
CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction


Abstract
:1. Introduction
2. Materials and Methods
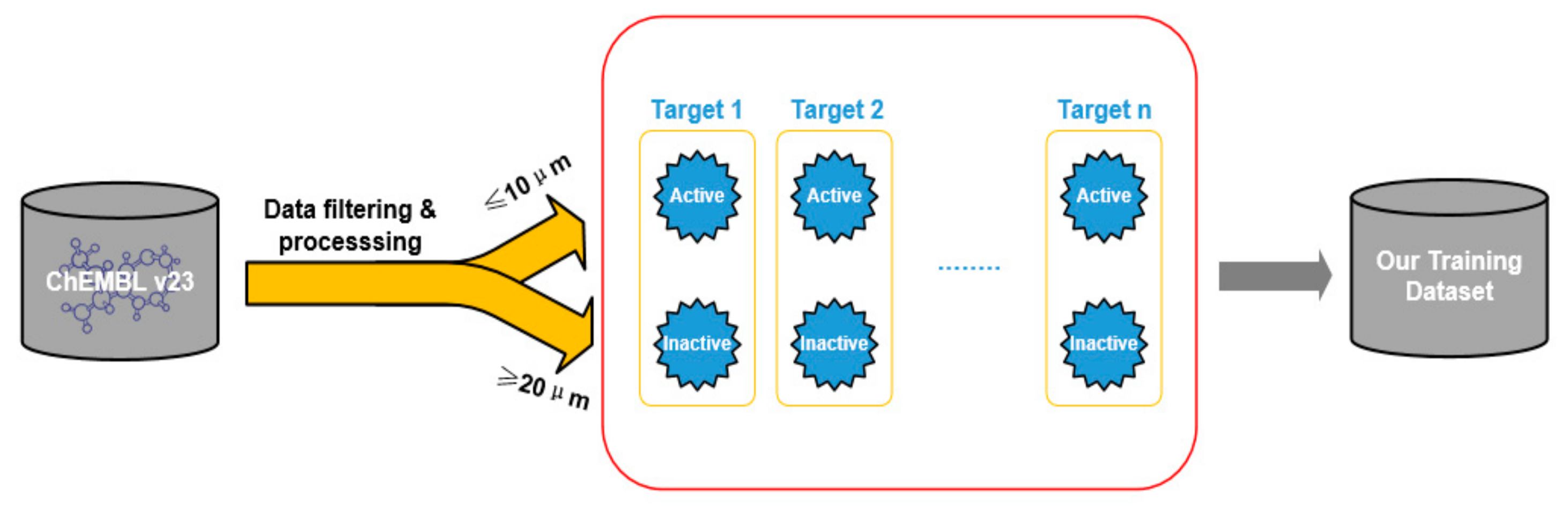
2.1. Dataset
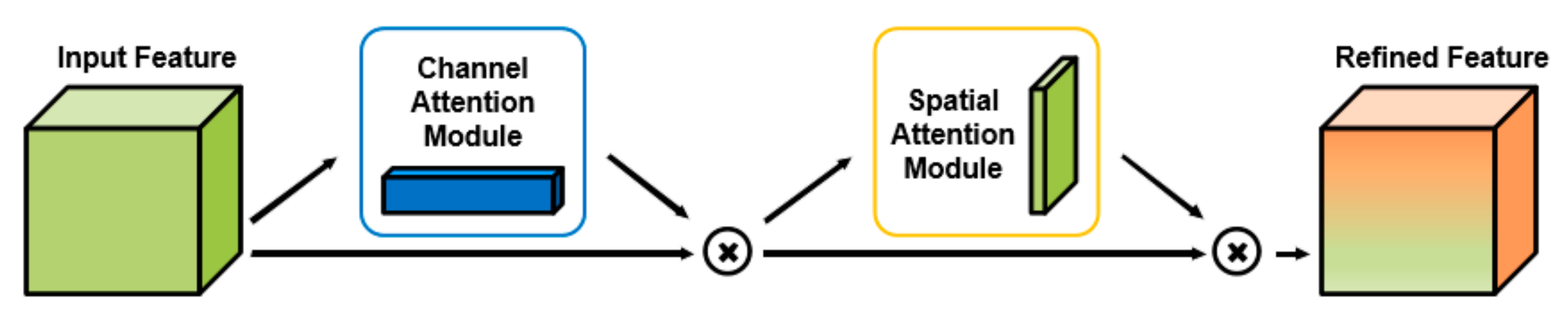
2.2. Attention Module
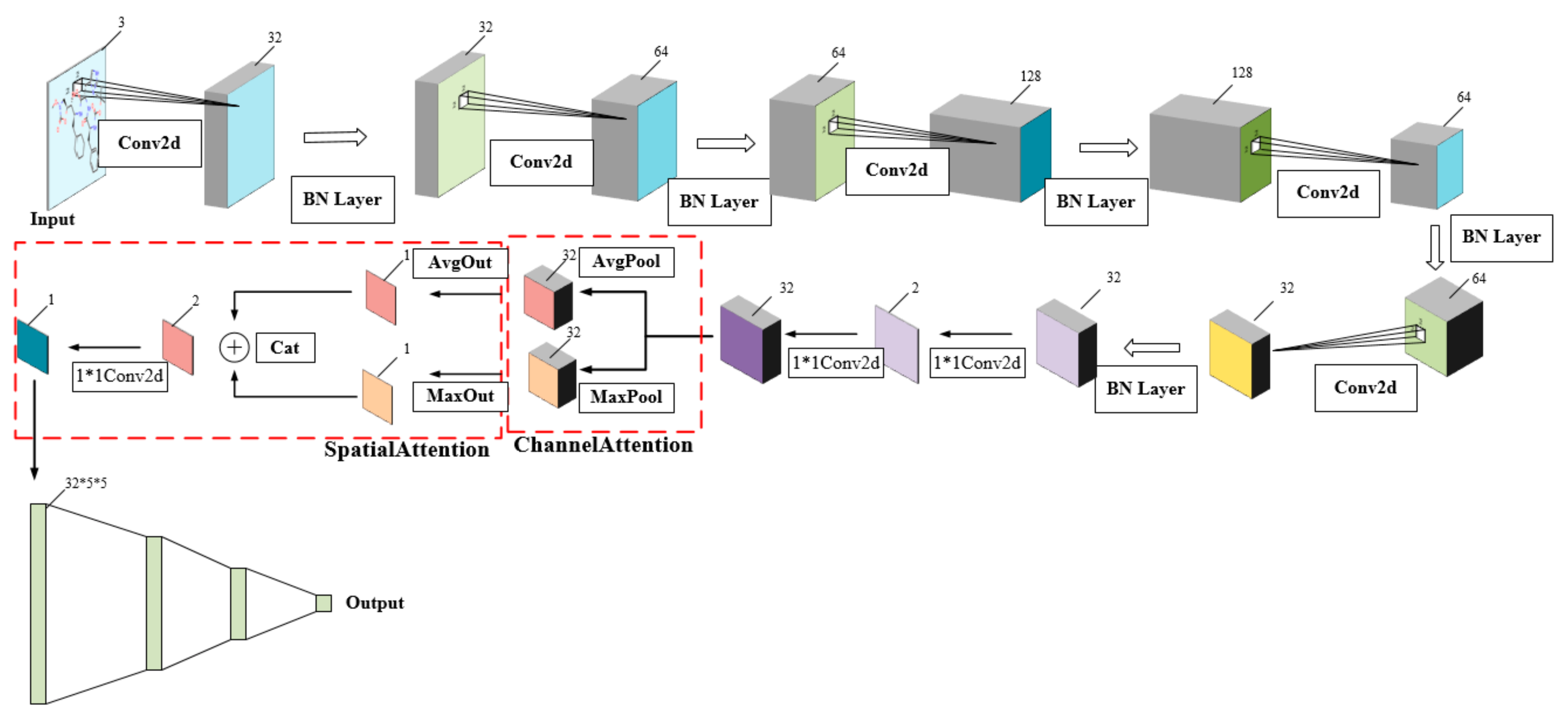
2.3. Our CSConv2d Model
2.4. Evaluation Metrics
3. Results
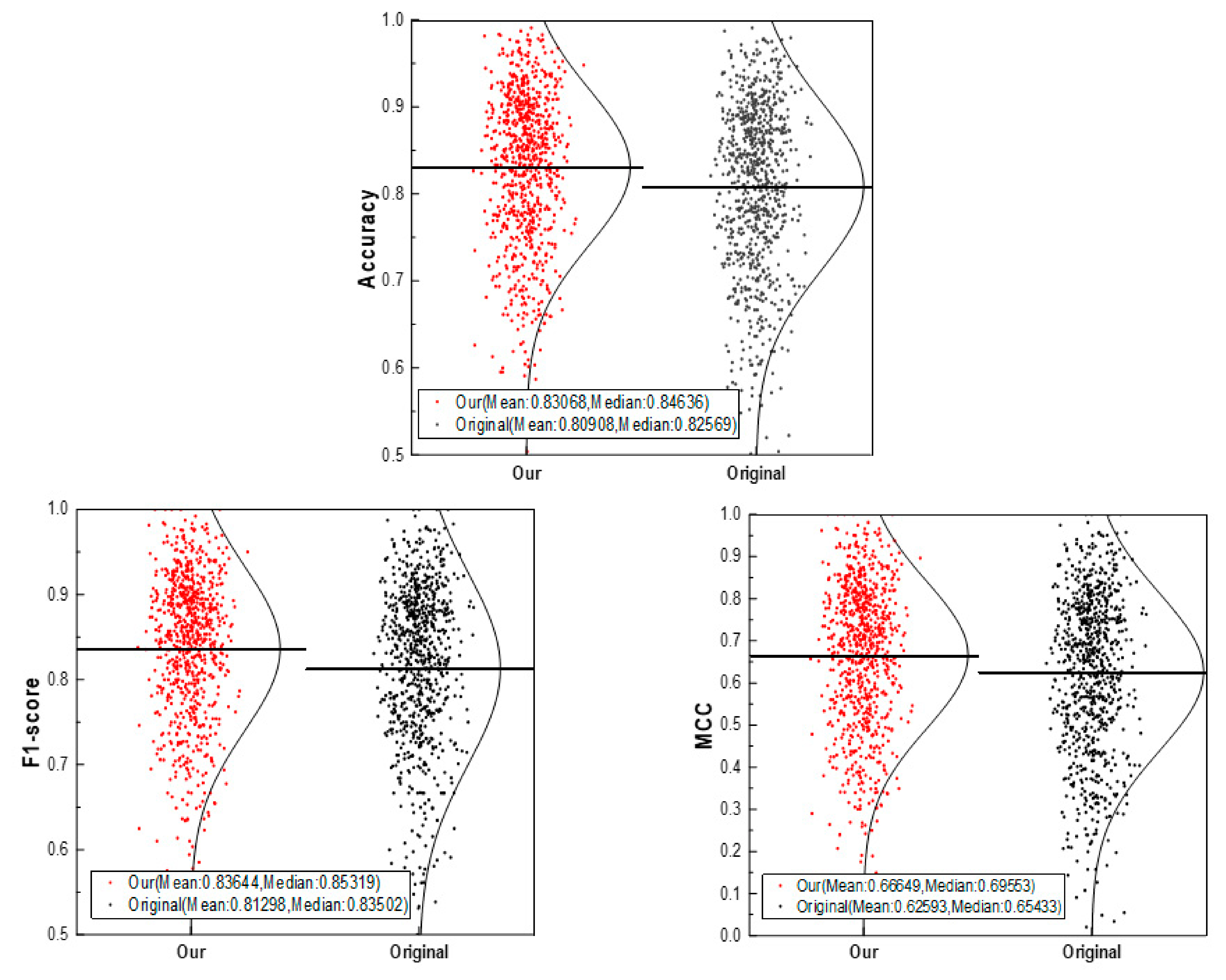
3.1. Performance of the CSConv2d Model
3.2. Comparison with Different Models
3.3. The Robustness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, 1202–1213. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008, 24, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y. Random Forest for Bioinformatics, Ensemble Machine Learning; Springer: Boston, MA, USA, 2012; pp. 307–323. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, D.A.; Ponce, J. Computer Vision: A Modern Approach, 2/E[M]; Prentice Hall: Hoboken, NJ, USA, 2002. [Google Scholar]
- Halle, M.; Stevens, K. Speech recognition: A model and a program for research. IEEE Trans. Inf. Theory 1962, 8, 155–159. [Google Scholar] [CrossRef]
- Chowdhury, G.G. Natural language processing. Annu. Rev. Inf. Sci. Technol. 2003, 37, 51–89. [Google Scholar] [CrossRef] [Green Version]
- Hinton, G.E. Deep belief networks. Scholarpedia 2009, 4, 5947. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Le, H.; Quinn, T.P.; Venkatesh, S. GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 2020, btaa921. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformartics 2021, 36, 5545–5547. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhao, K.; Xiao, T. DeepGS: Deep representation learning of graphs and sequences for drug-target binding affinity prediction. arXiv 2020, arXiv:2003.13902. [Google Scholar]
- Woo, S.; Park, J.; Lee, J.-Y.; Kweon, I.S. CBAM: Convolutional block attention module. In Proceedings of the European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 3–19. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Modeling 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Lowe, R.; KalantarMotamedi, Y.; Mussa, H.Y.; Klaffke, W.; Mitchell, J.B.; Glen, R.C.; Bender, A. In Silico target predictions: Defining a benchmarking data set and com-parison of performance of the multiclass Naïve Bayes and Parzen-Rosenblatt window. J. Chem. Inf. Modeling 2013, 53, 1957–1966. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Nalbat, E.; Atalay, V.; Martin, M.J.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 2020, 11, 2531–2557. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Ten Dijke, N.; Bongers, B.; Papadatos, G.; Van Vlijmen, H.W.; Kowalczyk, W.; IJzerman, A.P.; Van Westen, G.J. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set (Dataset). 4TU.ResearchData. J. Cheminformatics 2017, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Harris, S.F.; Gibbons, P.; Deshmukh, G.; Gustafson, A.; Kellar, T.; Lin, H.; Liu, X.; Liu, Y.; Liu, Y.; et al. Scaffold-hopping and structure-based discovery of potent, selective, and brain penetrant N-(1 H-pyrazol-3-yl) pyridin-2-amine inhibitors of dual leucine zipper kinase (DLK, MAP3K12). J. Med. Chem. 2015, 58, 8182–8199. [Google Scholar] [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2010, 39, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient op-timization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | ACC | F1-Score | MCC |
|---|---|---|---|
| CSConv2d (our) | 0.83 | 0.84 | 0.67 |
| DEEPScreen (Original) | 0.81 | 0.81 | 0.63 |
| Model | ACC | F1-Score | MCC |
|---|---|---|---|
| DeepConv-DTI | 0.79 | 0.80 | 0.64 |
| CPI-Prediction CPI-Prediction+CS DeepGS DeepGS+CS CSConv2d | 0.76 0.80 0.83 0.85 0.87 | 0.81 0.83 0.85 0.86 0.89 | 0.63 0.67 0.66 0.70 0.72 |
| Dataset | Model | MCC |
|---|---|---|
| ChEMBL Bioactivity Benchmark Set [23] | Feed-forward DNN PCM | 0.38 |
| CPI-Prediction+CS | 0.42 | |
| DeepGS+CS | 0.47 | |
| DEEPScreen | 0.47 | |
| CSConv2d | 0.57 |
| Compound ID | Affinity (kcal/mol) | Pred (CSConv2d) | Pred (DEEPScreen) | Label |
|---|---|---|---|---|
| 50D | −9.2 | 1 | 0 | 1 |
| CHEMBL3731242 | −8.7 | 1 | 1 | 1 |
| CHEMBL3729274 | −8.1 | 1 | 1 | 1 |
| CHEMBL3355005 | −7.9 | 1 | 1 | 1 |
| CHEMBL3727745 | −7.9 | 1 | 0 | 1 |
| CHEMBL1242663 | −7.5 | 0 | 0 | 0 |
| CHEMBL1767275 | −7.4 | 0 | 1 | 0 |
| CHEMBL598911 | −6.8 | 1 | 0 | 0 |
| CHEMBL206659 | −6.2 | 0 | 1 | 0 |
| CHEMBL25829 | −4.7 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Liu, D.; Zhu, J.; Rodriguez-Paton, A.; Song, T. CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction. Biomolecules 2021, 11, 643. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050643
Wang X, Liu D, Zhu J, Rodriguez-Paton A, Song T. CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction. Biomolecules. 2021; 11(5):643. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050643
Chicago/Turabian StyleWang, Xun, Dayan Liu, Jinfu Zhu, Alfonso Rodriguez-Paton, and Tao Song. 2021. "CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction" Biomolecules 11, no. 5: 643. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050643