
Aortic Aneurysms, Chronic Kidney Disease and Metalloproteinases

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Study Selection
3.2. Association between Metalloproteinases and Aortic Aneurysms
3.3. Chronic Kidney Disease and Increased Risk for Aortic Aneurysms
3.4. Role of Metalloproteinases in Chronic Kidney Disease Progression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnston, K.; Rutherford, R.B.; Tilson, M.; Shah, D.M.; Hollier, L.; Stanley, J.C. Suggested standards for reporting on arterial aneurysms. J. Vasc. Surg. 1991, 13, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederle, F.A. Abdominal Aortic Aneurysm. Ann. Intern. Med. 2009, 150, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Bickerstaff, L.K.; Pairolero, P.C.; Hollier, L.H.; Melton, L.J.; Van Peenen, H.J.; Cherry, K.J.; Joyce, J.W.; Lie, J.T. Thoracic aortic aneurysms: A population-based study. Surgery 1982, 92, 1103–1108. [Google Scholar] [PubMed]
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Acosta, S.; Ögren, M.; Bengtsson, H.; Bergqvist, D.; Lindblad, B.; Zdanowski, Z. Increasing incidence of ruptured abdominal aortic aneurysm: A population-based study. J. Vasc. Surg. 2006, 44, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Filardo, G.; Powell, J.T.; Martinez, M.A.-M.; Ballard, D.J. Surgery for small asymptomatic abdominal aortic aneurysms. Cochrane Database Syst. Rev. 2015, 2015, CD001835. [Google Scholar] [CrossRef]
- Lederle, F.A. Abdominal aortic aneurysm: Still no pill. Ann. Intern. Med. 2013, 159, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Kwak, L.; Ballew, S.H.; Grams, M.E.; Selvin, E.; Folsom, A.R.; Coresh, J.; Tang, W. Chronic kidney disease measures and the risk of abdominal aortic aneurysm. Atherosclerosis 2018, 279, 107–113. [Google Scholar] [CrossRef]
- Provenzano, M.; Rotundo, S.; Chiodini, P.; Gagliardi, I.; Michael, A.; Angotti, E.; Borrelli, S.; Serra, R.; Foti, D.; De Sarro, G.; et al. Contribution of Predictive and Prognostic Biomarkers to Clinical Research on Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 5846. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Coppolino, G.; Faga, T.; Garofalo, C.; Serra, R.; Andreucci, M. Epidemiology of cardiovascular risk in chronic kidney disease patients: The real silent killer. Rev. Cardiovasc. Med. 2019, 20, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Coppolino, G.; De Nicola, L.; Serra, R.; Garofalo, C.; Andreucci, M.; Bolignano, D. Unraveling Cardiovascular Risk in Renal Patients: A New Take on Old Tale. Front. Cell Dev. Biol. 2019, 7, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Faga, T.; Michael, A.; Ielapi, N.; Grande, R.; Sapienza, P.; De Franciscis, S.; Mastroroberto, P.; et al. The Association of Matrix Metalloproteinases with Chronic Kidney Disease and Peripheral Vascular Disease: A Light at the End of the Tunnel? Biomolecules 2020, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin Proteases and Their Inhibitors: Foes or Friends in Nervous System Physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butrico, L.; Barbetta, A.; Ciranni, S.; Andreucci, M.; Mastroroberto, P.; De Franciscis, S. Role of metalloproteinases and their inhibitors in the development of abdominal aortic aneurysm: Current insights and systematic review of the literature. Chirurgia 2017, 30, 151–159. [Google Scholar]
- De Franciscis, S.; Mastroroberto, P.; Gallelli, L.; Buffone, G.; Montemurro, R.; Serra, R. Increased Plasma Levels of Metalloproteinase-9 and Neutrophil Gelatinase–Associated Lipocalin in a Rare Case of Multiple Artery Aneurysm. Ann. Vasc. Surg. 2013, 27, 1185.e5–1185.e7. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Grande, R.; Montemurro, R.; Butrico, L.; Caliò, F.G.; Mastrangelo, D.; Scarcello, E.; Gallelli, L.; Buffone, G.; De Franciscis, S. The role of matrix metalloproteinases and neutrophil gelatinase-associated lipocalin in central and peripheral arterial aneurysms. Surgery 2015, 157, 155–162. [Google Scholar] [CrossRef] [PubMed]
- De Caridi, G.; Massara, M.; Spinelli, F.; Grande, R.; Butrico, L.; Rende, P.; Amato, M.; Compagna, R.; Amato, B.; De Franciscis, S.; et al. An uncommon case of arterial aneurysms association with high plasma levels of Matrix Metalloproteinase-9 and Neutrophil Gelatinase-Associated Lipocalin. Open Med. 2015, 10, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Fukami, K.; Yamagishi, S.-I.; Ueda, S.; Kaida, Y.; Matsumoto, T.; Yoshimura, J.; Hazama, T.; Takamiya, Y.; Kusumoto, T.; et al. Circulating Matrix Metalloproteinase-2 Is an Independent Correlate of Proteinuria in Patients with Chronic Kidney Disease. Am. J. Nephrol. 2009, 29, 109–115. [Google Scholar] [CrossRef]
- Zakiyanov, O.; Kalousová, M.; Zima, T.; Tesař, V. Matrix Metalloproteinases in Renal Diseases: A Critical Appraisal. Kidney Blood Press. Res. 2019, 44, 298–330. [Google Scholar] [CrossRef]
- Ohnishi, J.; Ohnishi, E.; Shibuya, H.; Takahashi, T. Functions for proteinases in the ovulatory process. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2005, 1751, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Limbu, M.H.; Wang, Z.; Liu, J.; Liu, L.; Zhang, X.; Chen, P.; Liu, B.-C. MMP-2 and 9 in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Amanzadeh, M.; Mota, A.; Zarghami, N.; Abedi-Azar, S.; Abroon, S.; Akbarian, N.; Mihanfar, A.; Rahmati-Yamchi, M. Association Between Matrix Metalloproteinase-3 Activity and Glomerular Filtration Rate and Albuminuria Status in Patients with Type 2 Diabetes Mellitus. Iran. J. Kidney Dis. 2018, 12, 40–47. [Google Scholar] [PubMed]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix Metalloproteinases-7 and Kidney Fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H., Jr.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Buültmann, A.; Fischel, S.; Gillitzer, A.; Cullen, P.; Walch, A.; Jost, P.; Ungerer, M.; Tolley, N.D.; Lindemann, S.; et al. Extracellular Matrix Metalloproteinase Inducer (CD147) Is a Novel Receptor on Platelets, Activates Platelets, and Augments Nuclear Factor κB–Dependent Inflammation in Monocytes. Circ. Res. 2008, 102, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008, 75, 346–359. [Google Scholar] [CrossRef] [Green Version]
- McMillan, W.D.; Tamarina, N.A.; Cipollone, M.; Johnson, D.A.; Parker, M.A.; Pearce, W.H. Size matters: The relationship between MMP-9 expression and aortic diameter. Circulation 1997, 96, 2228–2232. [Google Scholar] [CrossRef]
- Newman, K.M.; Jean-Claude, J.; Li, H.; Scholes, J.V.; Ogata, Y.; Nagase, H.; Tilson, M. Cellular localization of matrix metalloproteinases in the abdominal aortic aneurysm wall. J. Vasc. Surg. 1994, 20, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.H.; Georgescu, H.I.; Lin, C.-W.; Mendelow, D.; Steed, D.L.; Webster, M.W. Inducible synthesis of collagenase and other neutral metalloproteinases by cells of aortic origin. J. Surg. Res. 1991, 51, 399–404. [Google Scholar] [CrossRef]
- Wilson, W.R.W.; Anderton, M.; Schwalbe, E.C.; Jones, J.L.; Furness, P.N.; Bell, P.R.; Thompson, M.M. Matrix Metalloproteinase-8 and -9 Are Increased at the Site of Abdominal Aortic Aneurysm Rupture. Circulation 2006, 113, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, R.J.; McCarthy, W.J.; Dixit, S.N.; Lilly, M.P.; Shively, V.P.; Flinn, W.R.; Yao, J.S. Collagen types and matrix protein content in human abdominal aortic aneurysms. J. Vasc. Surg. 1989, 10, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Pasta, S.; Agnese, V.; Gallo, A.; Cosentino, F.; Di Giuseppe, M.; Gentile, G.; Raffa, G.M.; Maalouf, J.F.; Michelena, H.I.; Bellavia, D.; et al. Shear Stress and Aortic Strain Associations with Biomarkers of Ascending Thoracic Aortic Aneurysm. Ann. Thorac. Surg. 2020, 110, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Khanafer, K.; Ghosh, A.; Vafai, K. Correlation between MMP and TIMP levels and elastic moduli of ascending thoracic aortic aneurysms. Cardiovasc. Revascularization Med. 2019, 20, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mozo, P.; Madrigal-Matute, J.; Vega De Ceniga, M.V.; Blanco-Colio, L.M.; Meilhac, O.; Feldman, L.; Michel, J.-B.; Clancy, P.; Golledge, J.; Norman, P.E.; et al. Increased plasma levels of NGAL, a marker of neutrophil activation, in patients with abdominal aortic aneurysm. Atheroscler. 2012, 220, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, M.; Li, C.; Frebelius, S.; Swedenborg, J.; Wågsäter, D.; Williams, K.J.; Eriksson, P.; Roy, J.; Liu, M.-L. Proteolytically active ADAM10 and ADAM17 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb. Haemost. 2015, 114, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Andreucci, M.; De Nicola, L.; Garofalo, C.; Battaglia, Y.; Borrelli, S.; Gagliardi, I.; Faga, T.; Michael, A.; Mastroroberto, P.; et al. The Role of Prognostic and Predictive Biomarkers for Assessing Cardiovascular Risk in Chronic Kidney Disease Patients. BioMed Res. Int. 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nicola, L.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Russo, L.; Bellasi, A.; Santoro, D.; Conte, G.; Minutolo, R. Epidemiology of low-proteinuric chronic kidney disease in renal clinics. PLoS ONE 2017, 12, e0172241. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, L.; Minutolo, R.; Chiodini, P.; Zoccali, C.; Castellino, P.; Donadio, C.; Strippoli, M.; Casino, F.; Giannattasio, M.; Petrarulo, F.; et al. Global approach to cardiovascular risk in chronic kidney disease: Reality and opportunities for intervention. Kidney Int. 2006, 69, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, K.C.; Teng, K.Y.; Chavez, L.A.; Van Spyk, E.N.; Samadzadeh, K.M.; Carson, J.G.; Lee, E.S. Risk Factors Associated with the Diagnosis of Abdominal Aortic Aneurysm in Patients Screened at a Regional Veterans Affairs Health Care System. Ann. Vasc. Surg. 2014, 28, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, H.G.; Wolfson, S.K.; Sutton-Tyrrell, K.; Kuller, L.H.; O’Leary, D. Risk Factors for Abdominal Aortic Aneurysms in Older Adults Enrolled in the Cardiovascular Health Study. Arter. Thromb. Vasc. Biol. 1996, 16, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Reeps, C.; Maier, A.; Pelisek, J.; Härtl, F.; Grabher-Meier, V.; Wall, W.A.; Essler, M.; Eckstein, H.-H.; Gee, M.W. Measuring and modeling patient-specific distributions of material properties in abdominal aortic aneurysm wall. Biomech. Model. Mechanobiol. 2013, 12, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Pelisek, J.; Assadian, A.; Sarkar, O.; Eckstein, H.-H.; Frank, H. Carotid Plaque Composition in Chronic Kidney Disease: A Retrospective Analysis of Patients Undergoing Carotid Endarterectomy. Eur. J. Vasc. Endovasc. Surg. 2010, 39, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Lauhio, A.; Sorsa, T.; Srinivas, R.; Stenman, M.; Tervahartiala, T.; Stenman, U.-H.; Grönhagen-Riska, C.; Honkanen, E. Urinary matrix metalloproteinase -8, -9, -14 and their regulators (TRY-1, TRY-2, TATI) in patients with diabetic nephropathy. Ann. Med. 2008, 40, 312–320. [Google Scholar] [CrossRef]
- Gharagozlian, S.; Svennevig, K.; Bangstad, H.-J.; Winberg, J.-O.; Kolset, S.O. Matrix metalloproteinases in subjects with type 1 diabetes. BMC Clin. Pathol. 2009, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, K.; Koyanagi, I.; Ohara, I.; Ito, T.; Saitoh, A.; Horikoshi, S.; Tomino, Y. Levels of urinary matrix metalloproteinase-9 (MMP-9) and renal injuries in patients with type 2 diabetic nephropathy. J. Clin. Lab. Anal. 2004, 18, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B.; Mothu, N.; Nguyen, J.; Nguyen-Khoa, T.; Nöel, L.-H.; Jungers, P. Specific changes in plasma concentrations of matrix metalloproteinase-2 and -9, TIMP-1 and TGF- 1 in patients with distinct types of primary glomerulonephritis. Nephrol. Dial. Transplant. 2007, 22, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, K.A.; Bennett, M.; Devarajan, P. Distinct metalloproteinase excretion patterns in focal segmental glomerulosclerosis. Pediatr. Nephrol. 2011, 26, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Thrailkill, K.M.; Moreau, C.S.; Cockrell, G.E.; Jo, C.-H.; Bunn, R.C.; Morales-Pozzo, A.E.; Lumpkin, C.K.; Fowlkes, J.L. Disease and gender-specific dysregulation of NGAL and MMP-9 in type 1 diabetes mellitus. Endocrine 2010, 37, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Ivashkiv, L.B. Costimulation of Chemokine Receptor Signaling by Matrix Metalloproteinase-9 Mediates Enhanced Migration of IFN-α Dendritic Cells. J. Immunol. 2006, 176, 6022–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Park, P.W.; Wilson, C.L.; Parks, W.C. Matrilysin Shedding of Syndecan-1 Regulates Chemokine Mobilization and Transepithelial Efflux of Neutrophils in Acute Lung Injury. Cell 2002, 111, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [Green Version]
- Peiskerová, M.; Kalousová, M.; Kratochvílová, M.; Dusilová-Sulková, S.; Uhrová, J.; Bandúr, S.; Malbohan, I.M.; Zima, T.; Tesař, V. Fibroblast growth factor 23 and matrix-metalloproteinases in patients with chronic kidney disease: Are they associated with cardiovascular disease? Kidney Blood Press. Res. 2009, 32, 276–283. [Google Scholar] [CrossRef]
- Henger, A.; Kretzler, M.; Doran, P.P.; Bonrouhi, M.; Schmid, H.; Kiss, E.; Cohen, C.D.; Madden, S.F.; Porubsky, S.; Gröne, E.F.; et al. Gene expression fingerprints in human tubulointerstitial inflammation and fibrosis as prognostic markers of disease progression11See Editorial by Roneo, p. 1107. Kidney Int. 2004, 65, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKDWork Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013, 3 (Suppl. 2013), 1–150. [Google Scholar]
- Palau, V.; Pascual, J.; Soler, M.J.; Riera, M. Role of ADAM17 in kidney disease. Am. J. Physiol. Physiol. 2019, 317, F333–F342. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hagiyama, M.; Ito, A. Renal ADAM10 and 17: Their Physiological and Medical Meanings. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zimpelmann, J.; Agaybi, S.; Gurley, S.B.; Puente, L.; Burns, K.D. Characterization of Angiotensin-Converting Enzyme 2 Ectodomain Shedding from Mouse Proximal Tubular Cells. PLoS ONE 2014, 9, e85958. [Google Scholar] [CrossRef] [PubMed]
- Isermann, B.; Vinnikov, I.A.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.A.F.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.-K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.-C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR Signaling Promotes TGFβ-Dependent Renal Fibrosis. J. Am. Soc. Nephrol. 2011, 23, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Hagiyama, M.; Takashima, Y.; Yoneshige, A.; Ito, A. Cell adhesion molecule-1 shedding induces apoptosis of renal epithelial cells and exacerbates human nephropathies. Am. J. Physiol. Physiol. 2018, 314, F388–F398. [Google Scholar] [CrossRef]
- Xu, J.X.; Lu, T.-S.; Li, S.; Wu, Y.; Ding, L.; Denker, B.M.; Bonventre, J.V.; Kong, T. Polycystin-1 and Gα12 regulate the cleavage of E-cadherin in kidney epithelial cells. Physiol. Genom. 2015, 47, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Xie, C.; Wang, H.W.; Zhou, X.J.; Schwartz, N.; Calixto, S.; Mackay, M.; Aranow, C.; Putterman, C.; Mohan, C. Elevated Urinary VCAM-1, P-Selectin, Soluble TNF Receptor-1, and CXC Chemokine Ligand 16 in Multiple Murine Lupus Strains and Human Lupus Nephritis. J. Immunol. 2007, 179, 7166–7175. [Google Scholar] [CrossRef] [Green Version]
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. von Willebrand Factor–Cleaving Protease in Thrombotic Thrombocytopenic Purpura and the Hemolytic–Uremic Syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef]
- Bramham, K.; Hilton, R.; Horsfield, C.; McDonald, V.; Camilleri, R.; Hunt, B.J. ADAMTS-13 deficiency: Can it cause chronic renal failure? Nephrol. Dial. Transplant. 2011, 26, 742–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.S.; Shalhoub, J.; Gohel, M.S.; Shepherd, A.C.; Davies, A.H. Matrix Metalloproteinases in Vascular Disease - A Potential Therapeutic Target? Curr. Vasc. Pharmacol. 2010, 8, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Donato, V.; Coppolino, G.; Campo, S.; Buemi, A.; Lacquaniti, A.; Buemi, M. Neutrophil Gelatinase–Associated Lipocalin (NGAL) as a Marker of Kidney Damage. Am. J. Kidney Dis. 2008, 52, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ninomiya, T.; Sumiyoshi, S.; Fujii, H.; Doi, Y.; Hirakata, H.; Tsuruya, K.; Iida, M.; Kiyohara, Y.; Sueishi, K. Association of Kidney Function with Coronary Atherosclerosis and Calcification in Autopsy Samples from Japanese Elders: The Hisayama Study. Am. J. Kidney Dis. 2010, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Rivoli, L.; Garofalo, C.; Faga, T.; Pelagi, E.; Perticone, M.; Serra, R.; Michael, A.; Comi, N.; Andreucci, M. Renal resistive index in chronic kidney disease patients: Possible determinants and risk profile. PLoS ONE 2020, 15, e0230020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Chiodini, P.; Minutolo, R.; Zoccali, C.; Bellizzi, V.; Conte, G.; Locatelli, F.; Tripepi, G.; Del Vecchio, L.; Mallamaci, F.; et al. Reclassification of chronic kidney disease patients for end-stage renal disease risk by proteinuria indexed to estimated glomerular filtration rate: Multicentre prospective study in nephrology clinics. Nephrol. Dial. Transplant. 2018, 35, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Muntner, P.M.; Lloyd, A.; Manns, B.J.; Klarenbach, S.; Pannu, N.; James, M.T.; Hemmelgarn, B.R.; Alberta Kidney Disease Network. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: A population-level cohort study. Lancet 2012, 380, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; De Nicola, L.; Pena, M.J.; Capitoli, G.; Garofalo, C.; Borrelli, S.; Gagliardi, I.; Antolini, L.; Andreucci, M. Precision Nephrology Is a Non-Negligible State of Mind in Clinical Research: Remember the Past to Face the Future. Nephron 2020, 144, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Ermolli, M.; Schumacher, M.; Lods, N.; Hammoud, M.; Marti, H.P. Di_erential expression of MMP-2/MMP-9 and potential benefit of an MMP inhibitor in experimental acute kidney allograft rejection. Transpl. Immunol. 2003, 11, 137–145. [Google Scholar] [CrossRef]
- Aggarwal, H.K.; Jain, D.; Talapatra, P.; Yadav, R.K.; Gupta, T.; Kathuria, K.L. Evaluation of role of doxycycline (a matrix metalloproteinase inhibitor) on renal functions in patients of diabetic nephropathy. Ren. Fail. 2010, 32, 941–946. [Google Scholar] [CrossRef]
- Steinmann-Niggli, K.; Ziswiler, R.; Küng, M.; Marti, H.P. Inhibition of matrix metalloproteinases attenuates anti-Thy1.1 nephritis. J. Am. Soc. Nephrol. 1998, 9, 397–407. [Google Scholar]
- Provenzano, M.; Garofalo, C.; Chiodini, P.; Mancuso, C.; Barbato, E.; De Nicola, L.; Andreucci, M. Ruolo della proteinuria nella ricerca clinica: Per ogni vecchia risposta, una nuova domanda. (Role of proteinuria in clinical research: For each old-answer, a new key-question). Recenti. Prog. Med. 2020, 111, 74–81. (In Italian) [Google Scholar] [CrossRef] [PubMed]
- Baxter, B.T.; Pearce, W.H.; Waltke, E.A.; Littooy, F.N.; Hallett, J.W., Jr.; Kent, K.C.; Upchurch, G.R., Jr.; Chaikof, E.L.; Mills, J.L.; Fleckten, B.; et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: Report of a prospective (Phase II) multicenter study. J. Vasc. Surg 2002, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hackmann, A.E.; Rubin, B.G.; Sanchez, L.A.; Geraghty, P.A.; Thompson, R.W.; Curci, J.A. A randomized, placebo-controlled trial of doxycycline after endoluminal aneurysm repair. J. Vasc. Surg. 2008, 48, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Minutolo, R.; Serra, R.; De Nicola, L. Selective endothelin A receptor antagonism in patients with proteinuric chronic kidney disease. Expert Opin. Investig. Drugs 2020. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ielapi, N.; Andreucci, M.; Licastro, N.; Faga, T.; Grande, R.; Buffone, G.; Mellace, S.; Sapienza, P.; Serra, R. Precision Medicine and Precision Nursing: The Era of Biomarkers and Precision Health. Int. J. Gen. Med. 2020, 13, 1705–1711. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
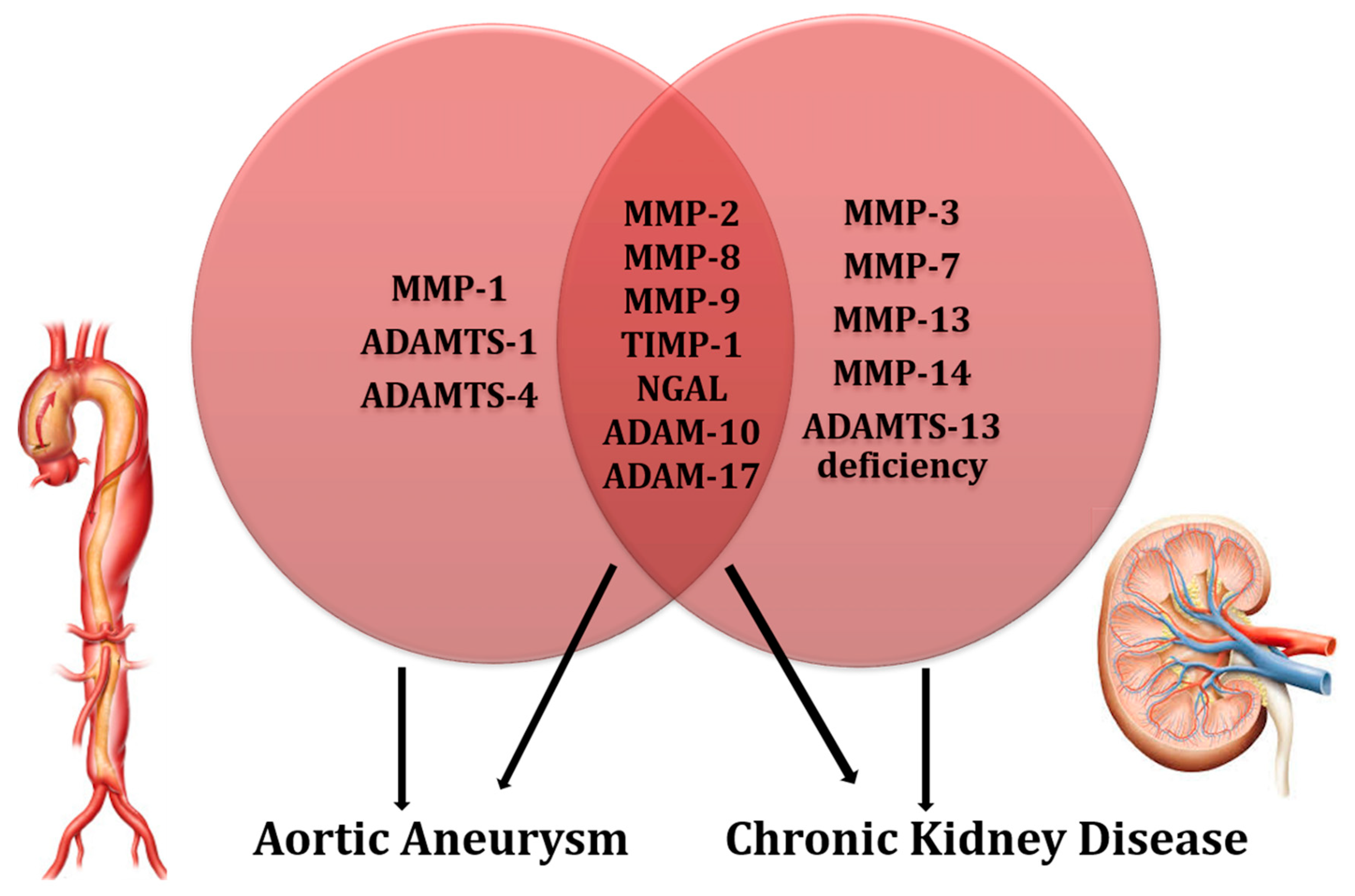
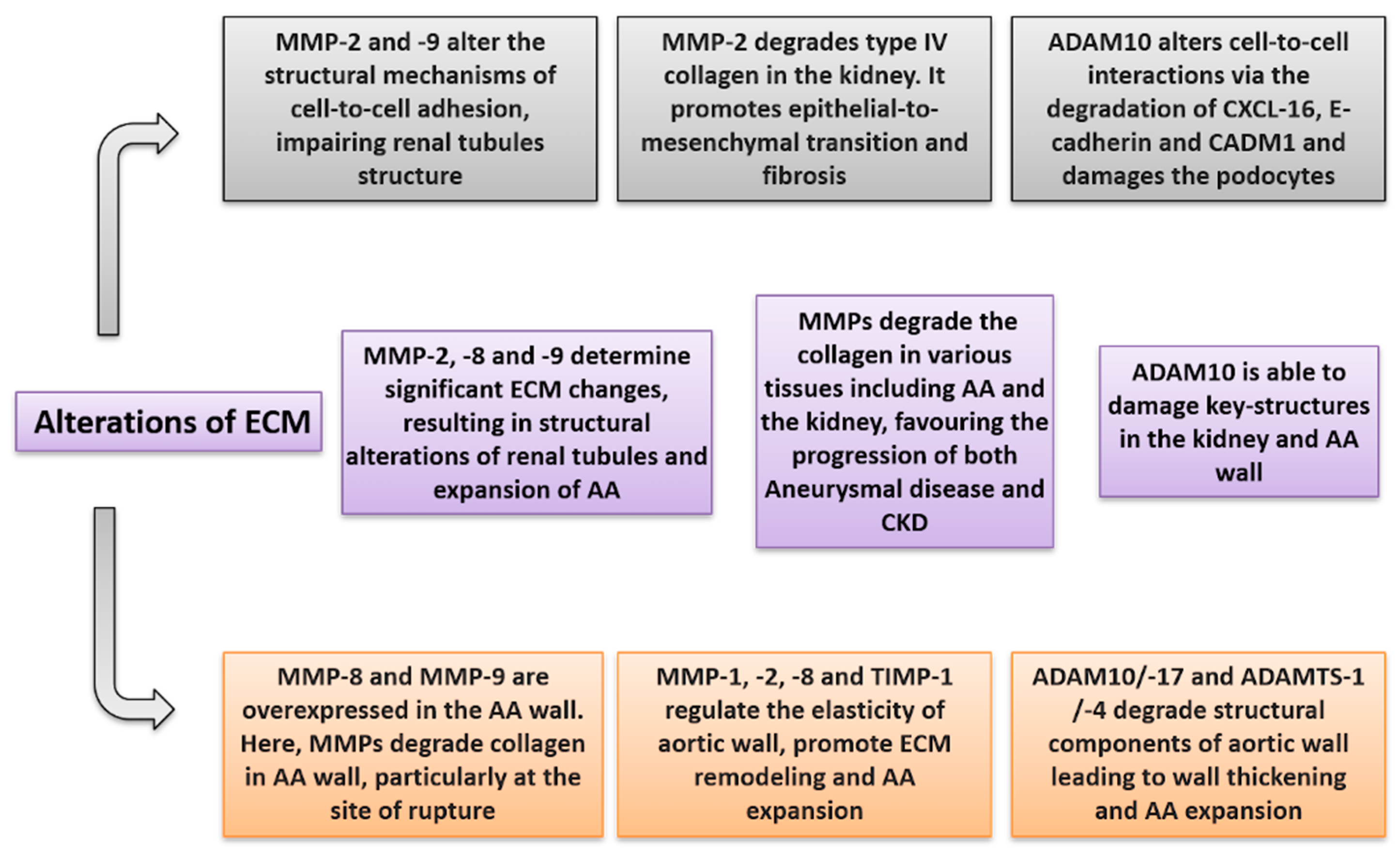
| Key-Concepts | |
|---|---|
| MPs and Aortic Aneurysm | • MMP-2 and MMP-9 are activated and overexpressed in the Aortic Aneurysm wall. Moreover, inflammatory cells producing MMP-2 and -9 are increased in the adventitial-medial junction of aortic wall. • MMP-8 and MMP-9 are increased in the site of aortic rupture where they degrade collagen, weakening the aortic wall. • MMP-1, -2, and TIMP-1 are associated with AA expansion and aortic shear stress in the AA wall. • Circulating levels of NGAL are increased in patients with AA. • ADAM10 and ADAM17 are expressed in the oldest layer of intraluminal thrombus and degrade AA collagen, favoring AA expansion. • ADAMTS1 and -4 degrade versican in the AA wall, leading to neo-intima thickening. They determine pro-inflammatory alterations on the arterial wall. |
| MPs and Chronic Kidney Disease | • Circulating MMP-2, -7, -8, -9, and NGAL levels are increased in Chronic Kidney Disease patients • MMP-9 is directly related to albuminuria levels. • MMP-2 and -9 have been found to be increased in several CKD etiologies and promote tubular damage, renal inflammation, and fibrosis. • ADAM17 cleaves ACE2, leading to an imbalance of the RAAS system; it degrades protein C receptor on the endothelium, blocking the anti-inflammatory properties of protein C. • ADAM10 degrades CADM1 and E-cadherin, determining alterations in cell-to-cell adhesions. • ADAM10 and ADAM17 degrade CXCL-16, damaging podocyte and tubular cells in the kidney. • ADAMTS13 deficiency has been associated with thrombotic thrombocytopenic purpura, glomerular, and tubular damage. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreucci, M.; Provenzano, M.; Faga, T.; Michael, A.; Patella, G.; Mastroroberto, P.; Serraino, G.F.; Bracale, U.M.; Ielapi, N.; Serra, R. Aortic Aneurysms, Chronic Kidney Disease and Metalloproteinases. Biomolecules 2021, 11, 194. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020194
Andreucci M, Provenzano M, Faga T, Michael A, Patella G, Mastroroberto P, Serraino GF, Bracale UM, Ielapi N, Serra R. Aortic Aneurysms, Chronic Kidney Disease and Metalloproteinases. Biomolecules. 2021; 11(2):194. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020194
Chicago/Turabian StyleAndreucci, Michele, Michele Provenzano, Teresa Faga, Ashour Michael, Gemma Patella, Pasquale Mastroroberto, Giuseppe Filiberto Serraino, Umberto Marcello Bracale, Nicola Ielapi, and Raffaele Serra. 2021. "Aortic Aneurysms, Chronic Kidney Disease and Metalloproteinases" Biomolecules 11, no. 2: 194. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020194