
Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID-19

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods:
2.1. Acquisition of Data
2.2. Nucleotide Composition of SARS-CoV-2
2.3. Codon Preference Characteristics
2.4. Analysis of Codon Usage in SARS-CoV-2
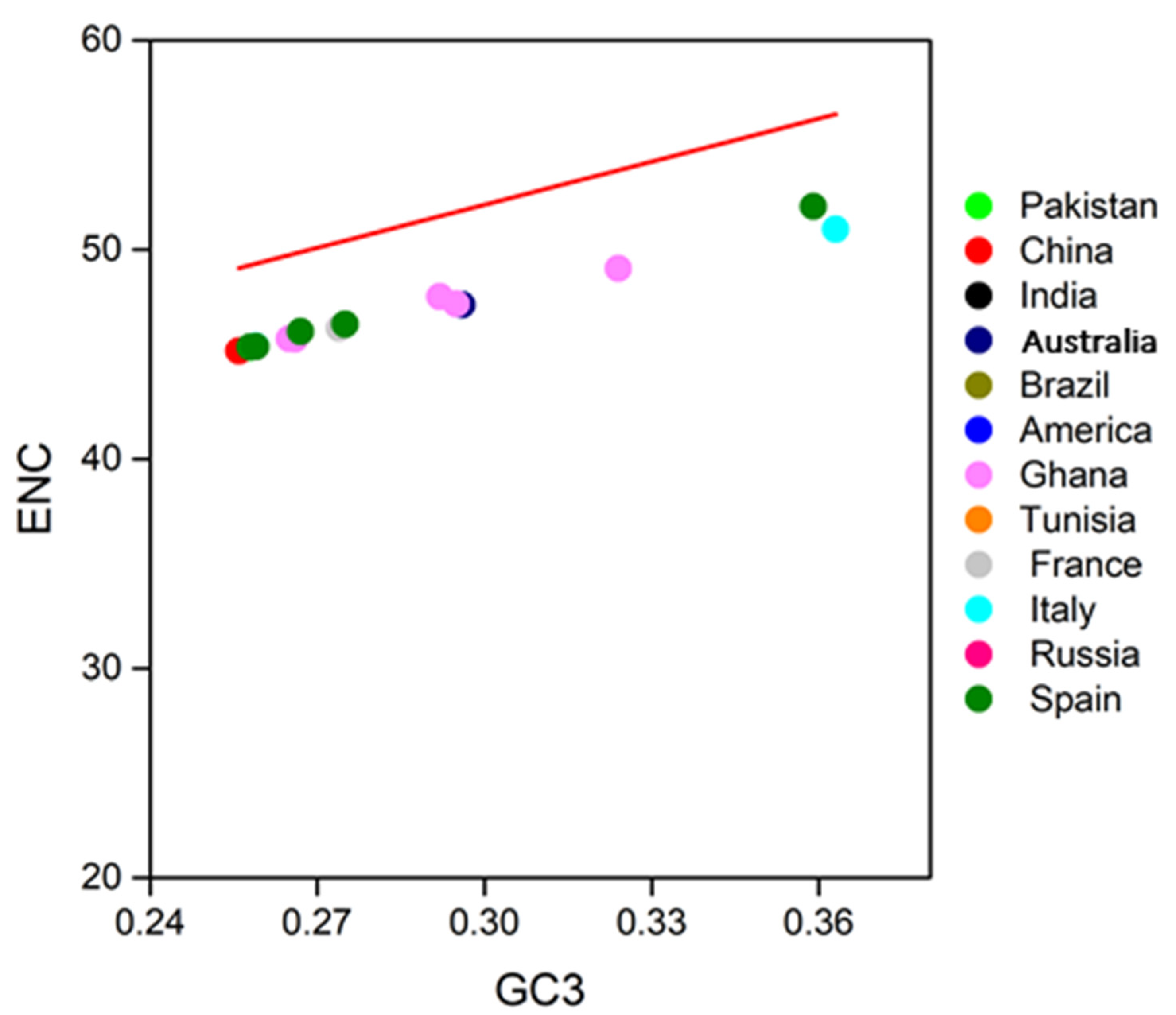
2.5. ENC-Plot Analysis
2.6. Neutral Evolution Analysis
2.7. Codon Adaptation Analysis
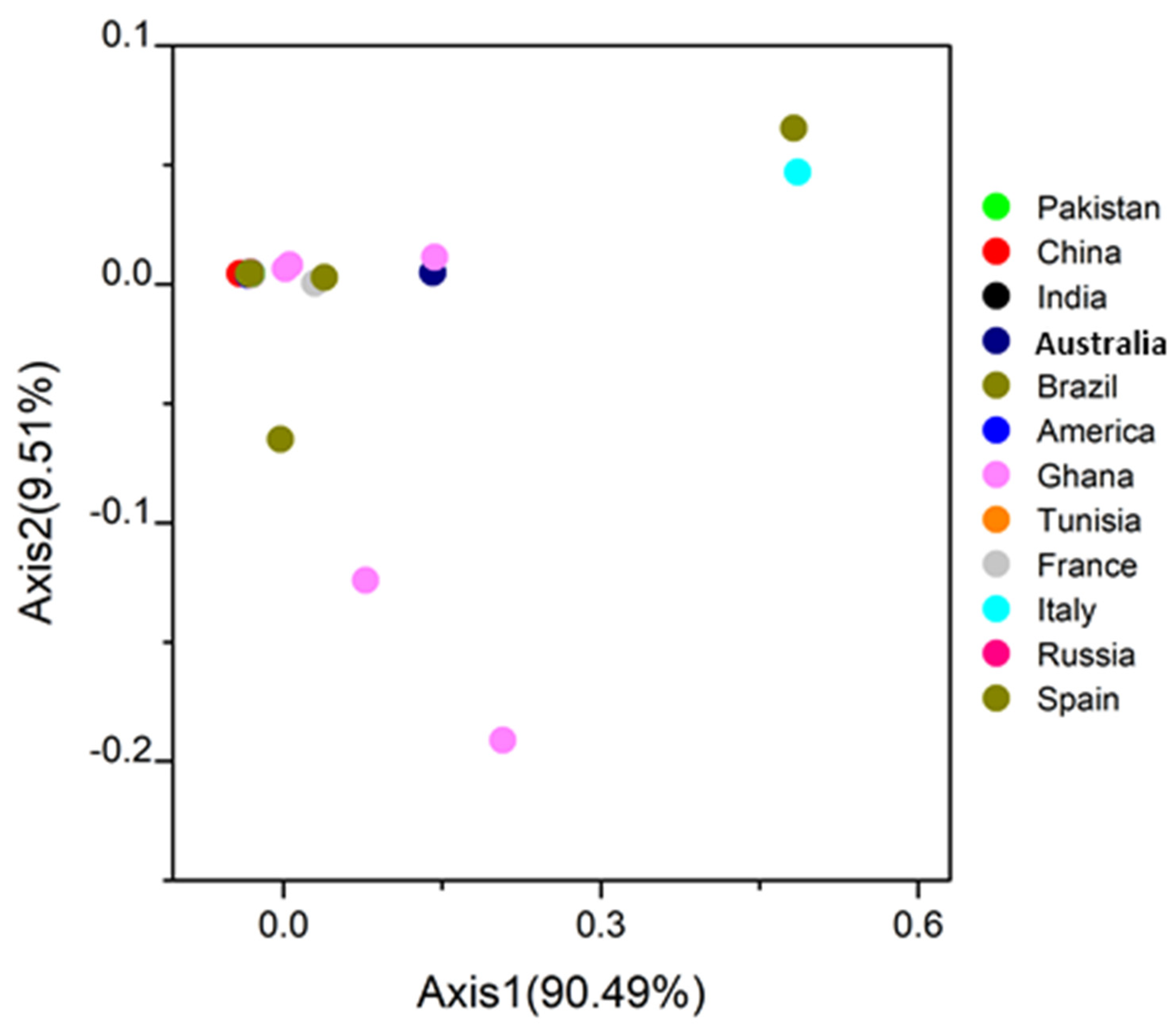
2.8. Correspondence Analysis (COA)
2.9. Phylogenetic Analysis
3. Results and Discussion
3.1. Nucleotide Composition Analysis of SARS-CoV-2
3.2. Relative Synonymous Codon Usage (RSCU) Analysis
3.3. Codon Usage Bias Analysis of SARS-CoV-2 Genomes
3.4. Neutrality Plot
3.5. Codon Adaptation Analysis
3.6. COA Analysis
3.7. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More Than Just the Common Cold. JAMA 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Ahmad, S.; Navid, A.; Farid, R.; Abbas, G.; Ahmad, F.; Zaman, N.; Parvaiz, N.; Azam, S.S. Design of a Novel Multi Epitope-Based Vaccine for Pandemic Coronavirus Disease (COVID-19) by Vaccinomics and Probable Prevention Strategy against Avenging Zoonotics. Eur. J. Pharm. Sci. 2020, 151, 105387. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Akter, S.; Rubayet Ul Alam, A.S.M.; Siddique, M.A.; Saha, O.; Rahaman, M.M.; Sultana, M.; Crandall, K.A.; et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2, the etiologic agent of COVID-19 pandemic: An in silico approach. PeerJ 2020, 8, e9572. [Google Scholar] [CrossRef]
- Chiara, M.; Horner, D.S.; Gissi, C.; Pesole, G. Comparative genomics suggests limited variability and similar evolutionary patterns between major clades of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Armijos-Jaramillo, V.; Yeager, J.; Muslin, C.; Perez-Castillo, Y. SARS-CoV-2, an evolutionary perspective of interaction with human ACE2 reveals undiscovered amino acids necessary for complex stability. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Sardar, R.; Satish, D.; Birla, S.; Gupta, D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Dilucca, M.; Forcelloni, S.; Georgakilas, A.G.; Giansanti, A.; Pavlopoulou, A. Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses 2020, 12, 498. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [Green Version]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Kaelin, E.A.; Maqsood, R.; Estifanos, B.; Wu, L.I.; Varsani, A.; Halden, R.U.; Hogue, B.G.; Scotch, M.; Lim, E.S. An 81-Nucleotide Deletion in SARS-CoV-2 ORF7a Identified from Sentinel Surveillance in Arizona (January to March 2020). J. Virol. 2020, 94, e00711–e00720. [Google Scholar] [CrossRef]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Marburg virus evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.U.; Yao, X.; Li, X.; Chen, D.; Tao, S. Analysis of codon usage bias of Crimean-Congo hemorrhagic fever virus and its adaptation to hosts. Infect. Genet. Evol. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Tort, F.L.; Castells, M.; Cristina, J. A comprehensive analysis of genome composition and codon usage patterns of emerging coronaviruses. Virus Res. 2020, 283, 197976. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, S.; Norenburg, J.L.; Sundberg, P. Mutation and Selection Cause Codon Usage and Bias in Mitochondrial Genomes of Ribbon Worms (Nemertea). PLoS ONE 2014, 9, e85631. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981, 146, 1–21. [Google Scholar] [CrossRef]
- Gu, H.; Chu, D.K.W.; Peiris, M.; Poon, L.L.M. Multivariate analyses of codon usage of SARS-CoV-2 and other betacoronaviruses. Virus Evol. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Higgs, P.G. Codon Usage in Mitochondrial Genomes: Distinguishing Context-Dependent Mutation from Translational Selection. Mol. Biol. Evol. 2007, 25, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Bailes, E.; Grocock, R.J.; Peden, J.F.; Sockett, R.E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005, 33, 1141–1153. [Google Scholar] [CrossRef]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces that influence the evolution of codon bias. Sharp P M, Emery L R, Zeng K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. B 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, N.; Sardar, R.; Gupta, D. Comparative analysis of codon usage patterns in SARS-CoV-2, its mutants and other respiratory viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hou, W. Characterization of codon usage pattern in SARS-CoV-2. Virol. J. 2020, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.M.; Diambra, L. SARS-CoV-2 Codon Usage Bias Downregulates Host Expressed Genes With Similar Codon Usage. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Moratorio, G.; Iriarte, A.; Moreno, P.; Musto, H.; Cristina, J. A detailed comparative analysis on the overall codon usage patterns in West Nile virus. Infect. Genet. Evol. 2013, 14, 396–400. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary Basis of Codon Usage and Nucleotide Composition Bias in Vertebrate DNA Viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.K.; Tyagi, A.; Kaur, R.; Verma, R.; Gupta, P.K. Characterization of codon usage pattern and influencing factors in Japanese encephalitis virus. Virus Res. 2016, 221, 58–65. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhäuser, M. Wilcoxon–Mann–Whitney Test. In International Encyclopedia of Statistical Science; Lovric, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1656–1658. [Google Scholar]
- Tao, P.; Dai, L.; Luo, M.; Tang, F.; Tien, P.; Pan, Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes 2009, 38, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.M.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L.M. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Berkhout, B.; van Hemert, F. On the biased nucleotide composition of the human coronavirus RNA genome. Virus Res. 2015, 202, 41–47. [Google Scholar] [CrossRef]
- Kindler, E.; Thiel, V. To sense or not to sense viral RNA--essentials of coronavirus innate immune evasion. Curr. Opin. Microbiol. 2014, 20, 69–75. [Google Scholar] [CrossRef]
- van Hemert, F.; van der Kuyl, A.C.; Berkhout, B. On the nucleotide composition and structure of retroviral RNA genomes. Virus Res. 2014, 193, 16–23. [Google Scholar] [CrossRef]
- Yao, X.; Fan, Q.; Yao, B.; Lu, P.; Rahman, S.U.; Chen, D.; Tao, S. Codon Usage Bias Analysis of Bluetongue Virus Causing Livestock Infection. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Buragohain, L.; Borah, P. Analysis of codon usage of severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) and its adaptability in dog. Virus Res. 2020, 288, 198113. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhou, T.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004, 101, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Victoria, M.; Colina, R.; Musto, H.; Cristina, J. Genome-wide analysis of codon usage bias in Bovine Coronavirus. Virol. J. 2017, 14, 115. [Google Scholar] [CrossRef] [Green Version]
- Bente, D.A.; Forrester, N.L.; Watts, D.M.; McAuley, A.J.; Whitehouse, C.A.; Bray, M. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 2013, 100, 159–189. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Codon | SARS-CoV-2 | Human | Dog | Cat | Cattle | AA | Codon | SARS-CoV-2 | Human | Dog | Cat | Cattle |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 1.31 | 0.97 | 1.09 | 0.77 | 0.85 | Ala | GCU | 1.78 | 1.01 | 1.07 | 0.69 | 1.12 |
| UUC | 0.69 | 1.03 | 0.91 | 1.33 | 1.15 | GCC | 0.69 | 1.12 | 0.27 | 1.79 | 0.57 | ||
| Leu | UUA | 1.51 | 0.50 | 1.32 | 0.35 | 0.38 | GCA | 1.26 | 1.18 | 1.18 | 0.76 | 0.94 | |
| UUG | 1.25 | 1.00 | 0.51 | 0.76 | 0.71 | GCG | 0.27 | 0.7 | 0.48 | 0.50 | 0.35 | ||
| CUU | 1.31 | 0.81 | 1.22 | 0.67 | 0.70 | Tyr | UAU | 1.06 | 0.71 | 1.15 | 0.78 | 0.87 | |
| CUC | 0.50 | 1.07 | 1.09 | 1.09 | 1.09 | UAC | 0.93 | 1.29 | 0.85 | 1.22 | 1.12 | ||
| CUA | 0.80 | 0.46 | 1.42 | 0.36 | 0.36 | His | CAU | 1.04 | 0.85 | 1.2 | 0.74 | 0.81 | |
| CUG | 0.63 | 2.33 | 0.51 | 2.57 | 2.59 | CAC | 0.96 | 1.15 | 0.80 | 1.26 | 1.18 | ||
| Iie | AUU | 1.44 | 1.13 | 1.05 | 0.95 | 0.98 | Gln | CAA | 1.35 | 0.49 | 1.25 | 0.56 | 0.49 |
| AUC | 0.72 | 1.37 | 0.94 | 1.58 | 1.57 | CAG | 0.65 | 1.51 | 0.75 | 1.44 | 1.50 | ||
| AUA | 0.84 | 0.50 | 1.01 | 0.47 | 0.45 | Asn | AAU | 1.10 | 0.98 | 1.18 | 0.82 | 0.92 | |
| Val | GUU | 1.58 | 0.79 | 1.12 | 0.62 | 0.64 | AAC | 0.90 | 1.02 | 0.82 | 1.18 | 1.07 | |
| GUC | 0.66 | 0.90 | 0.57 | 1.13 | 1.01 | Lys | AAA | 1.36 | 0.88 | 1.37 | 0.86 | 0.84 | |
| GUA | 0.91 | 0.52 | 1.67 | 0.38 | 0.40 | AAG | 0.64 | 1.12 | 0.63 | 1.14 | 1.15 | ||
| GUG | 0.85 | 1.79 | 0.64 | 1.87 | 1.95 | Asp | GAU | 1.13 | 0.99 | 1.13 | 0.84 | 0.98 | |
| Ser | UCU | 1.44 | 1.15 | 1.35 | 1.12 | 1.04 | GAC | 0.87 | 1.01 | 0.87 | 1.16 | 1.01 | |
| UCC | 0.55 | 1.17 | 1.04 | 1.48 | 1.37 | Glu | GAA | 1.33 | 0.85 | 1.17 | 0.86 | 1.88 | |
| UCA | 1.37 | 0.93 | 1.27 | 0.74 | 0.79 | GAG | 0.67 | 1.15 | 0.83 | 1.14 | 1.05 | ||
| UCG | 0.28 | 0.36 | 0.39 | 0.38 | 0.39 | Cys | UGU | 1.21 | 0.95 | 0.89 | 0.87 | 0.92 | |
| AGU | 1.42 | 0.98 | 1.91 | 0.8 | 0.87 | UGC | 0.79 | 1.05 | 1.11 | 1.13 | 1.07 | ||
| AGC | 0.93 | 1.42 | 1.05 | 1.47 | 1.53 | Arg | CGU | 0.68 | 0.54 | 1.17 | 0.41 | 0.49 | |
| Pro | CCU | 1.50 | 1.20 | 1.41 | 1.03 | 1.08 | CGC | 0.43 | 1.11 | 0.92 | 1.09 | 0.94 | |
| CCC | 0.56 | 1.22 | 1.24 | 1.51 | 1.39 | CGA | 0.87 | 0.76 | 0.71 | 0.55 | 0.74 | ||
| CCA | 1.58 | 1.14 | 0.92 | 0.97 | 1.00 | CGG | 0.35 | 1.31 | 0.48 | 1.19 | 1.18 | ||
| CCG | 0.14 | 0.45 | 0.43 | 0.50 | 0.53 | AGA | 2.81 | 1.18 | 1.29 | 1.33 | 1.26 | ||
| Thr | ACU | 1.32 | 1.03 | 1.35 | 0.84 | 0.89 | AGG | 1.36 | 1.1 | 1.42 | 1.41 | 1.36 | |
| ACC | 0.82 | 1.32 | 1.06 | 1.59 | 1.55 | Gly | GGU | 1.54 | 0.71 | 1.02 | 0.58 | 0.71 | |
| ACA | 1.52 | 1.19 | 1.16 | 0.94 | 1.01 | GGC | 0.84 | 1.35 | 1.05 | 1.42 | 1.36 | ||
| ACG | 0.34 | 0.46 | 0.43 | 0.63 | 0.56 | GGA | 1.10 | 1.01 | 1.27 | 1.01 | 0.96 | ||
| GGG | 0.52 | 0.93 | 0.66 | 0.99 | 0.96 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khattak, S.; Rauf, M.A.; Zaman, Q.; Ali, Y.; Fatima, S.; Muhammad, P.; Li, T.; Khan, H.A.; Khan, A.A.; Ngowi, E.E.; et al. Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID-19. Biomolecules 2021, 11, 912. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060912
Khattak S, Rauf MA, Zaman Q, Ali Y, Fatima S, Muhammad P, Li T, Khan HA, Khan AA, Ngowi EE, et al. Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID-19. Biomolecules. 2021; 11(6):912. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060912
Chicago/Turabian StyleKhattak, Saadullah, Mohd Ahmar Rauf, Qamar Zaman, Yasir Ali, Shabeen Fatima, Pir Muhammad, Tao Li, Hamza Ali Khan, Azhar Abbas Khan, Ebenezeri Erasto Ngowi, and et al. 2021. "Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID-19" Biomolecules 11, no. 6: 912. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060912