
Lower Gene Expression of Angiotensin Converting Enzyme 2 Receptor in Lung Tissues of Smokers with COVID-19 Pneumonia

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Molecular Analyses
2.3. Statistical Analysis
3. Results
3.1. Study Population
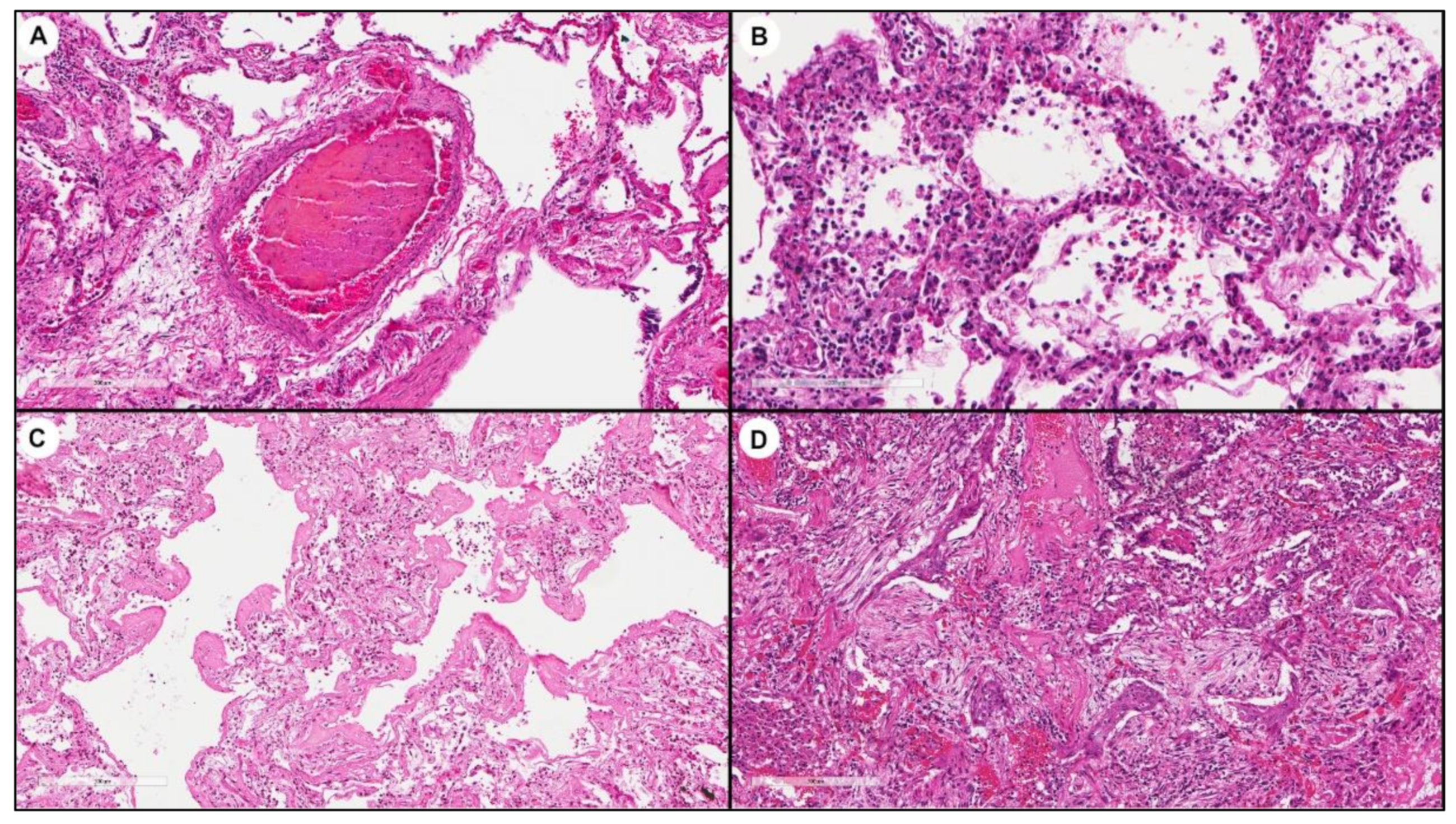
3.2. Molecular Analyses
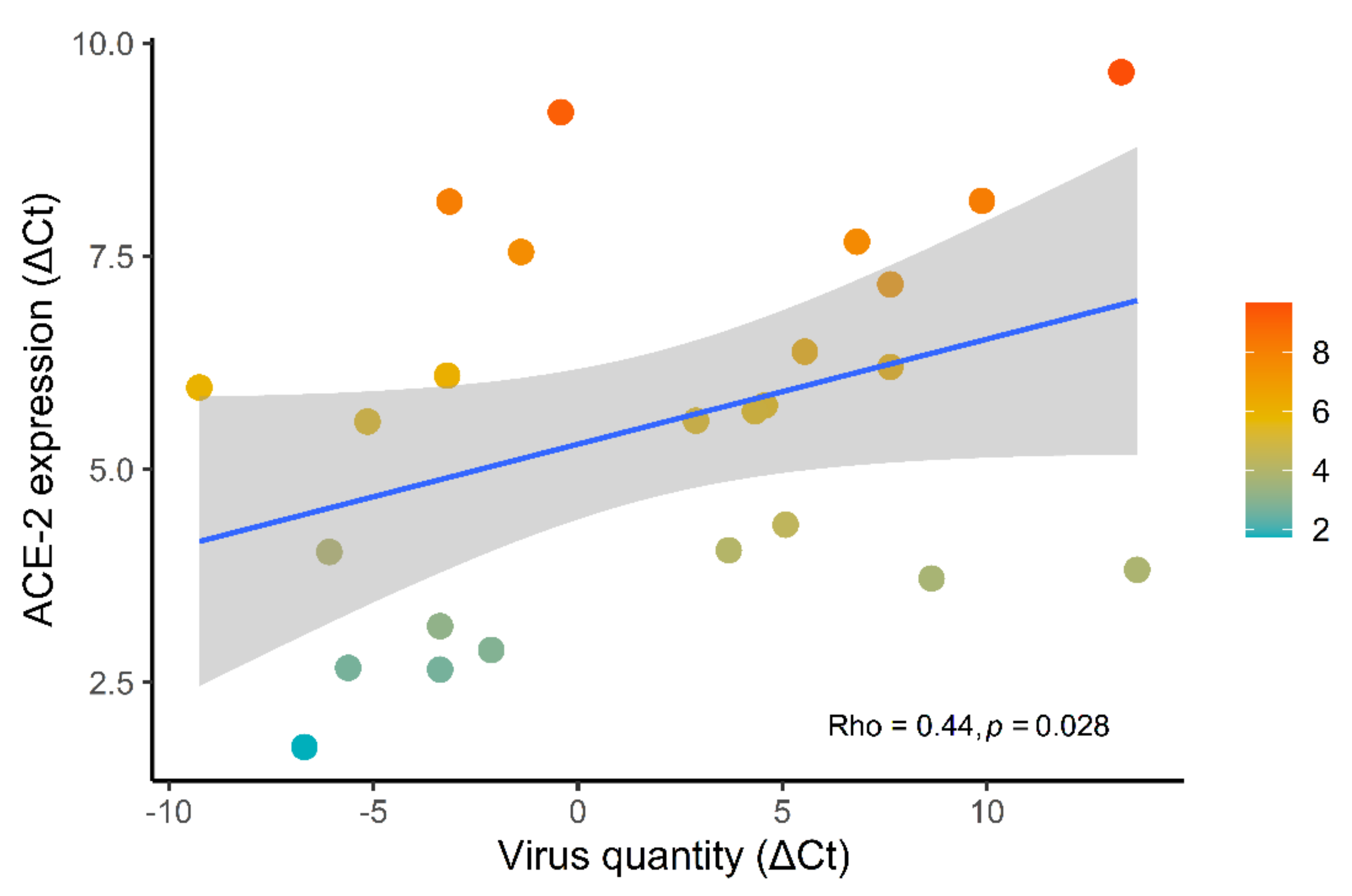
3.3. Correlations between ACE-2 Expression and Clinicopathological Data
- The effect of smoking (Median = −0.73, 95% CI [−1.41, −0.06]) has a 98.17% probability of being negative (<0), 97.63% of being significant (<−0.05), and 90.42% of being large (<−0.30).
- The effect of age (Median = −0.12, 95% CI [−0.45, 0.22]) has a 76.37% probability of being negative, 65.70% of being significant, and 14.38% of being large.
- The effect of gender (female) (Median = −0.23, 95% CI [−0.90, 0.40]) has a 75.82% probability of being negative, 70.98% of being significant, and 40.55% of being large.
- The effect of hypertension (Median = 0.38, 95% CI [−0.25, 1.03]) has a 88.28% probability of being positive (>0), 84.58% of being significant (>0.05), and 58.97% of being large (>0.30).
- The effect of virus quantity (Median = 0.33, 95% CI [−0.05, 0.72]) has a 95.37% probability of being positive, 92.63% of being significant, and 56.38% of being large.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johns Hopkins University Coronavirus Resource Center. COVID-19 Dash-Board by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. Available online: https://0-coronavirus-jhu-edu.brum.beds.ac.uk/map.html (accessed on 29 December 2020).
- Phua, J.; Weng, L.; Ling, L.; Egi, M.; Lim, C.-M.; Divatia, J.V.; Shrestha, B.R.; Arabi, Y.M.; Ng, J.; Gomersall, C.D.; et al. Intensive Care Management of Coronavirus Disease 2019 (COVID-19): Challenges and Recommendations. Lancet Respir. Med. 2020, 8, 506–517. [Google Scholar] [CrossRef]
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV-2 spike protein with altered human ACE-2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 538, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE-2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue Distribution of ACE-2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Salvatore, S.P.; Seshan, S.V.; Patel, S.S.; Bussel, J.B.; Mostyka, M.; Elsoukkary, S.; He, B.; Del Vecchio, C.; Fortarezza, F.; et al. COVID-19 Pulmonary Pathology: A Multi-Institutional Autopsy Cohort from Italy and New York City. Mod. Pathol. 2020, 33, 2156–2168. [Google Scholar] [CrossRef]
- Calabrese, F.; Fortarezza, F.; Giraudo, C.; Pezzuto, F.; Faccioli, E.; Rea, F.; Pittarello, D.; Correale, C.; Navalesi, P. Two Sorts of Microthrombi in a Patient with Coronavirus Disease 2019 and Lung Cancer. J. Thorac. Oncol. 2020, 15, 1782–1785. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE-2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High Expression of ACE-2 Receptor of 2019-NCoV on the Epithelial Cells of Oral Mucosa. Int. J. Oral Sci. 2020, 12, 1–5. [Google Scholar] [CrossRef]
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on Receptors for Coronaviruses with Special Reference to Angiotensin—Converting Enzyme 2 as a Potential Drug Target—A Perspective. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 807–811. [Google Scholar] [CrossRef]
- Li, J.; Gao, J.; Xu, Y.P.; Zhou, T.L.; Jin, Y.Y.; Lou, J.N. Expression of severe acute respiratory syndrome coronavirus receptors, ACE-2 and CD209L in different organ derived microvascular endothelial cells. Zhonghua Yi Xue Za Zhi 2007, 87, 833–837. [Google Scholar]
- Hung, Y.H.; Hsieh, W.Y.; Hsieh, J.S.; Liu, F.C.; Tsai, C.H.; Lu, L.C.; Huang, C.Y.; Wu, C.L.; Lin, C.S. Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE-2 Knockout Mice. Int. J. Biol. Sci. 2016, 12, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Nawijn, M.C.; Timens, W. Can ACE 2 expression explain SARS -CoV-2 infection of the respiratory epithelia in COVID -19? Mol. Syst. Biol. 2020, 16, 1–3. [Google Scholar] [CrossRef]
- Ortiz, M.E.; Thurman, A.; Pezzulo, A.A.; Leidinger, M.R.; Klesney-Tait, J.A.; Karp, P.H.; Tan, P.; Wohlford-Lenane, C.; McCray, P.B., Jr.; Meyerholz, D.K. Heterogeneous Expression of the SARS-Coronavirus-2 Receptor ACE-2 in the Human Respiratory Tract. EBioMedicine 2020, 60, 102976. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal Histopathological Analysis of 26 Postmortem Findings of Patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.-L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-Converting Enzyme 2 (ACE-2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Leung, J.M.; Sin, D.D. Smoking, ACE-2 and COVID-19: Ongoing Controversies. Eur. Respir. J. 2020, 56, 1–8. [Google Scholar] [CrossRef]
- Farsalinos, K.; Angelopoulou, A.; Alexandris, N.; Poulas, K. COVID-19 and the Nicotinic Cholinergic System. Eur. Respir. J. 2020, 56, 1–8. [Google Scholar] [CrossRef]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and Smoking: Is Nicotine the Hidden Link? Eur. Respir. J. 2020, 55, 1–5. [Google Scholar] [CrossRef]
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Alqahtani, J.S.; Oyelade, T.; Aldhahir, A.M.; Alghamdi, S.M.; Almehmadi, M.; Alqahtani, A.S.; Quaderi, S.; Mandal, S.; Hurst, J.R. Prevalence, Severity and Mortality Associated with COPD and Smoking in Patients with COVID-19: A Rapid Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0233147. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.-L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 Expression in the Small Airway Epithelia of Smokers and COPD Patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Calabrese, F.; Sbaraglia, M.; Del Vecchio, C.; Carretta, G.; Saieva, A.; Donato, D.; Flor, L.; Crisanti, A.; Dei Tos, A.P. Feasibility of postmortem examination in the era of COVID-19 pandemic: The experience of a Northeast Italy University Hospital. Virchows Arch. 2020, 477, 341–347. [Google Scholar] [CrossRef]
- World Health Organization. Clinical Management of Severe Acute Respiratory Infection when Novel Coronavirus (nCoV) Infection is Suspected. Who. 2020. Available online: https://www.who.int/publications/i/item/10665-332299 (accessed on 12 January 2020).
- Calabrese, F.; Pezzuto, F.; Fortarezza, F.; Hofman, P.; Kern, I.; Panizo, A.; von der Thüsen, J.; Timofeev, S.; Gorkiewicz, G.; Lunardi, F. Pulmonary pathology and COVID-19: Lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020, 477, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Pezzuto, F.; Fortarezza, F.; Boscolo, A.; Lunardi, F.; Giraudo, C.; Cattelan, A.; Del Vecchio, C.; Lorenzoni, G.; Vedovelli, L.; et al. Machine learning-based analysis of alveolar and vascular injury in SARS-CoV-2 acute respiratory failure. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654–1665. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 18 May 2021).
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Yu, T.; Cai, S.; Zheng, Z.; Cai, X.; Liu, Y.; Yin, S.; Peng, J.; Xu, X. Association Between Clinical Manifestations and Prognosis in Patients with COVID-19. Clin. Ther. 2020, 42, 964–972. [Google Scholar] [CrossRef]
- Patanavanich, R.; Glantz, S.A. Smoking Is Associated With COVID-19 Progression: A Meta-analysis. Nicotine Tob. Res. 2020, 22, 1653–1656. [Google Scholar] [CrossRef]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Lian, N.; Deng, Y.; Lin, S. The impact of COPD and smoking history on the severity of COVID-19: A systemic review and meta-analysis. J. Med. Virol. 2020, 92, 1915–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020, 81, 16–25. [Google Scholar] [CrossRef]
- Guo, F.R. Active smoking is associated with severity of coronavirus disease 2019 (COVID-19): An update of a meta-analysis. Tob. Induc. Dis. 2020, 18, 37–39. [Google Scholar] [CrossRef]
- Lowe, K.E.; Zein, J.; Hatipoglu, U.; Attaway, A. Association of Smoking and Cumulative Pack-Year Exposure With COVID-19 Outcomes in the Cleveland Clinic COVID-19 Registry. JAMA Intern. Med. 2021, 25, 1–3. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, S.; Han, T.; Qu, G.; Cheng, C.; Uy, J.P.; Shaikh, M.B.; Zhou, Q.; Song, E.J.; Sun, C. Association of smoking history with severe and critical outcome in COVID-19 patients: A systemic review and meta-analysis. Eur. J. Integr. Med. 2021, 43, 1–11. [Google Scholar] [CrossRef]
- Umnuaypornlert, A.; Kanchanasurakit, S.; Lucero-Prisno, D.E.I.; Saokaew, S. Smoking and risk of negative outcomes among COVID-19 patients: A systematic review and meta-analysis. Tob. Induc. Dis. 2021, 19, 9–22. [Google Scholar] [CrossRef]
- Hopkinson, N.S.; Rossi, N.; El-Sayed, M.J.; Laverty, A.A.; Quint, J.K.; Freidin, M.; Visconti, A.; Murray, B.; Modat, M.; Ourselin, S.; et al. Current smoking and COVID-19 risk: Results from a population symptom app in over 2.4 million people. Thorax 2021, 1–9. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Suri, R.; Emeruwa, I.O.; Stretch, R.J.; Cortes-Lopez, R.Y.; Sherman, A.; Lindsay, C.C.; Fulcher, J.A.; Goodman-Meza, D.; Sapru, A.; et al. Obesity and smoking as risk factors for invasive mechanical ventilation in COVID-19: A retrospective, observational cohort study. PLoS ONE 2020, 15, 1–13. [Google Scholar] [CrossRef]
- Farsalinos, K.; Barbouni, A.; Niaura, R. Systematic review of the prevalence of current smoking among hospitalized COVID-19 patients in China: Could nicotine be a therapeutic option? Reply. Intern. Emerg. Med. 2021, 16, 235–236. [Google Scholar] [CrossRef]
- Emami, A.; Javanmardi, F.; Pirbonyeh, N.; Akbari, A. Prevalence of Underlying Diseases in Hospitalized Patients with COVID-19: A Systematic Review and Meta-Analysis. Arch. Acad. Emerg. Med. 2020, 8, 1–14. [Google Scholar]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.; Chernyak, Y.; Tobin, K.A.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: Prospective cohort study. BMJ 2020, 369, 1–15. [Google Scholar] [CrossRef]
- Lippi, G.; Henry, B.M. Active smoking is not associated with severity of coronavirus disease 2019 (COVID-19). Eur. J. Intern. Med. 2020, 75, 107–108. [Google Scholar] [CrossRef]
- Farsalinos, K.; Bagos, P.G.; Giannouchos, T.; Niaura, R.; Barbouni, A.; Poulas, K. Smoking prevalence among hospitalized COVID-19 patients and its association with disease severity and mortality: An expanded re-analysis of a recent publication. Harm Reduct. J. 2021, 18, 1–9. [Google Scholar] [CrossRef]
- Paleiron, N.; Mayet, A.; Marbac, V.; Perisse, A.; Barazzutti, H.; Brocq, F.X.; Janvier, F.; Bertrand, D.; Bylicki, O. Impact of Tobacco Smoking on the risk of COVID-19. A large scale retrospective cohort study. Nicotine Tob. Res. 2021, 1–7. [Google Scholar] [CrossRef]
- Shastri, M.D.; Shukla, S.D.; Chong, W.C.; Kc, R.; Dua, K.; Patel, R.P.; Peterson, G.M.; O’Toole, R.F. Smoking and COVID-19: What we know so far. Respir. Med. 2020, 176, 1–7. [Google Scholar] [CrossRef]
- Rossato, M.; Russo, L.; Mazzocut, S.; Di Vincenzo, A.; Fioretto, P.; Vettor, R. Current smoking is not associated with COVID-19. Eur. Respir. J. 2020, 55, 1–3. [Google Scholar] [CrossRef]
- Cai, G.; Bossé, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco smoking increases the lung gene expression of ACE-2, the receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE-2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.; Van Eeckhoutte, H.P.; Wijnant, S.R.A.; Janssens, W.; Joos, G.F.; Brusselle, G.G.; Bracke, K.R. Increased expression of ACE-2, the SARS-CoV-2 entry receptor, in alveolar and bronchial epithelium of smokers and COPD subjects. Eur. Respir. J. 2020, 56, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE-2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529. [Google Scholar] [CrossRef]
- Aguiar, J.A.; Tremblay, B.J.; Mansfield, M.J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A.; et al. Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 2020, 56, 1–17. [Google Scholar] [CrossRef]
- Sifat, A.E.; Nozohouri, S.; Villalba, H.; Vaidya, B.; Abbruscato, T.J. The Role of Smoking and Nicotine in the Transmission and Pathogenesis of COVID-19. J. Pharmacol. Exp. Ther. 2020, 375, 498–509. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Copeland, R.L.; Aschner, M. Nicotine and the nicotinic cholinergic system in COVID-19. FEBS J. 2020, 287, 3656–3663. [Google Scholar] [CrossRef]
- Lupacchini, L.; Maggi, F.; Tomino, C.; De Dominicis, C.; Mollinari, C.; Fini, M.; Bonassi, S.; Merlo, D.; Russo, P. Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity. Molecules 2020, 26, 101. [Google Scholar] [CrossRef]
- Olds, J.L.; Kabbani, N. Is nicotine exposure linked to cardiopulmonary vulnerability to COVID-19 in the general population? FEBS J. 2020, 287, 3651–3655. [Google Scholar] [CrossRef] [Green Version]
- Diabasana, Z.; Perotin, J.M.; Belgacemi, R.; Ancel, J.; Mulette, P.; Delepine, G.; Gosset, P.; Maskos, U.; Polette, M.; Deslée, G.; et al. Nicotinic Receptor Subunits Atlas in the Adult Human Lung. Int. J. Mol. Sci. 2020, 21, 7446. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE-2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef]
- Polverino, F. Cigarette Smoking and COVID-19: A Complex Interaction. Am. J. Respir. Crit. Care Med. 2020, 202, 471–472. [Google Scholar] [CrossRef]
- Tomchaney, M.; Contoli, M.; Mayo, J.; Baraldo, S.; Shuaizhi, L.; Cabel, C.R.; Bull, D.A.; Lick, S.; Malo, J.; Knoper, S.; et al. Paradoxical effects of cigarette smoke and COPD on SARS-CoV2 infection and disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Haitao, T.; Vermunt, J.V.; Abeykoon, J.; Ghamrawi, R.; Gunaratne, M.; Jayachandran, M.; Narang, K.; Parashuram, S.; Suvakov, S.; Garovic, V.D. COVID-19 and Sex Differences: Mechanisms and Biomarkers. Mayo Clin. Proc. 2020, 95, 2189–2203. [Google Scholar] [CrossRef]
- Viveiros, A.; Rasmuson, J.; Vu, J.; Mulvagh, S.L.; Yip, C.Y.Y.; Norris, C.M.; Oudit, G.Y. Sex differences in COVID-19: Candidate pathways, genetics of ACE2, and sex hormones. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, 296–304. [Google Scholar] [CrossRef]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE-2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE-2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun. 2020, 112, 1–7. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactio.s identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Rockx, B.; Baas, T.; Zornetzer, G.A.; Haagmans, B.; Sheahan, T.; Frieman, M.; Dyer, M.D.; Teal, T.H.; Proll, S.; van den Brand, J.; et al. Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J. Virol. 2009, 83, 7062–7074. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE-2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006, 210, 288–297. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE-2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Luo, W.; Huang, L.; Xiao, J.; Li, F.; Qin, S.; Song, X.; Wu, Y.; Zeng, Q.; et al. A comprehensive investigation of the mRNA and protein level of ACE-2, the putative receptor of SARS-CoV-2, in human tissues and blood cells. Int. J. Med. Sci. 2020, 17, 1522–1531. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE-2 in human tissues. Mol. Syst. Biol. 2020, 16, 1–16. [Google Scholar] [CrossRef]
- Luo, F.; Darwiche, K.; Singh, S.; Torrego, A.; Steinfort, D.P.; Gasparini, S.; Liu, D.; Zhang, W.; Fernandez-Bussy, S.; Herth, F.J.F.; et al. Performing Bronchoscopy in Times of the COVID-19 Pandemic: Practice Statement from an International Expert Panel. Respiration 2020, 99, 417–422. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | N = 29 1 |
|---|---|
| Age (years) | 82 (75–87) |
| Gender | |
| Male | 17 (59%) |
| Female | 12 (41%) |
| Smokers | 10 (38%) |
| (Missing) | 3 |
| Comorbidities (overall) | 28 (97%) |
| Cardiovascular comorbidities | 25 (86%) |
| Hypertension | 17 (59%) |
| Other infections | 11 (38%) |
| Symptoms (overall) | |
| Yes | 29 (100%) |
| Cough | 17 (59%) |
| Dyspnea | 26 (90%) |
| Fever | 21 (78%) |
| (Missing) | 2 |
| Intensive care unit admission | 13 (45%) |
| Disease duration (days) | 9 (7–22) |
| Characteristic | High ACE-2 Expressors (ΔCt < 5.6), N = 16 1 | Low ACE-2 Expressors (ΔCt ≥ 5.6), N = 13 1 | p-Value 2 | q-Value 3 |
|---|---|---|---|---|
| Age (years) | 86 (77–90) | 79 (74–83) | 0.072 | 0.3 |
| Age | 0.2 | 0.4 | ||
| <82years | 6 (38%) | 8 (62%) | ||
| ≥82years | 10 (62%) | 5 (38%) | ||
| Gender | 0.3 | 0.4 | ||
| Male | 8 (50%) | 9 (69%) | ||
| Female | 8 (50%) | 4 (31%) | ||
| Smokers | 2 (14%) | 8 (67%) | 0.014 | 0.11 |
| (Missing) | 2 | 1 | ||
| ACE-2 expression (ΔCt) | 3.92 (2.87–4.37) | 7.17 (6.10–8.14) | <0.001 | <0.001 |
| Virus quantity (ΔCt) | −2.7 (−5.3–4.0) | 4.6 (−1.4–7.6) | 0.13 | 0.4 |
| Disease duration (days) | 8 (6–12) | 12 (9–24) | 0.2 | 0.4 |
| Comorbidities (overall) | 15 (94%) | 10 (77%) | 0.3 | 0.4 |
| Hypertension | 11 (69%) | 6 (46%) | 0.2 | 0.4 |
| Other infections | 7 (44%) | 4 (31%) | 0.7 | 0.8 |
| Cardiovascular comorbidities | 15 (94%) | 10 (77%) | 0.3 | 0.4 |
| Morphological diagnosis | 0.049 | 0.3 | ||
| Vascular | 9 (56%) | 4 (31%) | ||
| Mixed | 7 (44%) | 5 (38%) | ||
| ALI | 0 (0%) | 4 (31%) | ||
| Symptoms (overall) | 16 (100%) | 13 (100%) | ||
| Cough | 9 (56%) | 8 (62%) | 0.8 | 0.8 |
| Dyspnea | 13 (81%) | 13 (100%) | 0.2 | 0.4 |
| Fever | 11 (79%) | 10 (77%) | >0.9 | >0.9 |
| (Missing) | 2 | 0 | ||
| Intensive care unit admission | 5 (31%) | 8 (62%) | 0.10 | 0.3 |
| Characteristic | Smokers N = 10 1 | Non-Smokers N = 16 1 | p-Value 2 | q-Value 3 |
|---|---|---|---|---|
| Age (years) | 80 (77–84) | 81 (74–87) | 0.9 | >0.9 |
| Age | >0.9 | >0.9 | ||
| <82years | 5 (50%) | 8 (50%) | ||
| ≥82years | 5 (50%) | 8 (50%) | ||
| Gender | 0.4 | >0.9 | ||
| Male | 8 (80%) | 9 (56%) | ||
| Female | 2 (20%) | 7 (44%) | ||
| ACE-2 expression (ΔCt) | 6.15 (5.75–6.97) | 4.20 (3.58–5.61) | 0.012 | 0.11 |
| Virus quantity (ΔCt) | 4.3 (−0.4–6.8) | 3.7 (−3.4–8.7) | >0.9 | >0.9 |
| ACE-2 expression | 0.014 | 0.11 | ||
| High (ΔCt < 5.6) | 2 (20%) | 12 (75%) | ||
| Low (ΔCt ≥ 5.6) | 8 (80%) | 4 (25%) | ||
| Disease duration (days) | 12 (7–20) | 10 (7–24) | >0.9 | >0.9 |
| Comorbidities (overall) | 9 (90%) | 14 (88%) | >0.9 | >0.9 |
| Hypertension | 5 (50%) | 11 (69%) | 0.4 | >0.9 |
| Other infections | 2 (20%) | 8 (50%) | 0.2 | 0.9 |
| Cardiovascular comorbidities | 9 (90%) | 14 (88%) | >0.9 | >0.9 |
| Morphological diagnosis | 0.4 | >0.9 | ||
| Vascular | 4 (40%) | 8 (50%) | ||
| Mixed | 3 (30%) | 7 (44%) | ||
| ALI | 3 (30%) | 1 (6.2%) | ||
| Symptoms (overall) | 10 (100%) | 16 (100%) | ||
| Cough | 6 (60%) | 9 (56%) | >0.9 | >0.9 |
| Dyspnea | 10 (100%) | 14 (88%) | 0.5 | >0.9 |
| Fever | 10 (100%) | 11 (73%) | 0.12 | 0.7 |
| (Missing) | 0 | 1 | ||
| Intensive care unit admission | 5 (50%) | 7 (44%) | >0.9 | >0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunardi, F.; Fortarezza, F.; Vedovelli, L.; Pezzuto, F.; Boscolo, A.; Rossato, M.; Vettor, R.; Cattelan, A.M.; Del Vecchio, C.; Crisanti, A.; et al. Lower Gene Expression of Angiotensin Converting Enzyme 2 Receptor in Lung Tissues of Smokers with COVID-19 Pneumonia. Biomolecules 2021, 11, 796. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060796
Lunardi F, Fortarezza F, Vedovelli L, Pezzuto F, Boscolo A, Rossato M, Vettor R, Cattelan AM, Del Vecchio C, Crisanti A, et al. Lower Gene Expression of Angiotensin Converting Enzyme 2 Receptor in Lung Tissues of Smokers with COVID-19 Pneumonia. Biomolecules. 2021; 11(6):796. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060796
Chicago/Turabian StyleLunardi, Francesca, Francesco Fortarezza, Luca Vedovelli, Federica Pezzuto, Annalisa Boscolo, Marco Rossato, Roberto Vettor, Anna Maria Cattelan, Claudia Del Vecchio, Andrea Crisanti, and et al. 2021. "Lower Gene Expression of Angiotensin Converting Enzyme 2 Receptor in Lung Tissues of Smokers with COVID-19 Pneumonia" Biomolecules 11, no. 6: 796. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060796