
Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology

 , , ,
, , ,  and
and 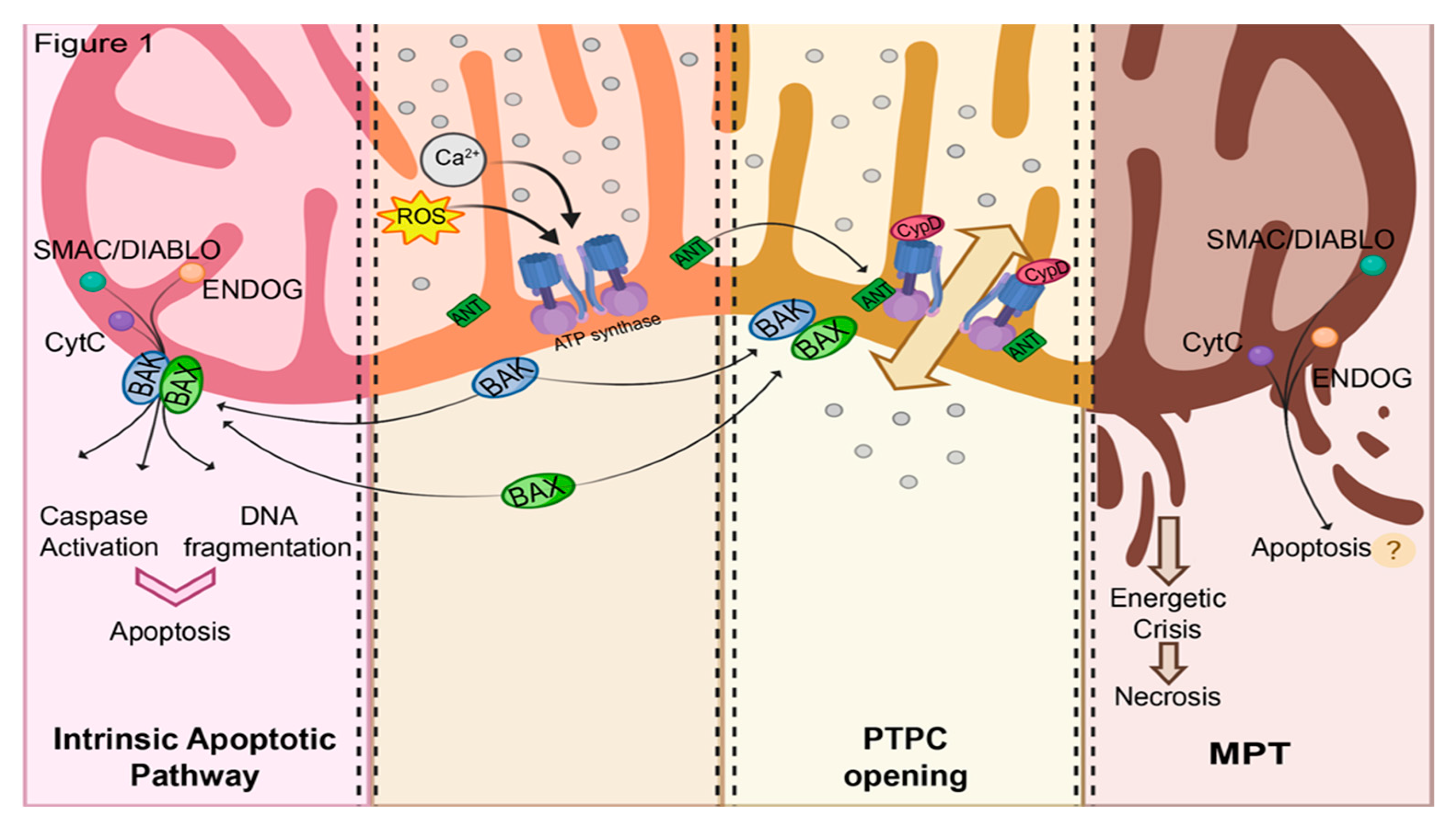{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Mitochondrial Routes of Cell Death
1.2. Current Hypotheses on PTPC Structure
1.3. Involvement of MPT in RCD Subroutines
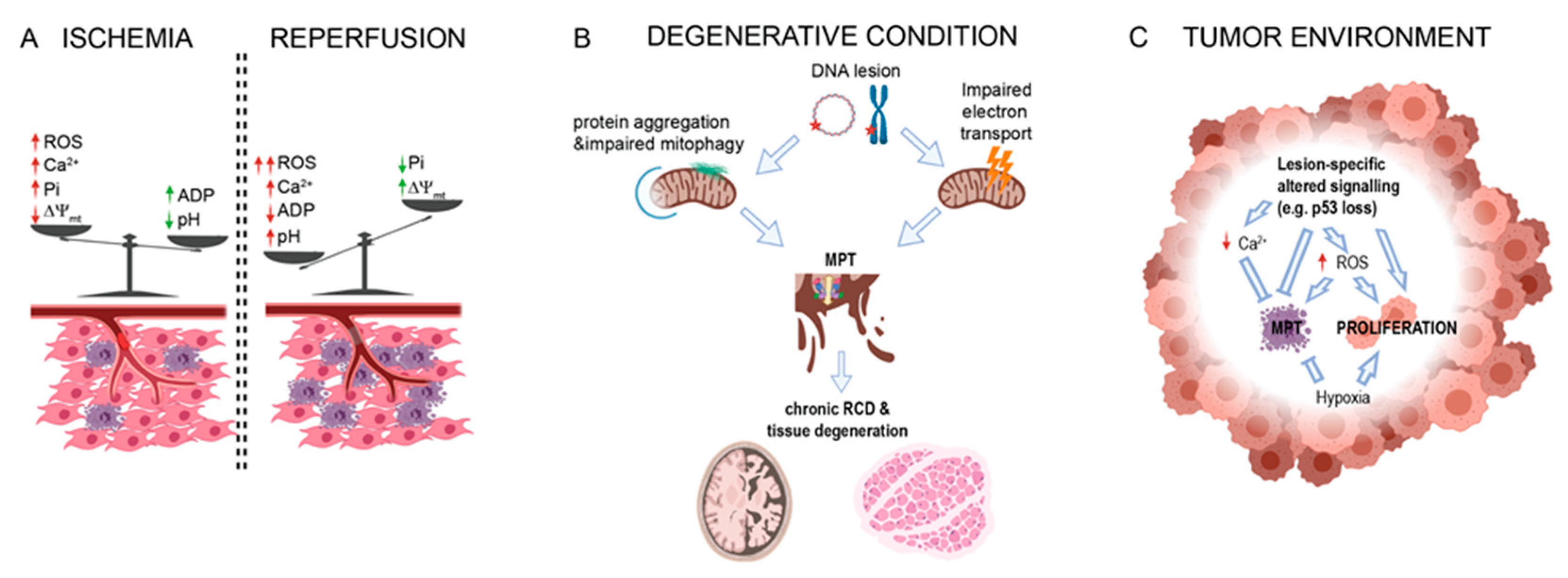
2. PTPC in Acute Conditions
2.1. MPT during Ischemia
2.2. Role of MPT in Reperfusion Injury
2.3. Role of MPT in Acute Kidney Injury
3. PTPC in Degenerative Conditions
3.1. Role of the PTPC in Protein-Aggregation-Related Neurodegenerative Diseases (NDs)
3.2. Amyotrophic Lateral Sclerosis (ALS) and PTPC
3.3. Multiple Sclerosis (MS) and PTPC
3.4. PTPC in Muscular Dystrophies (MDs)
4. Mitochondrial Disorders
5. PTPC in Nonalcoholic Fatty Liver Disease
6. PTPC in Cancer
7. Potential of PTPC Targeting and Concluding Remarks
Funding
Conflicts of Interest
References
- Dejean, L.M.; Martinez-Caballero, S.; Guo, L.; Hughes, C.; Teijido, O.; Ducret, T.; Ichas, F.; Korsmeyer, S.J.; Antonsson, B.; Jonas, E.A.; et al. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol. Biol. Cell 2005, 16, 2424–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejean, L.M.; Martinez-Caballero, S.; Manon, S.; Kinnally, K.W. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim. Biophys. Acta 2006, 1762, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; Garcia-Saez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantrow, S.P.; Piantadosi, C.A. Release of cytochrome c from liver mitochondria during permeability transition. Biochem. Biophys. Res. Commun. 1997, 232, 669–671. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. The mitochondrial permeability transition pore and cancer: Molecular mechanisms involved in cell death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Schroers, A.; Kramer, R.; Wohlrab, H. The reversible antiport-uniport conversion of the phosphate carrier from yeast mitochondria depends on the presence of a single cysteine. J. Biol. Chem. 1997, 272, 10558–10564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Cola, C.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+. J. Biol. Chem. 1993, 268, 1011–1016. [Google Scholar] [PubMed]
- Takeyama, N.; Matsuo, N.; Tanaka, T. Oxidative damage to mitochondria is mediated by the Ca(2+)-dependent inner-membrane permeability transition. Biochem. J. 1993, 294, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Wieckowski, M.R.; Brdiczka, D.; Wojtczak, L. Long-chain fatty acids promote opening of the reconstituted mitochondrial permeability transition pore. FEBS Lett. 2000, 484, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996, 378, 150–152. [Google Scholar] [CrossRef] [Green Version]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Preti, D.; Pedriali, G.; Aquila, G.; Missiroli, S.; Fantinati, A.; Caroccia, N.; Pacifico, S.; Bonora, M.; Talarico, A.; et al. Discovery of Novel 1,3,8-Triazaspiro[4.5]decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J. Med. Chem. 2018, 61, 7131–7143. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca(2+) binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Johnson, N.; Capano, M.; Edwards, M.; Crompton, M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 2004, 383, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Han, Y.; Shi, H.; Chen, J.; Zhang, X.; Wang, X.; Zhou, L.; Liu, J.; Zhang, J.; Ji, Z.; et al. Swine acute diarrhea syndrome coronavirus-induced apoptosis is caspase- and cyclophilin D- dependent. Emerg. Microbes Infect. 2020, 9, 439–456. [Google Scholar] [CrossRef]
- Garcia-Alvarado, F.; Govoni, G.; de Pascual, R.; Ruiz-Ruiz, C.; Munoz-Montero, A.; Gandia, L.; de Diego, A.M.G.; Garcia, A.G. Otilonium and pinaverium trigger mitochondrial-mediated apoptosis in rat embryo cortical neurons in vitro. Neurotoxicology 2019, 70, 99–111. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, C. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Zhai, N.; Chen, X.; Gan, F.; Li, H.; Huang, K. Ochratoxin A-Induced Apoptosis of IPEC-J2 Cells through ROS-Mediated Mitochondrial Permeability Transition Pore Opening Pathway. J. Agric. Food. Chem. 2017, 65, 10630–10637. [Google Scholar] [CrossRef]
- Naserzadeh, P.; Ansari Esfeh, F.; Kaviani, M.; Ashtari, K.; Kheirbakhsh, R.; Salimi, A.; Pourahmad, J. Single-walled carbon nanotube, multi-walled carbon nanotube and Fe2O3 nanoparticles induced mitochondria mediated apoptosis in melanoma cells. Cutan. Ocul. Toxicol. 2018, 37, 157–166. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, J.; Liu, M.; Chen, D.; Qiu, C.; Sun, K. CC-223 blocks mTORC1/C2 activation and inhibits human hepatocellular carcinoma cells in vitro and in vivo. PLoS ONE 2017, 12, e0173252. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Wang, B.; Gao, J.; Zhang, Y.; Xu, W.; Tao, L. Spinosad induces programmed cell death involves mitochondrial dysfunction and cytochrome C release in Spodoptera frugiperda Sf9 cells. Chemosphere 2017, 169, 155–161. [Google Scholar] [CrossRef]
- Liu, G.; Zou, H.; Luo, T.; Long, M.; Bian, J.; Liu, X.; Gu, J.; Yuan, Y.; Song, R.; Wang, Y.; et al. Caspase-Dependent and Caspase-Independent Pathways Are Involved in Cadmium-Induced Apoptosis in Primary Rat Proximal Tubular Cell Culture. PLoS ONE 2016, 11, e0166823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Fakharnia, F.; Khodagholi, F.; Dargahi, L.; Ahmadiani, A. Prevention of Cyclophilin D-Mediated mPTP Opening Using Cyclosporine-A Alleviates the Elevation of Necroptosis, Autophagy and Apoptosis-Related Markers Following Global Cerebral Ischemia-Reperfusion. J. Mol. Neurosci. 2017, 61, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Jiang, T.; Gao, Q.; Tian, Y.Y.; Zhou, J.S.; Wu, L.; Shi, J.Q.; Zhang, Y.D. Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. J. Renin. Angiotensin Aldosterone Syst. 2016, 17, 1470320316672349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.Y.; Zhuang, Y.H.; Cai, W.J.; Liu, Y.; Zou, W.B. The mitochondrion interfering compound NPC-26 exerts potent anti-pancreatic cancer cell activity in vitro and in vivo. Tumour Biol. 2016, 37, 15053–15063. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, M.; Li, Z.; Gao, P.; Zhou, X.; Zhang, J. Bufalin induces apoptosis in the U2OS human osteosarcoma cell line via triggering the mitochondrial pathway. Mol. Med. Rep. 2016, 13, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Borutaite, V.; Jekabsone, A.; Morkuniene, R.; Brown, G.C. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J. Mol. Cell. Cardiol. 2003, 35, 357–366. [Google Scholar] [CrossRef]
- Precht, T.A.; Phelps, R.A.; Linseman, D.A.; Butts, B.D.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Heidenreich, K.A. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005, 12, 255–265. [Google Scholar] [CrossRef]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Chandel, N.S.; Schumacker, P.T.; Thompson, C.B. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell 1999, 3, 159–167. [Google Scholar] [CrossRef]
- Belzacq, A.S.; Vieira, H.L.; Verrier, F.; Vandecasteele, G.; Cohen, I.; Prevost, M.C.; Larquet, E.; Pariselli, F.; Petit, P.X.; Kahn, A.; et al. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 2003, 63, 541–546. [Google Scholar] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Temkin, V.; Huang, Q.; Liu, H.; Osada, H.; Pope, R.M. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol. Cell. Biol. 2006, 26, 2215–2225. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol. Cell. Biol. 2011, 31, 3745–3758. [Google Scholar] [CrossRef] [Green Version]
- Tischner, D.; Manzl, C.; Soratroi, C.; Villunger, A.; Krumschnabel, G. Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 2012, 17, 1197–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, H. A medicine of human relationships. Pharos. Alpha Omega Alpha Honor Med. Soc. 1989, 52, 2–6. [Google Scholar] [PubMed]
- Krawczak, M.; Bockel, B. The formal analysis of multilocus DNA fingerprints. EXS 1993, 67, 249–255. [Google Scholar] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, H.; Wang, Y.; Peng, X.; Shao, H.; Li, H.; Bai, S.; Xu, C. Exogenous spermine inhibits hypoxia/ischemia-induced myocardial apoptosis via regulation of mitochondrial permeability transition pore and associated pathways. Exp. Biol. Med. (Maywood) 2016, 241, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Zahrebelski, G.; Nieminen, A.L.; al-Ghoul, K.; Qian, T.; Herman, B.; Lemasters, J.J. Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: A laser scanning confocal microscopic study. Hepatology 1995, 21, 1361–1372. [Google Scholar]
- Fang, Y.D.; Xu, X.; Dang, Y.M.; Zhang, Y.M.; Zhang, J.P.; Hu, J.Y.; Zhang, Q.; Dai, X.; Teng, M.; Zhang, D.X.; et al. MAP4 mechanism that stabilizes mitochondrial permeability transition in hypoxia: Microtubule enhancement and DYNLT1 interaction with VDAC1. PLoS ONE 2011, 6, e28052. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Evora, P.; Castro, E.S.O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.R.; Murphy, T.H. Reversible cyclosporin A-sensitive mitochondrial depolarization occurs within minutes of stroke onset in mouse somatosensory cortex in vivo: A two-photon imaging study. J. Biol. Chem. 2009, 284, 36109–36117. [Google Scholar] [CrossRef] [Green Version]
- Adachi, M.; Takahashi, K.; Nishikawa, M.; Miki, H.; Uyama, M. High intraocular pressure-induced ischemia and reperfusion injury in the optic nerve and retina in rats. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 445–451. [Google Scholar] [CrossRef]
- Hwang, J.H.; Lee, J.H.; Lee, K.H.; Bae, E.J.; Sung, D.K.; Chang, Y.S.; Park, W.S. Cyclosporine A attenuates hypoxic-ischemic brain injury in newborn rats. Brain Res. 2010, 1359, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.K.; Brown, P.A.; Cuzzocrea, S.; Zacharowski, K.; Stewart, K.N.; Mota-Filipe, H.; McDonald, M.C.; Thiemermann, C. Calpain inhibitor-1 reduces renal ischemia/reperfusion injury in the rat. Kidney Int. 2001, 59, 2073–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [PubMed] [Green Version]
- Shintani-Ishida, K.; Yoshida, K. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int. J. Cardiol. 2015, 197, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Piper, H.M.; Meuter, K.; Schafer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Barzyc, A.; Lysik, W.; Slyk, J.; Kuszewski, M.; Zarebinski, M.; Wojciechowska, M.; Cudnoch-Jedrzejewska, A. Reperfusion injury as a target for diminishing infarct size. Med. Hypotheses 2020, 137, 109558. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [PubMed]
- Upadhaya, S.; Madala, S.; Baniya, R.; Subedi, S.K.; Saginala, K.; Bachuwa, G. Impact of cyclosporine A use in the prevention of reperfusion injury in acute myocardial infarction: A meta-analysis. Cardiol. J. 2017, 24, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottecher, J.; Kindo, M.; Chamaraux-Tran, T.N.; Charles, A.L.; Lejay, A.; Kemmel, V.; Vogel, T.; Chakfe, N.; Zoll, J.; Diemunsch, P.; et al. Skeletal muscle ischemia-reperfusion injury and cyclosporine A in the aging rat. Fundam. Clin. Pharmacol. 2016, 30, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Minamikawa-Tachino, R.; Kristian, T.; Perkins, G.; Narazaki, M.; Siesjo, B.K.; Shibasaki, F. Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol. Dis. 2002, 10, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konukoglu, D.; Tasci, I.; Cetinkale, O. Effects of cyclosporin A and ibuprofen on liver ischemia-reperfusion injury in the rat. Clin. Chim. Acta 1998, 275, 1–8. [Google Scholar] [CrossRef]
- Li, J.; Yan, Z.; Fang, Q. A Mechanism Study Underlying the Protective Effects of Cyclosporine-A on Lung Ischemia-Reperfusion Injury. Pharmacology 2017, 100, 83–90. [Google Scholar] [CrossRef]
- Yazdani, I.; Majdani, R.; Ghasemnejad-Berenji, M.; Dehpour, A.R. Comparison of multiple doses of cyclosporine A on germ cell apoptosis and epididymal sperm parameters after testicular ischemia/reperfusion in rats. Exp. Mol. Pathol. 2019, 110, 104271. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Inserte, J.; Fernandez-Sanz, C.; Hernando, V.; Miro-Casas, E.; Barba, I.; Garcia-Dorado, D. The role of mitochondrial permeability transition in reperfusion-induced cardiomyocyte death depends on the duration of ischemia. Basic Res. Cardiol. 2011, 106, 1259–1268. [Google Scholar] [CrossRef]
- Imahashi, K.; Pott, C.; Goldhaber, J.I.; Steenbergen, C.; Philipson, K.D.; Murphy, E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ. Res. 2005, 97, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imahashi, K.; Schneider, M.D.; Steenbergen, C.; Murphy, E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ. Res. 2004, 95, 734–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Wang, S.; Li, Y.; Wang, P.; Li, S.; Guo, Y.; Yao, R. The role of the mitochondrial calcium uniporter in cerebral ischemia/reperfusion injury in rats involves regulation of mitochondrial energy metabolism. Mol. Med. Rep. 2013, 7, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Campo, G.; Morciano, G.; Pavasini, R.; Bonora, M.; Sbano, L.; Biscaglia, S.; Bovolenta, M.; Pinotti, M.; Punzetti, S.; Rizzo, P.; et al. Fo ATP synthase C subunit serum levels in patients with ST-segment Elevation Myocardial Infarction: Preliminary findings. Int. J. Cardiol. 2016, 221, 993–997. [Google Scholar] [CrossRef]
- Eirin, A.; Li, Z.; Zhang, X.; Krier, J.D.; Woollard, J.R.; Zhu, X.Y.; Tang, H.; Herrmann, S.M.; Lerman, A.; Textor, S.C.; et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 2012, 60, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- Tanno, M.; Kuno, A.; Ishikawa, S.; Miki, T.; Kouzu, H.; Yano, T.; Murase, H.; Tobisawa, T.; Ogasawara, M.; Horio, Y.; et al. Translocation of glycogen synthase kinase-3beta (GSK-3beta), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J. Biol. Chem. 2014, 289, 29285–29296. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Ge, Y.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ge, Y.; Bao, H.; Dworkin, L.; Peng, A.; Gong, R. Redox-sensitive glycogen synthase kinase 3beta-directed control of mitochondrial permeability transition: Rheostatic regulation of acute kidney injury. Free Radic. Biol. Med. 2013, 65, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; De Zen, F.; Weinberg, J.; Kunzendorf, U.; Krautwald, S. Programmed necrosis in acute kidney injury. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2012, 27, 3412–3419. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Honarpisheh, M.M.; Foresto-Neto, O.; Shi, C.; Desai, J.; Zhao, Z.B.; Marschner, J.A.; Popper, B.; Buhl, E.M.; Boor, P.; et al. Mitochondria Permeability Transition versus Necroptosis in Oxalate-Induced AKI. J. Am. Soc. Nephrol. 2019, 30, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Ueda, K.; Chan, P.; Yu, S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef]
- Zhu, Y.; Duan, C.; Lu, L.; Gao, H.; Zhao, C.; Yu, S.; Ueda, K.; Chan, P.; Yang, H. alpha-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int. J. Biochem. Cell Biol. 2011, 43, 732–741. [Google Scholar] [CrossRef]
- Tillement, L.; Lecanu, L.; Yao, W.; Greeson, J.; Papadopoulos, V. The spirostenol (22R, 25R)-20alpha-spirost-5-en-3beta-yl hexanoate blocks mitochondrial uptake of Abeta in neuronal cells and prevents Abeta-induced impairment of mitochondrial function. Steroids 2006, 71, 725–735. [Google Scholar] [CrossRef]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol. Aging 2014, 35, 975–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol. Aging 2014, 35, 1132–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.; Fan, C.; Gu, L.; Gao, H.; Liu, Q.; Zhang, T.; Qi, Z.; Zhao, C.; Zhao, H.; Cai, Q.; et al. Silencing of PINK1 induces mitophagy via mitochondrial permeability transition in dopaminergic MN9D cells. Brain Res. 2011, 1394, 1–13. [Google Scholar] [CrossRef]
- Gautier, C.A.; Giaime, E.; Caballero, E.; Nunez, L.; Song, Z.; Chan, D.; Villalobos, C.; Shen, J. Regulation of mitochondrial permeability transition pore by PINK1. Mol. Neurodegener. 2012, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Das Banerjee, T.; Janda, E. How Parkinsonian toxins dysregulate the autophagy machinery. Int. J. Mol. Sci. 2013, 14, 22163–22189. [Google Scholar] [CrossRef]
- Rasheed, M.Z.; Tabassum, H.; Parvez, S. Mitochondrial permeability transition pore: A promising target for the treatment of Parkinson’s disease. Protoplasma 2017, 254, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.J.; Clemensson, L.E.; Schioth, H.B.; Nguyen, H.P. Olesoxime in neurodegenerative diseases: Scrutinising a promising drug candidate. Biochem. Pharmacol. 2019, 168, 305–318. [Google Scholar] [CrossRef]
- Bordet, T.; Berna, P.; Abitbol, J.L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals. 2010, 3, 345–368. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Yamashita, S.; Ueyama, H.; Ishizaki, M.; Maeda, Y.; Ando, Y. Long-term effects of edaravone on survival of patients with amyotrophic lateral sclerosis. eNeurologicalSci 2018, 11, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Inarrea, P.; Alarcia, R.; Alava, M.A.; Capablo, J.L.; Casanova, A.; Iniguez, C.; Iturralde, M.; Larrode, P.; Martin, J.; Mostacero, E.; et al. Mitochondrial complex enzyme activities and cytochrome C expression changes in multiple sclerosis. Mol. Neurobiol. 2014, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Tranah, G.J.; Santaniello, A.; Caillier, S.J.; D’Alfonso, S.; Martinelli Boneschi, F.; Hauser, S.L.; Oksenberg, J.R. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 2015, 85, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflammation 2019, 16, 131. [Google Scholar] [CrossRef]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef] [Green Version]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lenneras, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Marchi, S.; Rimessi, A.; Bonora, M.; Giorgi, C.; Mehta, K.D.; Pinton, P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy 2013, 9, 1367–1385. [Google Scholar] [CrossRef] [Green Version]
- Su, K.G.; Savino, C.; Marracci, G.; Chaudhary, P.; Yu, X.; Morris, B.; Galipeau, D.; Giorgio, M.; Forte, M.; Bourdette, D. Genetic inactivation of the p66 isoform of ShcA is neuroprotective in a murine model of multiple sclerosis. Eur. J. Neurosci. 2012, 35, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Savino, C.; Pelicci, P.; Giorgio, M. The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxidative Med. Cell. Longev. 2013, 2013, 719407. [Google Scholar] [CrossRef] [Green Version]
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Wang, E.T.; Wagers, A.J. Skeletal muscle degenerative diseases and strategies for therapeutic muscle repair. Annu. Rev. Pathol. 2013, 8, 441–475. [Google Scholar] [CrossRef] [Green Version]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Durbeej, M.; Campbell, K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: An overview of current mouse models. Curr. Opin. Genet. Dev. 2002, 12, 349–361. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Monaco, A.P.; Feener, C.C.; Kunkel, L.M. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 1987, 238, 347–350. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Giacomotto, J.; Brouilly, N.; Walter, L.; Mariol, M.C.; Berger, J.; Segalat, L.; Becker, T.S.; Currie, P.D.; Gieseler, K. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Hum. Mol. Genet. 2013, 22, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Stupka, N.; Gregorevic, P.; Plant, D.R.; Lynch, G.S. The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol. 2004, 107, 299–310. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Harrison, M.A.; Carbonetto, S.; Chin, E.; Michel, R.N.; Jasmin, B.J. Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum. Mol. Genet. 2004, 13, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Tiepolo, T.; Angelin, A.; Sabatelli, P.; Maraldi, N.M.; Basso, E.; Forte, M.A.; Bernardi, P.; Bonaldo, P. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum. Mol. Genet. 2009, 18, 2024–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampe, A.K.; Bushby, K.M. Collagen VI related muscle disorders. J. Med Genet. 2005, 42, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, L.; Martoni, E.; Grumati, P.; Sabatelli, P.; Squarzoni, S.; Urciuolo, A.; Ferlini, A.; Gualandi, F.; Bonaldo, P. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008, 71, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Braghetta, P.; Zanetti, M.; Piccolo, S.; Volpin, D.; Bressan, G.M. Collagen VI deficiency induces early onset myopathy in the mouse: An animal model for Bethlem myopathy. Hum. Mol. Genet. 1998, 7, 2135–2140. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Angelin, A.; Tiepolo, T.; Sabatelli, P.; Grumati, P.; Bergamin, N.; Golfieri, C.; Mattioli, E.; Gualandi, F.; Ferlini, A.; Merlini, L.; et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc. Natl. Acad. Sci. USA 2007, 104, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Tagliavini, F.; Sardone, F.; Squarzoni, S.; Maraldi, N.M.; Merlini, L.; Faldini, C.; Sabatelli, P. Ultrastructural changes in muscle cells of patients with collagen VI-related myopathies. Muscles Ligaments Tendons J. 2013, 3, 281–286. [Google Scholar] [CrossRef] [Green Version]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Cortopassi, G. mtDNA mutations confer cellular sensitivity to oxidant stress that is partially rescued by calcium depletion and cyclosporin A. Biochem. Biophys. Res. Commun. 1997, 239, 139–145. [Google Scholar] [CrossRef]
- Haroon, M.F.; Fatima, A.; Scholer, S.; Gieseler, A.; Horn, T.F.; Kirches, E.; Wolf, G.; Kreutzmann, P. Minocycline, a possible neuroprotective agent in Leber’s hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11,778 mutation. Neurobiol. Dis. 2007, 28, 237–250. [Google Scholar] [CrossRef]
- Cotan, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodriguez-Hernandez, A.; Gomez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, L.; Ren, X.; Zhang, Y.; Zhang, H. LRPPRC: A Multifunctional Protein Involved in Energy Metabolism and Human Disease. Front. Physiol. 2019, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Burelle, Y.; Bemeur, C.; Rivard, M.E.; Thompson Legault, J.; Boucher, G.; Consortium, L.; Morin, C.; Coderre, L.; Des Rosiers, C. Mitochondrial vulnerability and increased susceptibility to nutrient-induced cytotoxicity in fibroblasts from leigh syndrome French canadian patients. PLoS ONE 2015, 10, e0120767. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef] [Green Version]
- Piquereau, J.; Caffin, F.; Novotova, M.; Prola, A.; Garnier, A.; Mateo, P.; Fortin, D.; Huynh le, H.; Nicolas, V.; Alavi, M.V.; et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 2012, 94, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lu, C.; Whiteman, M.; Chance, B.; Armstrong, J.S. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 2008, 283, 3476–3486. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.; Baggett, A.; Csordas, G.; Sheu, S.S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca(2+) influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 2019, 294, 10807–10818. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Kozakova, M.; Hojlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; Balkau, B.; Investigators, R. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009, 49, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Rolo, A.P.; Duarte, F.V.; Simoes, A.M.; Palmeira, C.M. Differential alterations in mitochondrial function induced by a choline-deficient diet: Understanding fatty liver disease progression. Mitochondrion 2008, 8, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.F.; Wang, C.H. The relationship between the opening of mitochondrial permeability transition pores of cultured hepatocytes with their apoptoses in a non-alcoholic fatty liver disease model. Chin. J. Hepatol. 2007, 15, 837–839. [Google Scholar]
- Kang, M.; Li, S.; Zhong, D.; Yang, Z.; Li, P. Hepatocyte apoptosis and mitochondrial permeability transition pore opening in rats with nonalcoholic fatty liver. J. South. Med Univ. 2013, 33, 1062–1066. [Google Scholar]
- Wang, X.; Du, H.; Shao, S.; Bo, T.; Yu, C.; Chen, W.; Zhao, L.; Li, Q.; Wang, L.; Liu, X.; et al. Cyclophilin D deficiency attenuates mitochondrial perturbation and ameliorates hepatic steatosis. Hepatology 2018, 68, 62–77. [Google Scholar] [CrossRef]
- Lazarin Mde, O.; Ishii-Iwamoto, E.L.; Yamamoto, N.S.; Constantin, R.P.; Garcia, R.F.; da Costa, C.E.; Vitoriano Ade, S.; de Oliveira, M.C.; Salgueiro-Pagadigorria, C.L. Liver mitochondrial function and redox status in an experimental model of non-alcoholic fatty liver disease induced by monosodium L-glutamate in rats. Exp. Mol. Pathol. 2011, 91, 687–694. [Google Scholar] [CrossRef]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340. [Google Scholar] [CrossRef]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef] [Green Version]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Bittremieux, M.; Parys, J.B.; Pinton, P.; Bultynck, G. ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca(2+) signaling. Biochim. Biophys. Acta 2016, 1863, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Rimessi, A.; Giorgi, C.; Baldini, C.; Ferroni, L.; Rizzuto, R.; Pinton, P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 2008, 375, 501–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzym. Res. 2011, 2011, 329098. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Giorgi, C. Novel function of the tumor suppressor PML at ER-mitochondria sites in the control of autophagy. Oncotarget 2017, 8, 81723–81724. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [Green Version]
- Luo, J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Morganti, C.; Morciano, G.; Wieckowski, M.R.; Pinton, P. Alterations in Mitochondrial and Endoplasmic Reticulum Signaling by p53 Mutants. Front. Oncol. 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Pinton, P. Novel frontiers in calcium signaling: A possible target for chemotherapy. Pharmacol. Res. 2015, 99, 82–85. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Pinton, P. Inside the tumor: p53 modulates calcium homeostasis. Cell Cycle 2015, 14, 933–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Poletti, F.; Ramirez, F.G.; Morciano, G.; Morganti, C.; Pandolfi, P.P.; Mammano, F.; Pinton, P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 2015, 6, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; D’Silva, P. Chaperoning mitochondrial permeability transition: Regulation of transition pore complex by a J-protein, DnaJC15. Cell Death Dis. 2014, 5, e1101. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Ishida, T.; Hino, M.; Yamazaki, N.; Baba, Y.; Terada, H. Characterization of porin isoforms expressed in tumor cells. Eur. J. Biochem. 2000, 267, 6067–6073. [Google Scholar] [CrossRef] [PubMed]
- Beinlich, A.; Strohmeier, R.; Kaufmann, M.; Kuhl, H. Relation of cell proliferation to expression of peripheral benzodiazepine receptors in human breast cancer cell lines. Biochem. Pharmacol. 2000, 60, 397–402. [Google Scholar] [CrossRef]
- Maaser, K.; Hopfner, M.; Jansen, A.; Weisinger, G.; Gavish, M.; Kozikowski, A.P.; Weizman, A.; Carayon, P.; Riecken, E.O.; Zeitz, M.; et al. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human colorectal cancer cells. Br. J. Cancer 2001, 85, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Faure Vigny, H.; Heddi, A.; Giraud, S.; Chautard, D.; Stepien, G. Expression of oxidative phosphorylation genes in renal tumors and tumoral cell lines. Mol. Carcinog. 1996, 16, 165–172. [Google Scholar] [CrossRef]
- Rempel, A.; Mathupala, S.P.; Griffin, C.A.; Hawkins, A.L.; Pedersen, P.L. Glucose catabolism in cancer cells: Amplification of the gene encoding type II hexokinase. Cancer Res. 1996, 56, 2468–2471. [Google Scholar]
- Gudnason, V.; Ingvarsson, S.; Jonasdottir, A.; Andresdottir, V.; Egilsson, V. Isoenzyme pattern and subcellular localization of hexokinases in human breast cancer and nonpathological breast tissue. Int. J. Cancer 1984, 34, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. New Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Schaller, S.; Paradis, S.; Ngoh, G.A.; Assaly, R.; Buisson, B.; Drouot, C.; Ostuni, M.A.; Lacapere, J.J.; Bassissi, F.; Bordet, T.; et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J. Pharmacol. Exp. Ther. 2010, 333, 696–706. [Google Scholar] [CrossRef]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [Green Version]
- Atar, D.; Arheden, H.; Berdeaux, A.; Bonnet, J.L.; Carlsson, M.; Clemmensen, P.; Cuvier, V.; Danchin, N.; Dubois-Rande, J.L.; Engblom, H.; et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 2015, 36, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leruez, S.; Verny, C.; Bonneau, D.; Procaccio, V.; Lenaers, G.; Amati-Bonneau, P.; Reynier, P.; Scherer, C.; Prundean, A.; Orssaud, C.; et al. Cyclosporine A does not prevent second-eye involvement in Leber’s hereditary optic neuropathy. Orphanet J. Rare Dis. 2018, 13, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valasani, K.R.; Vangavaragu, J.R.; Day, V.W.; Yan, S.S. Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J. Chem. Inf. Modeling 2014, 54, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small Molecule Inhibitors of Cyclophilin D To Protect Mitochondrial Function as a Potential Treatment for Acute Pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Panel, M.; Ruiz, I.; Brillet, R.; Lafdil, F.; Teixeira-Clerc, F.; Nguyen, C.T.; Calderaro, J.; Gelin, M.; Allemand, F.; Guichou, J.F.; et al. Small-Molecule Inhibitors of Cyclophilins Block Opening of the Mitochondrial Permeability Transition Pore and Protect Mice From Hepatic Ischemia/Reperfusion Injury. Gastroenterology 2019, 157, 1368–1382. [Google Scholar] [CrossRef]
- Roy, S.; Sileikyte, J.; Schiavone, M.; Neuenswander, B.; Argenton, F.; Aube, J.; Hedrick, M.P.; Chung, T.D.; Forte, M.A.; Bernardi, P.; et al. Discovery, Synthesis, and Optimization of Diarylisoxazole-3-carboxamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2015, 10, 1655–1671. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Sileikyte, J.; Neuenswander, B.; Hedrick, M.P.; Chung, T.D.; Aube, J.; Schoenen, F.J.; Forte, M.A.; Bernardi, P. N-Phenylbenzamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2016, 11, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63, e12413. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10070998
Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, Bouhamida E, Giorgi C, Wieckowski MR, Pinton P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules. 2020; 10(7):998. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10070998
Chicago/Turabian StyleBonora, Massimo, Simone Patergnani, Daniela Ramaccini, Giampaolo Morciano, Gaia Pedriali, Asrat Endrias Kahsay, Esmaa Bouhamida, Carlotta Giorgi, Mariusz R. Wieckowski, and Paolo Pinton. 2020. "Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology" Biomolecules 10, no. 7: 998. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10070998