
Valency and Binding Affinity Variations Can Regulate the Multilayered Organization of Protein Condensates with Many Components

 , and
, and
Abstract
:1. Introduction
2. Methods
3. Results
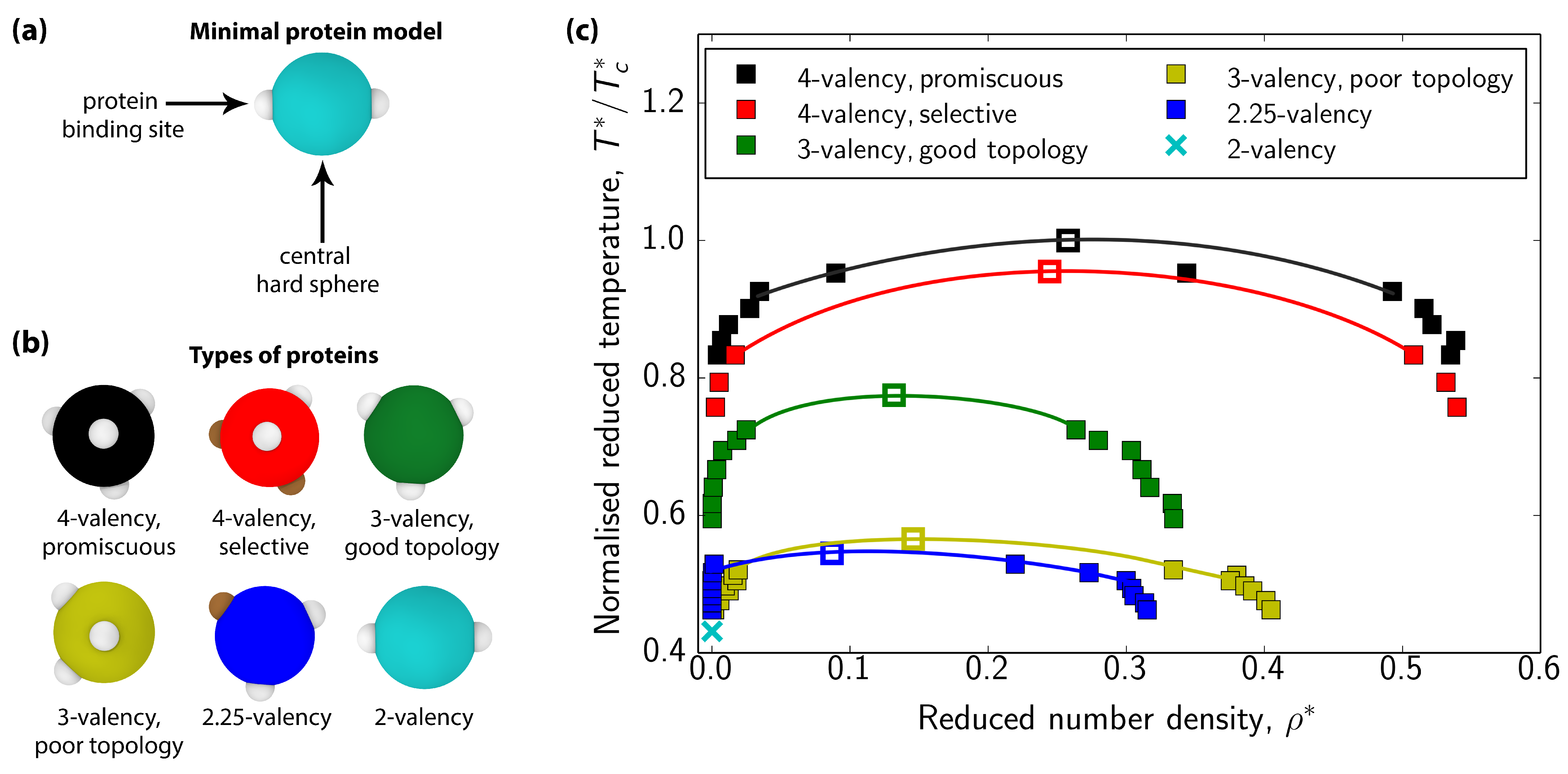
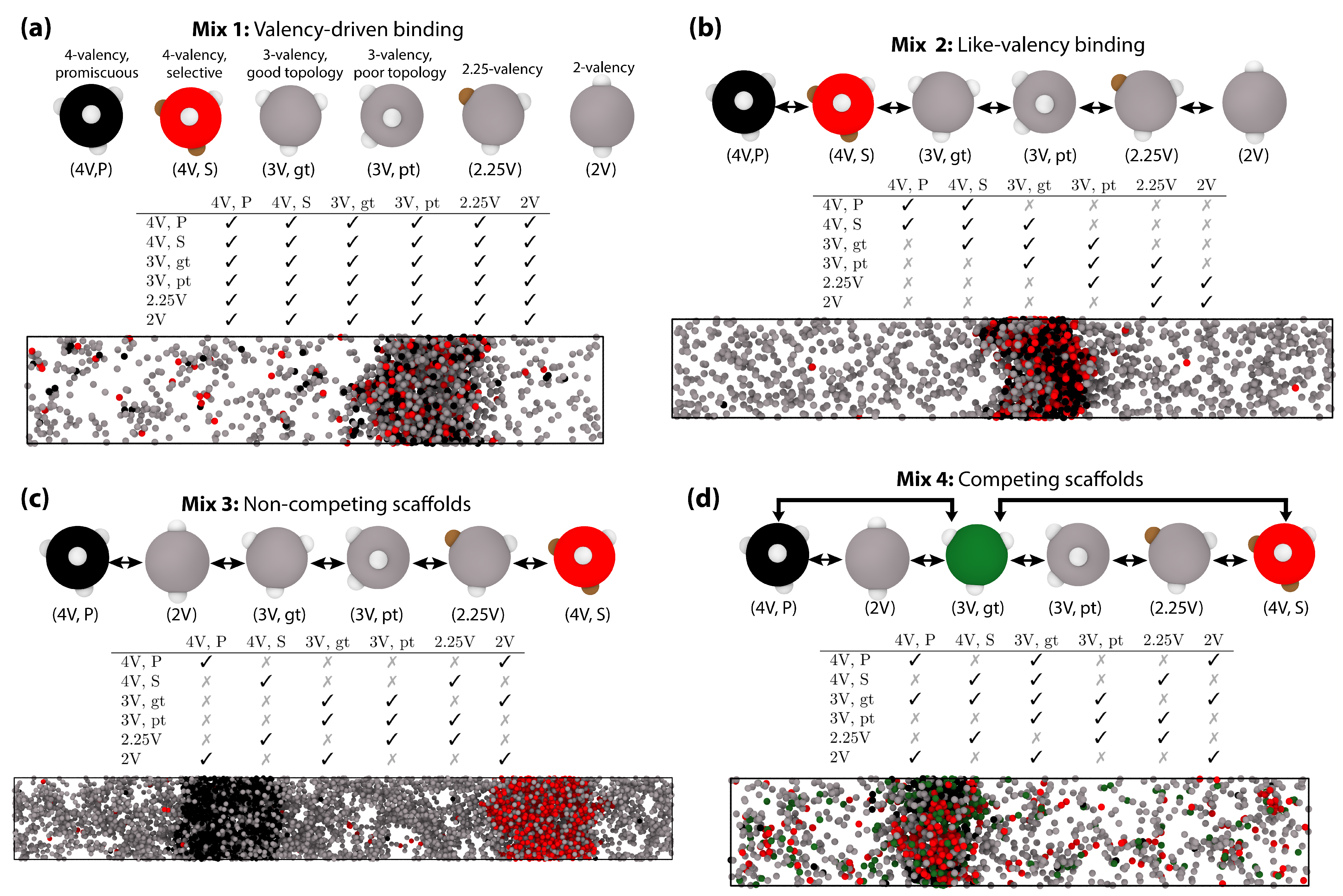
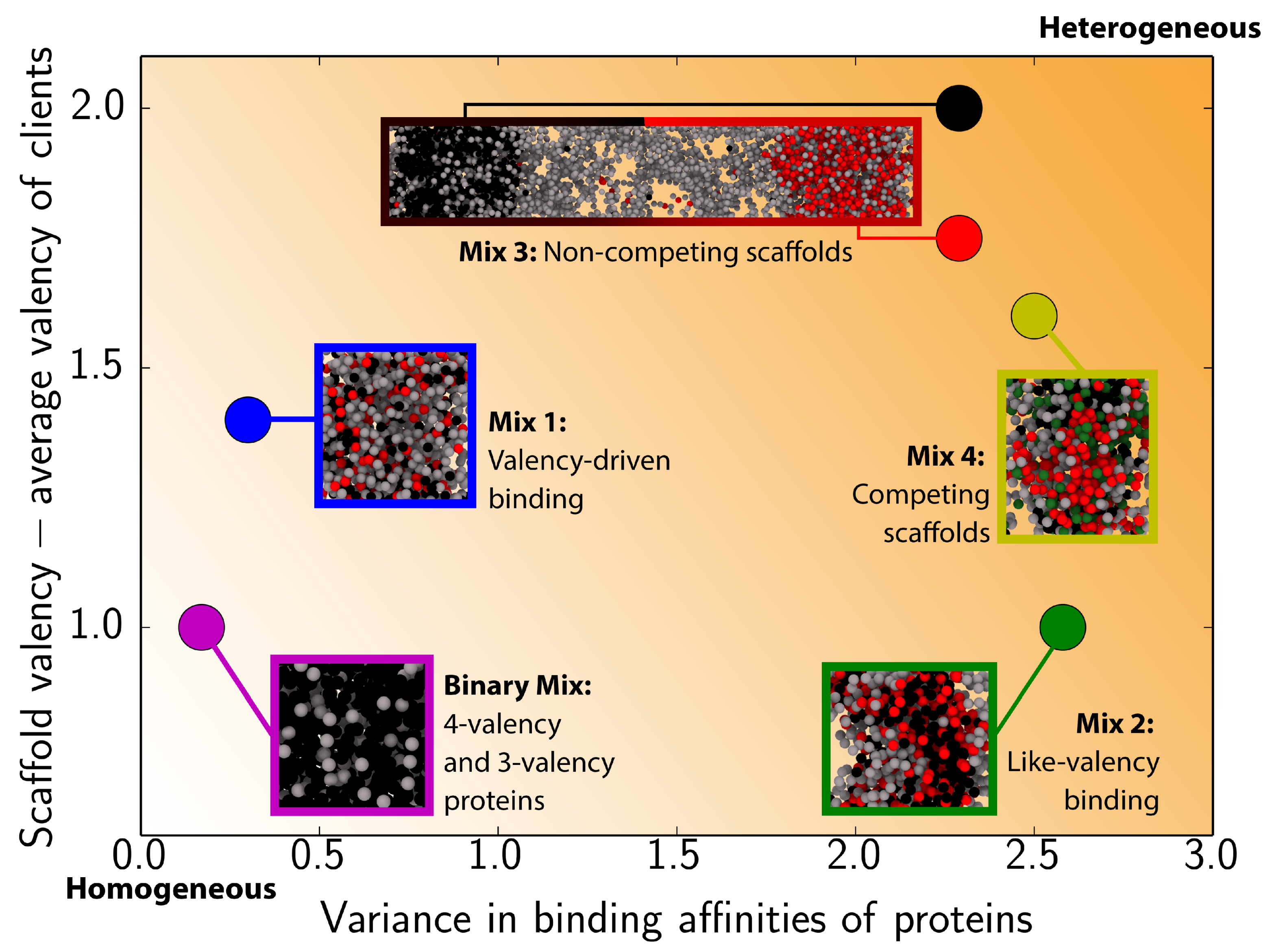
3.1. Impact of Protein Valency and Binding Affinity in the Molecular Organization of Multicomponent Condensates
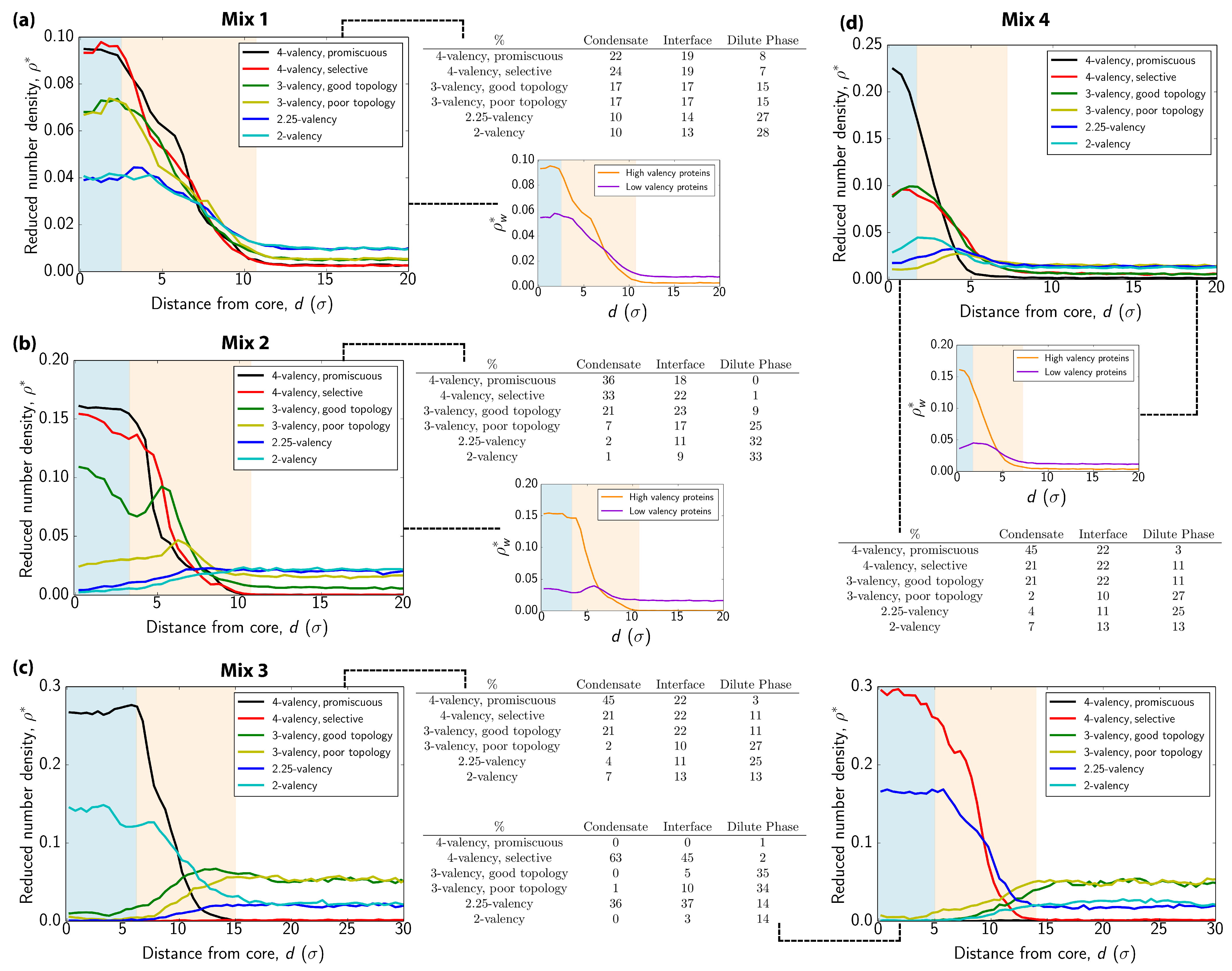
3.2. Interfacial Free Energy as a Driving Force for Multilayered Condensate Organization
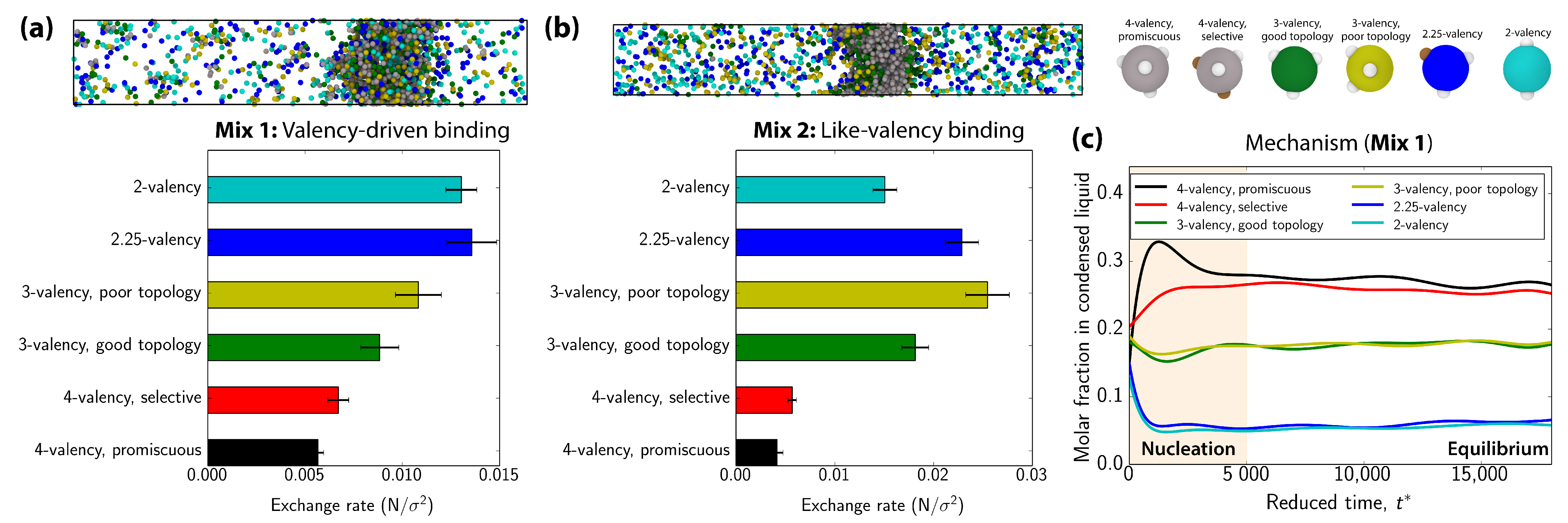
3.3. Exchange of Species in and Out of Condensates
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Simulation Details
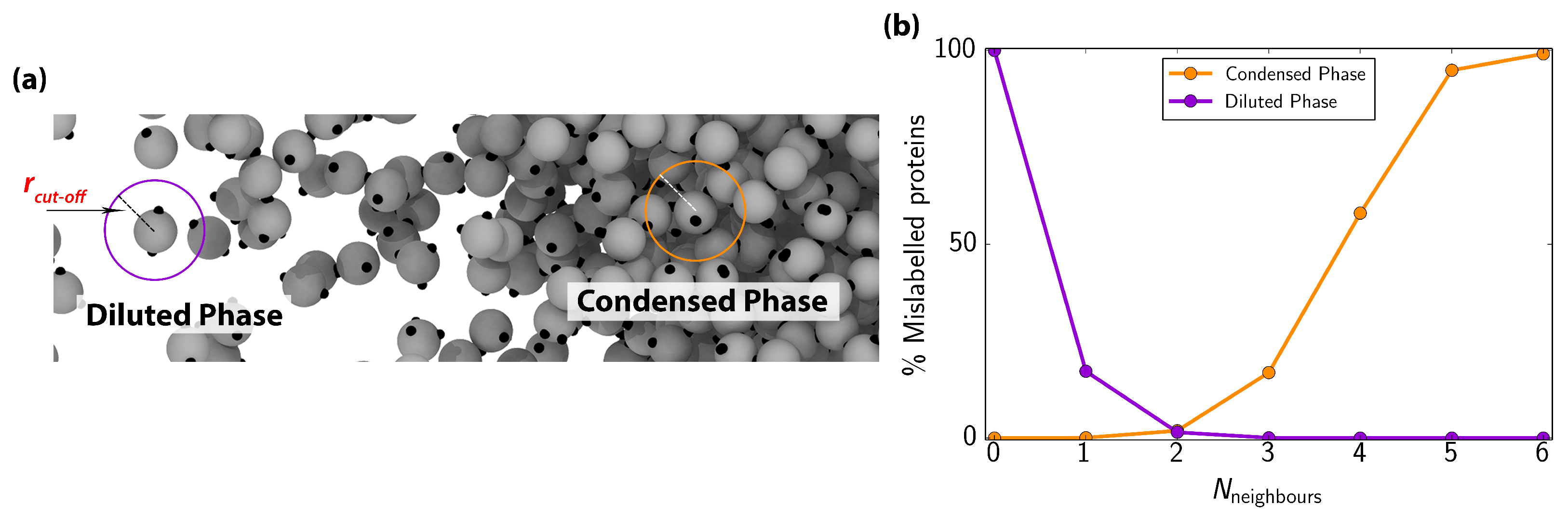
Appendix B. Local Order Parameter

References
- Sear, R.P. The cytoplasm of living cells: A functional mixture of thousands of components. J. Phys. Condens. Matter 2005, 17, S3587–S3595. [Google Scholar] [CrossRef] [Green Version]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Shin, Y.; Chang, Y.C.; Lee, D.S.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 2018, 175, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klosin, A.; Oltsch, F.; Harmon, T.; Honigmann, A.; Jülicher, F.; Hyman, A.A.; Zechner, C. Phase separation provides a mechanism to reduce noise in cells. Science 2020, 367, 464–468. [Google Scholar] [CrossRef]
- Sheu-Gruttadauria, J.; MacRae, I.J. Phase Transitions in the Assembly and Function of Human miRISC. Cell 2018, 173, 946–957.e16. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Alberti, S. Prion-like low-complexity sequences: Key regulators of protein solubility and phase behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef] [Green Version]
- Kroschwald, S.; Munder, M.C.; Maharana, S.; Franzmann, T.M.; Richter, D.; Ruer, M.; Hyman, A.A.; Alberti, S. Different Material States of Pub1 Condensates Define Distinct Modes of Stress Adaptation and Recovery. Cell Rep. 2018, 23, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.J.; Otero, J.H.; Scott, D.C.; Szulc, E.; Martin, E.W.; Sabri, N.; Granata, D.; Marzahn, M.R.; Lindorff-Larsen, K.; Salvatella, X.; et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 2018, 72, 19–36.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Carra, S. Quality Control of Membraneless Organelles. J. Mol. Biol. 2018, 430, 4711–4729. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting liquid phases underlie nucleolar subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [Green Version]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [CrossRef]
- Woodruff, J.B.; Gomes, B.F.; Widlund, P.O.; Mahamid, J.; Honigmann, A.; Hyman, A.A. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell 2017, 169, 1066–1077.e10. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid–liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Melki, R.; Kopito, R.; Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s Disease: Genetics and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 193–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.; Kim, H.; Mittag, T.; Taylor, J. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Kato, M.; Wu, L.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrea, D.M.; Cika, J.A.; Stanley, C.B.; Nourse, A.; Onuchic, P.L.; Banerjee, P.R.; Phillips, A.H.; Park, C.G.; Deniz, A.A.; Kriwacki, R.W. Self-interaction of NPM1 modulates multiple mechanisms of liquid–liquid phase separation. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Asherie, N.; Pande, J.; Lomakin, A.; Ogun, O.; Hanson, S.R.; Smith, J.B.; Benedek, G.B. Oligomerization and phase separation in globular protein solutions. Biophys. Chem. 1998, 75, 213–227. [Google Scholar] [CrossRef]
- Sun, X.S.; Wang, D.; Zhang, L.; Mo, X.; Zhu, L. Morphology and phase separation of hydrophobic clusters of soy globular protein polymers. Macromol. Biosci. 2008, 8, 295–303. [Google Scholar] [CrossRef]
- Joseph, J.A.; Espinosa, J.R.; Sanchez-Burgos, I.; Garaizar, A.; Frenkel, D.; Collepardo-Guevara, R. Thermodynamics and kinetics of phase separation of protein–RNA mixtures by a minimal model. Biophys. J. 2021. [Google Scholar] [CrossRef]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Holehouse, A.S.; Weinhardt, V.; Kovacs, D.; Van Lindt, J.; Larabell, C.; Van Den Bosch, L.; Das, R.; Tompa, P.S.; Pappu, R.V.; et al. Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc. Natl. Acad. Sci. USA 2019, 116, 7889–7898. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing protein-RNA interaction networks control multiphase intracellular organization. Cell 2020, 181, 306–324. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Kuo, P.H.; Chu, L.Y.; Golzarroshan, B.; Jain, M.; Yuan, H.S. RNA recognition motifs of disease-linked RNA-binding proteins contribute to amyloid formation. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2020, 1–13. [Google Scholar] [CrossRef]
- Loughlin, F.E.; Wilce, J.A. TDP-43 and FUS—Structural insights into RNA recognition and self-association. Curr. Opin. Struct. Biol. 2019, 59, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M. The RNA face of phase separation. Science 2018, 360, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlüssler, R.; Kim, K.; Trussina, I.R.; Wang, J.; Mateju, D.; et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 2020, 181, 346–361. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.R.; Joseph, J.A.; Sanchez-Burgos, I.; Garaizar, A.; Frenkel, D.; Collepardo-Guevara, R. Liquid network connectivity regulates the stability and composition of biomolecular condensates with many components. Proc. Natl. Acad. Sci. USA 2020, 117, 13238–13247. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S. Phase separation in biology. Curr. Biol. 2017, 27, R1097–R1102. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nott, T.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.; Pawson, T.; Forman-Kay, J.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, J.; Jadaliha, M.; Harmon, T.S.; Li, I.T.S.; Hua, B.; Hao, Q.; Holehouse, A.S.; Reyer, M.; Sun, Q.; Freier, S.M.; et al. Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J. Cell Sci. 2017, 130, 4180–4192. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Spruijt, E. Multiphase complex coacervate droplets. J. Am. Chem. Soc. 2020, 142, 2905–2914. [Google Scholar] [CrossRef] [Green Version]
- Mountain, G.A.; Keating, C.D. Formation of Multiphase Complex Coacervates and Partitioning of Biomolecules within them. Biomacromolecules 2019, 21, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Raju, M.; Alshareedah, I.; Davis, R.B.; Potoyan, D.A.; Banerjee, P.R. Sequence-encoded and composition-dependent protein-RNA interactions control multiphasic condensate topologies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fisher, R.S.; Elbaum-Garfinkle, S. Tunable multiphase dynamics of arginine and lysine liquid condensates. Nat. Commun. 2020, 11, 4628. [Google Scholar] [CrossRef]
- Jacobs, W.M.; Frenkel, D. Phase transitions in biological systems with many components. Biophys. J. 2017, 112, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Dar, F.; Pappu, R.V. Multidimensional Phase Diagrams for Multicomponent Systems Comprising Multivalent Proteins. Biophys. J. 2020, 118, 213a. [Google Scholar] [CrossRef]
- Espinosa, J.R.; Garaizar, A.; Vega, C.; Frenkel, D.; Collepardo-Guevara, R. Breakdown of the law of rectilinear diameter and related surprises in the liquid-vapor coexistence in systems of patchy particles. J. Chem. Phys. 2019, 150, 224510. [Google Scholar] [CrossRef]
- Wheeler, R.J.; Hyman, A.A. Controlling compartmentalization by non-membrane-bound organelles. Philos. Trans. R. Soc. Biol. Sci. 2018, 373, 20170193. [Google Scholar] [CrossRef]
- Strom, A.R.; Brangwynne, C.P. The liquid nucleome—Phase transitions in the nucleus at a glance. J. Cell Sci. 2019, 132, jcs235093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloni, M.; Bailly, R.; Ciandrini, L.; Barducci, A. Unraveling Molecular Interactions in Liquid–Liquid Phase Separation of Disordered Proteins by Atomistic Simulations. J. Phys. Chem. B 2020, 124, 9009–9016. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Dignon, G.L.; Jovic, N.; Xu, X.; Regy, R.M.; Fawzi, N.L.; Kim, Y.C.; Best, R.B.; Mittal, J. Molecular Details of Protein Condensates Probed by Microsecond Long Atomistic Simulations. J. Phys. Chem. B 2020, 124, 11671–11679. [Google Scholar] [CrossRef]
- Welsh, T.J.; Krainer, G.; Espinosa, J.R.; Joseph, J.A.; Sridhar, A.; Jahnel, M.; Arter, W.E.; Saar, K.L.; Alberti, S.; Collepardo- Guevara, R.; et al. Single particle zeta-potential measurements reveal the role of electrostatics in protein condensate stability. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Nguemaha, V.; Zhou, H.X. Liquid-Liquid Phase Separation of Patchy Particles Illuminates Diverse Effects of Regulatory Components on Protein Droplet Formation. Sci. Rep. 2018, 8, 6728. [Google Scholar] [CrossRef] [Green Version]
- Dignon, G.L.; Zheng, W.; Mittal, J. Simulation methods for liquid–liquid phase separation of disordered proteins. Curr. Opin. Chem. Eng. 2019, 23, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Pak, A.J.; Voth, G.A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119–126. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V.; Holehouse, A.S. Conformational preferences and phase behavior of intrinsically disordered low complexity sequences: Insights from multiscale simulations. Curr. Opin. Struct. Biol. 2019, 56, 1–10. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Liu, H.; Kumar, S.K.; Sciortino, F. Vapor-liquid coexistence of patchy models: Relevance to protein phase behavior. J. Chem. Phys. 2007, 127, 084902. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.Y.; Aksimentiev, A. Single-Protein Collapse Determines Phase Equilibria of a Biological Condensate. J. Phys. Chem. Lett. 2020, 11, 4923–4929. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Harmon, T.S.; Pappu, R.V. CAMELOT: A machine learning approach for coarse-grained simulations of aggregation of block-copolymeric protein sequences. J. Chem. Phys. 2015, 143, 243123. [Google Scholar] [CrossRef] [Green Version]
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. eLife 2017, 6, e30294. [Google Scholar] [CrossRef]
- Choi, J.M.; Dar, F.; Pappu, R.V. LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput. Biol. 2019, 15, e1007028. [Google Scholar] [CrossRef] [Green Version]
- Garaizar, A.; Sanchez-Burgos, I.; Collepardo-Guevara, R.; Espinosa, J.R. Expansion of Intrinsically Disordered Proteins Increases the Range of Stability of Liquid–Liquid Phase Separation. Molecules 2020, 25, 4705. [Google Scholar] [CrossRef] [PubMed]
- Krainer, G.; Welsh, T.J.; Joseph, J.A.; Espinosa, J.R.; Wittmann, S.; de Csilléry, E.; Sridhar, A.; Toprakcioglu, Z.; Gudiškytė, G.; Czekalska, M.A.; et al. Reentrant liquid condensate phase of proteins is stabilized by hydrophobic and non-ionic interactions. Nat. Comms. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lichtinger, S.M.; Garaizar, A.; Collepardo-Guevara, R.; Reinhardt, A. Targeted modulation of protein liquid–liquid phase separation by evolution of amino-acid sequence. bioRxiv 2020. [Google Scholar] [CrossRef]
- Schreck, J.S.; Romano, F.; Zimmer, M.H.; Louis, A.A.; Doye, J.P. Characterizing DNA star-tile-based nanostructures using a coarse-grained model. ACS Nano 2016, 10, 4236–4247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doye, J.P.; Ouldridge, T.E.; Louis, A.A.; Romano, F.; Šulc, P.; Matek, C.; Snodin, B.E.; Rovigatti, L.; Schreck, J.S.; Harrison, R.M.; et al. Coarse-graining DNA for simulations of DNA nanotechnology. Phys. Chem. Chem. Phys. 2013, 15, 20395–20414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benayad, Z.; von Bülow, S.; Stelzl, L.S.; Hummer, G. Simulation of FUS protein condensates with an adapted coarse-grained model. J. Chem. Theory Comput. 2021, 17, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Largo, J.; Tartaglia, P.; Zaccarelli, E.; Sciortino, F. Phase diagram of patchy colloids: Towards empty liquids. Phys. Rev. Lett. 2006, 97, 168301. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Tartaglia, P.; Sciortino, F. Reversible gels of patchy particles: Role of the valence. J. Chem. Phys. 2009, 131, 014504. [Google Scholar] [CrossRef]
- Russo, J.; Tartaglia, P.; Sciortino, F. Association of limited valence patchy particles in two dimensions. Soft Matter 2010, 6, 4229–4236. [Google Scholar] [CrossRef]
- Statt, A.; Casademunt, H.; Brangwynne, C.P.; Panagiotopoulos, A.Z. Model for disordered proteins with strongly sequence-dependent liquid phase behavior. J. Chem. Phys. 2020, 152, 075101. [Google Scholar] [CrossRef] [Green Version]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Forman-Kay, J.D.; Chan, H.S. Theories for Sequence-Dependent Phase Behaviors of Biomolecular Condensates. Biochemistry 2018, 57, 2499–2508. [Google Scholar] [CrossRef]
- Hazra, M.; Levy, Y. Charge pattern affects the structure and dynamics of polyampholyte condensates. Phys. Chem. Chem. Phys. 2020, 22, 19368–19375. [Google Scholar] [CrossRef] [PubMed]
- Blas, F.J.; Galindo, A.; Vega, C. Study of the solid-liquid-vapour phase equilibria of flexible chain molecules using Wertheim’s thermodynamic perturbation theory. Mol. Phys. 2003, 101, 449–458. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Rowlinson, J.S.; Widom, B. Molecular Theory of Capillarity; Courier Corporation: North Chelmsford, MA, USA, 2013. [Google Scholar]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [Green Version]
- García Fernández, R.; Abascal, J.L.F.; Vega, C. The melting point of ice Ih for common water models calculated from direct coexistence of the solid-liquid interface. J. Chem. Phys. 2006, 124, 144506. [Google Scholar] [CrossRef]
- Espinosa, J.R.; Sanz, E.; Valeriani, C.; Vega, C. On fluid-solid direct coexistence simulations: The pseudo-hard sphere model. J. Chem. Phys. 2013, 139, 144502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, Z.; Chen, N.; Huang, Y.; Huang, S. Phase separation of Arabidopsis EMB1579 controls transcription, mRNA splicing, and development. PLoS Biol. 2020, 18, e3000782. [Google Scholar] [CrossRef]
- Pazhouhandeh, M.; Molinier, J.; Berr, A.; Genschik, P. MSI4/FVE interacts with CUL4–DDB1 and a PRC2-like complex to control epigenetic regulation of flowering time in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 3430–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkwood, J.G.; Buff, F.P. The statistical mechanical theory of surface tension. J. Chem. Phys. 1949, 17, 338–343. [Google Scholar] [CrossRef]
- Vega, C.; De Miguel, E. Surface tension of the most popular models of water by using the test-area simulation method. J. Chem. Phys. 2007, 126, 154707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Ijavi, M.; Style, R.W.; Emmanouilidis, L.; Kumar, A.; Meier, S.M.; Torzynski, A.L.; Allain, F.H.; Barral, Y.; Steinmetz, M.O.; Dufresne, E.R. Surface tensiometry of phase separated protein and polymer droplets by the sessile drop method. Soft Matter 2020. [Google Scholar] [CrossRef]
- Jover, J.; Haslam, A.J.; Galindo, A.; Jackson, G.; Müller, E.A. Pseudo hard-sphere potential for use in continuous molecular-dynamics simulation of spherical and chain molecules. J. Chem. Phys. 2012, 137, 144505. [Google Scholar] [CrossRef]
- Espinosa, J.; Vega, C.; Sanz, E. The mold integration method for the calculation of the crystal-fluid interfacial free energy from simulations. J. Chem. Phys. 2014, 141, 134709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- ten Wolde, P.R.; Frenkel, D. Computer simulation study of gas–iquid nucleation in a Lennard-Jones system. J. Chem. Phys. 1998, 109, 9901–9918. [Google Scholar] [CrossRef]
- ten Wolde, P.R.; Frenkel, D. Numerical study of gas–liquid nucleation in partially miscible binary mixtures. J. Chem. Phys. 1998, 109, 9919–9927. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Burgos, I.; de Hijes, P.M.; Rosales-Pelaez, P.; Vega, C.; Sanz, E. Condensation and boiling in a Lennard-Jones fluid. Phys. Rev. E 2020, 102, 062609. [Google Scholar] [CrossRef]
- Kashchiev, D. Nucleation; Elsevier: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Kelton, K.F. Crystal nucleation in liquids and glasses. In Solid State Physics; Elsevier: Amsterdam, The Netherlands, 1991; Volume 45, pp. 75–177. [Google Scholar]
- Espinosa, J.R.; Vega, C.; Valeriani, C.; Sanz, E. Seeding approach to crystal nucleation. J. Chem. Phys. 2016, 144, 034501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Breen, D. Calculating center of mass in an unbounded 2D environment. J. Graph. Tools 2008, 13, 53–60. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Tension, | ||
|---|---|---|
| System | ||
| Pure component 4-valency promiscuous | 1.44 ± 0.2 | 1.94 ± 0.2 |
| Valency-driven binding mixture | 0.05 ± 0.04 | - |
| Like-valency binding mixture | 0.33 ± 0.20 | - |
| Non-competing scaffolds mixture | - | 0.2 ± 0.15 & 0.21 ± 0.1 |
| Competing scaffolds mixture | 0.09 ± 0.04 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Burgos, I.; Espinosa, J.R.; Joseph, J.A.; Collepardo-Guevara, R. Valency and Binding Affinity Variations Can Regulate the Multilayered Organization of Protein Condensates with Many Components. Biomolecules 2021, 11, 278. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020278
Sanchez-Burgos I, Espinosa JR, Joseph JA, Collepardo-Guevara R. Valency and Binding Affinity Variations Can Regulate the Multilayered Organization of Protein Condensates with Many Components. Biomolecules. 2021; 11(2):278. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020278
Chicago/Turabian StyleSanchez-Burgos, Ignacio, Jorge R. Espinosa, Jerelle A. Joseph, and Rosana Collepardo-Guevara. 2021. "Valency and Binding Affinity Variations Can Regulate the Multilayered Organization of Protein Condensates with Many Components" Biomolecules 11, no. 2: 278. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020278