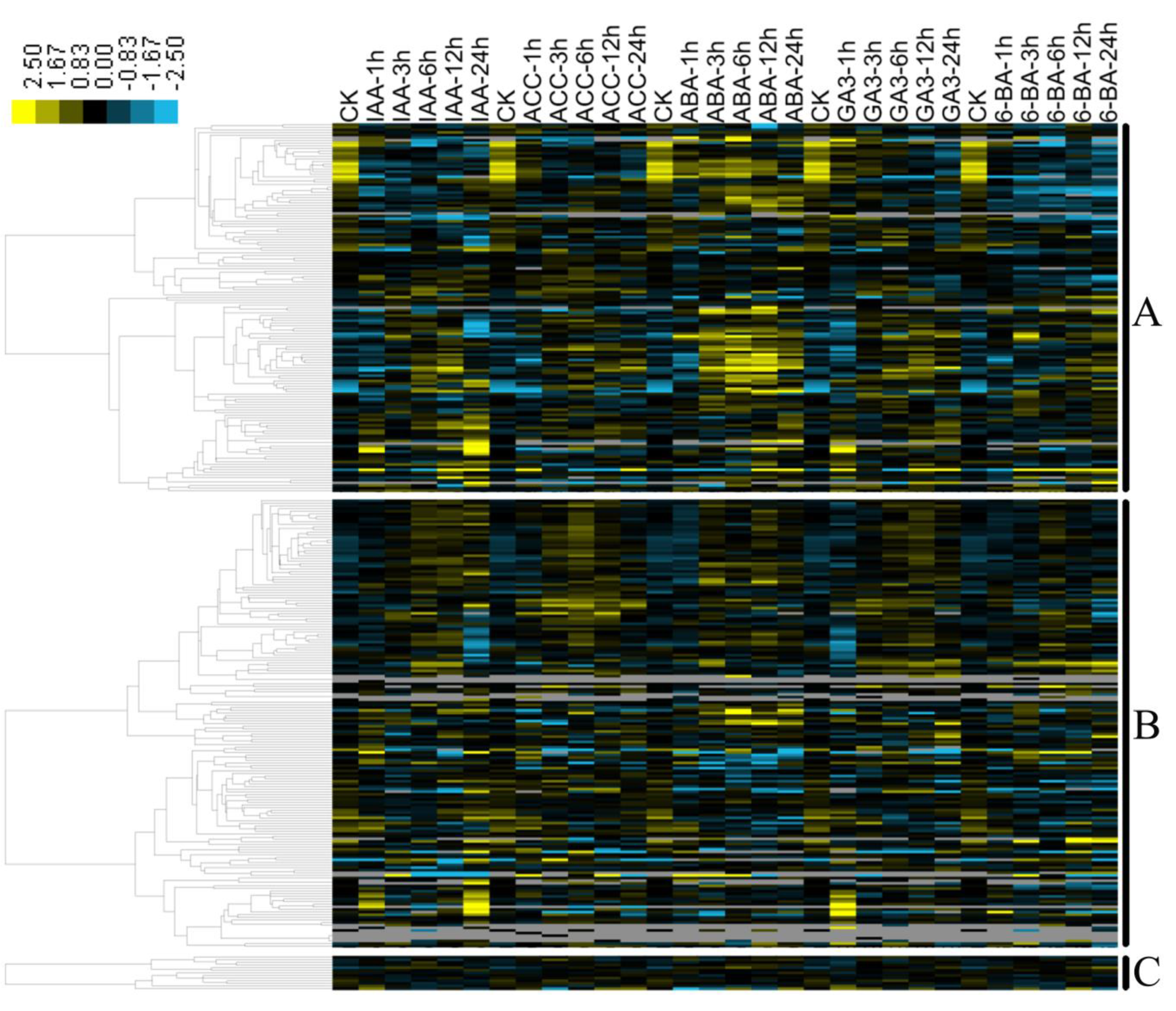
3.5. Hormone-Induced Expression Profiling of B. napus MYB Genes by RNA-Seq
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martin, C.; Paz-Ares, J. MYB transcription factors in plants. Trends Genet. 1997, 13, 67–73. [Google Scholar] [CrossRef]
- Lipsick, J.S. One billion years of Myb. Oncogene 1996, 13, 223–235. [Google Scholar]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Du, H.; Wang, Y.B.; Xie, Y.; Liang, Z.; Jiang, S.J.; Zhang, S.S.; Huang, Y.B.; Tang, Y.X. Genome-wide identification and evolutionary and expression analyses of MYB-related genes in land plants. DNA Res. 2013, 20, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Liang, Z.; Zhao, S.; Nan, M.G.; Tran, L.S.; Lu, K.; Huang, Y.B.; Li, J.N. The Evolutionary History of R2R3-MYB Proteins across 50 Eukaryotes: New Insights into Subfamily Classification and Expansion. Sci. Rep. 2015, 5, 11037. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yang, S.S.; Liang, Z.; Feng, B.R.; Liu, L.; Huang, Y.B.; Tang, Y.X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, W.; Gao, J.; Yin, K.; Wang, R.; Wang, C.; Petersen, M.; Mundy, J.; Qiu, J.L. MYB75 Phosphorylation by MPK4 Is Required for Light-Induced Anthocyanin Accumulation in Arabidopsis. Plant Cell 2016, 28, 2866–2883. [Google Scholar] [CrossRef] [Green Version]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, N.W.; Lewis, D.H.; Zhang, H.; Schwinn, K.E.; Jameson, P.E.; Davies, K.M. Members of an R2R3-MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning. Plant J. 2011, 65, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.M.; Schiefelbein, J. WEREWOLF, a MYB-Related Protein in Arabidopsis, Is a Position-Dependent Regulator of Epidermal Cell Patterning. Cell 1999, 99, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Nguyen, N.H.; Jeong, C.Y.; Nguyen, N.T.; Hong, S.W.; Lee, H. Loss of the R2R3 MYB, AtMyb73, causes hyper-induction of the SOS1 and SOS3 genes in response to high salinity in Arabidopsis. J. Plant Physiol. 2013, 170, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, R.; Ju, Q.; Li, W.; Tran, L.P.; Xu, J. The R2R3-MYB Transcription Factor MYB49 Regulates Cadmium Accumulation. Plant Physiol. 2019, 180, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Seo, J.S.; Han, S.W.; Koo, Y.J.; Kim, C.H.; Song, S.I.; Nahm, B.H.; Choi, Y.D.; Cheong, J.J. Overexpression of AtMYB44 enhances stomatal closure to confer abiotic stress tolerance in transgenic Arabidopsis. Plant Physiol. 2008, 146, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Zheng, Y.; Guo, Y. MYB30 transcription factor regulates oxidative and heat stress responses through ANNEXIN-mediated cytosolic calcium signaling in Arabidopsis. New Phytol. 2017, 216, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, D.H.; Doherty, C.J.; Pruneda-Paz, J.L.; Schmitz, R.J.; Ecker, J.R.; Kay, S.A. Genome-wide identification of CCA1 targets uncovers an expanded clock network in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, E4802–E4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; Lee, H. MYB-related transcription factors function as regulators of the circadian clock and anthocyanin biosynthesis in Arabidopsis. Plant Signal. Behav. 2016, 11, e1139278. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Tachibana, T.; Shimura, Y.; Okada, K. Epidermal cell differentiation in Arabidopsis determined by a Myb homolog, CPC. Science 1997, 277, 1113–1116. [Google Scholar] [CrossRef]
- Pant, B.D.; Burgos, A.; Pant, P.; Cuadros-Inostroza, A.; Willmitzer, L.; Scheible, W.R. The transcription factor PHR1 regulates lipid remodeling and triacylglycerol accumulation in Arabidopsis thaliana during phosphorus starvation. J. Exp. Bot. 2015, 66, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Ito, M. Conservation and diversification of three-repeat Myb transcription factors in plants. J. Plant Res. 2005, 118, 61–69. [Google Scholar] [CrossRef]
- Haga, N.; Kobayashi, K.; Suzuki, T.; Maeo, K.; Kubo, M.; Ohtani, M.; Mitsuda, N.; Demura, T.; Nakamura, K.; Jurgens, G.; et al. Mutations in MYB3R1 and MYB3R4 cause pleiotropic developmental defects and preferential down-regulation of multiple G2/M-specific genes in Arabidopsis. Plant Physiol. 2011, 157, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Xu, Y.; Ma, Q.; Xu, W.; Wang, T.; Xue, Y.; Chong, K. Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol. 2007, 143, 1739–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ma, R.; Xu, J.; Yan, J.; Guo, L.; Song, J.; Feng, R.; Yu, M. Genome-wide identification and classification of MYB superfamily genes in peach. PLoS ONE 2018, 13, e0199192. [Google Scholar] [CrossRef] [PubMed]
- Qing, J.; Dawei, W.; Jun, Z.; Yulan, X.; Bingqi, S.; Fan, Z. Genome-wide characterization and expression analyses of the MYB superfamily genes during developmental stages in Chinese jujube. PeerJ 2019, 7, e6353. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro Protein Families Database: The Classification Resource After 15 Years. Nucleic. Acids. Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef]
- Letunic, I.; Copley, R.R.; Schmidt, S.; Ciccarelli, F.D.; Doerks, T.; Schultz, J.; Ponting, C.P.; Bork, P. SMART 4.0: Towards genomic data integration. Nucleic. Acids. Res. 2004, 32, D142–D144. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic. Acids. Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjellqvist, B.; Hughes, G.J.; Pasquali, C.; Paquet, N.; Ravier, F.; Sanchez, J.C.; Frutiger, S.; Hochstrasser, D. The Focusing Positions of Polypeptides in Immobilized Ph Gradients Can Be Predicted from Their Amino-Acid-Sequences. Electrophoresis 1993, 14, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, E.; Pedersen, B.; Kane, J.; Alam, M.; Ming, R.; Tang, H.; Wang, X.; Bowers, J.; Paterson, A.; Lisch, D. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar and grape: CoGe with rosids. Plant Physiol. 2008, 148, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, A.J. Java Treeview--extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Xie, T.; Chen, C.; Luan, A.; Long, J.; Li, C.; Ding, Y.; He, Y. Genome-wide organization and expression profiling of the R2R3-MYB transcription factor family in pineapple (Ananas comosus). BMC Genom. 2017, 18, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Guo, C.; Ahmad, S.; Wang, Q.; Yu, J.; Liu, C.; Guo, Y. Systematic Analysis of MYB Family Genes in Potato and Their Multiple Roles in Development and Stress Responses. Biomolecules 2019, 9, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Yu, J.; Zhao, T.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. Dissecting the Genome-Wide Evolution and Function of R2R3-MYB Transcription Factor Family in Rosa chinensis. Genes 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mmadi, M.A.; Dossa, K.; Wang, L.; Zhou, R.; Wang, Y.; Cisse, N.; Sy, M.O.; Zhang, X. Functional Characterization of the Versatile MYB Gene Family Uncovered Their Important Roles in Plant Development and Responses to Drought and Waterlogging in Sesame. Genes 2017, 8, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yu, W.; Zhang, X.; Wang, G.; Cao, F.; Cheng, H. Identification and expression analysis under abiotic stress of the R2R3-MYB genes in Ginkgo biloba L. Physiol. Mol. Biol. Plants 2017, 23, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Cheng, Z.; Xie, L.; Li, X.; Li, J.; Mu, S.; Gao, J. The R2R3 MYB Gene Family in Phyllostachys edulis: Genome-Wide Analysis and Identification of Stress or Development-Related R2R3MYBs. Front. Plant Sci. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, M.; Camargo, E.L.; Carocha, V.; Cassan-Wang, H.; Clemente, H.S.; Savelli, B.; Hefer, C.A.; Paiva, J.A.; Myburg, A.A.; Grima-Pettenati, J. The Eucalyptus grandis R2R3-MYB transcription factor family: Evidence for woody growth-related evolution and function. New Phytol. 2015, 206, 1364–1377. [Google Scholar] [CrossRef]
- Bailey, P.C.; Dicks, J.; Wang, T.L.; Martin, C. IT3F: A web-based tool for functional analysis of transcription factors in plants. Phytochemistry 2008, 69, 2417–2425. [Google Scholar] [CrossRef]
- Sakaoka, S.; Mabuchi, K.; Morikami, A.; Tsukagoshi, H. MYB30 regulates root cell elongation under abscisic acid signaling. Commun. Integr. Biol. 2018, 11, e1526604. [Google Scholar] [CrossRef]
- Oh, J.E.; Kwon, Y.; Kim, J.H.; Noh, H.; Hong, S.W.; Lee, H. A dual role for MYB60 in stomatal regulation and root growth of Arabidopsis thaliana under drought stress. Plant Mol. Biol. 2011, 77, 91–103. [Google Scholar] [CrossRef]
- Liberman, L.M.; Sparks, E.E.; Moreno-Risueno, M.A.; Petricka, J.J.; Benfey, P.N. MYB36 regulates the transition from proliferation to differentiation in the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2015, 29, 12099–12104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, K.H.; Kang, Y.H.; Park, Y.H.; Hwang, I.; Schiefelbein, J.; Lee, M.M. The WEREWOLF MYB protein directly regulates CAPRICE transcription during cell fate specification in the Arabidopsis root epidermis. Development 2005, 132, 4765–4775. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Liu, Z.; Dai, X.; Xiang, F. Primary root growth in Arabidopsis thaliana is inhibited by the miR159 mediated repression of MYB33, MYB65 and MYB101. Plant Sci. 2017, 262, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Shin, R.; Burch, A.Y.; Huppert, K.A.; Tiwari, S.B.; Murphy, A.S.; Guilfoyle, T.J.; Schachtman, D.P. The Arabidopsis transcription factor MYB77 modulates auxin signal transduction. Plant Cell 2007, 19, 2440–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, D.J.; Voss, U.; Harding, S.A.; Fannon, J.; Moody, L.A.; Yamada, E.; Swarup, K.; Nibau, C.; Bassel, G.W.; Choudhary, A.; et al. AtMYB93 is a novel negative regulator of lateral root development in Arabidopsis. New Phytol. 2014, 203, 1194–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Z.; Yang, K.Z.; Zou, J.J.; Zhu, L.L.; Xie, Z.D.; Morita, M.T.; Tasaka, M.; Friml, J.; Grotewold, E.; Beeckman, T.; et al. Transcriptional regulation of PIN genes by FOUR LIPS and MYB88 during Arabidopsis root gravitropism. Nat. Commun. 2015, 6, 8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, R.L.; Cao, Y.R.; Liu, Y.F.; Lei, G.; Zou, H.F.; Liao, Y.; Wang, H.W.; Zhang, W.K.; Ma, B.; Du, J.Z.; et al. An R2R3-type transcription factor gene AtMYB59 regulates root growth and cell cycle progression in Arabidopsis. Cell Res. 2009, 19, 1291–1304. [Google Scholar] [CrossRef] [Green Version]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, J.; Du, M.L.; Li, L.; Wang, X.L.; Li, X.B. A cotton gene encoding MYB-like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant Cell Physiol. 2013, 54, 893–906. [Google Scholar] [CrossRef]
- Murray, F.; Kalla, R.; Jacobsen, J.; Gubler, F. A role for HvGAMYB in another development. Plant J. 2003, 33, 481–491. [Google Scholar] [CrossRef]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The Jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect Jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandaokar, A.; Browse, J. MYB108 acts together with MYB24 to regulate jasmonate-mediated stamen maturation in Arabidopsis. Plant Physiol. 2009, 149, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.G.; Mas, P.; Seo, P.J. MYB96 shapes the circadian gating of ABA signaling in Arabidopsis. Sci. Rep. 2016, 6, 17754. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Chen, P.; Liang, T.; Li, X.; Liu, H. UV-B photoreceptor UVR8 interacts with MYB73/MYB77 to regulate auxin responses and lateral root development. EMBO J. 2020, 39, e101928. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Wen, J.; Chen, P.; Guo, P.; Ke, Y.; Wang, M.; Liu, M.; Tran, L.-S.P.; Li, J.; Du, H. MYB Superfamily in Brassica napus: Evidence for Hormone-Mediated Expression Profiles, Large Expansion, and Functions in Root Hair Development. Biomolecules 2020, 10, 875. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060875
Li P, Wen J, Chen P, Guo P, Ke Y, Wang M, Liu M, Tran L-SP, Li J, Du H. MYB Superfamily in Brassica napus: Evidence for Hormone-Mediated Expression Profiles, Large Expansion, and Functions in Root Hair Development. Biomolecules. 2020; 10(6):875. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060875
Chicago/Turabian StyleLi, Pengfeng, Jing Wen, Ping Chen, Pengcheng Guo, Yunzhuo Ke, Mangmang Wang, Mingming Liu, Lam-Son Phan Tran, Jiana Li, and Hai Du. 2020. "MYB Superfamily in Brassica napus: Evidence for Hormone-Mediated Expression Profiles, Large Expansion, and Functions in Root Hair Development" Biomolecules 10, no. 6: 875. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060875