
Comparing Bayesian-Based Reconstruction Strategies in Topology-Based Pathway Enrichment Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Pre-Processing
2.2. Reconstruction of BN Structures
2.2.1. Reconstruction of BN Structures by BPA
2.2.2. Reconstruction of BN Structures by PROPS
2.2.3. Reconstruction of BN Structures by Clipper
2.2.4. Reconstruction of BN Structures by BNrich
2.2.5. Reconstruction of BN Structures by Ensemble Method
2.3. BN Parameter Learning by Non-Tumor Data
2.4. Pathway-Based Feature Engineering
2.5. Pathway Enrichment through Random Forest Classification
3. Results
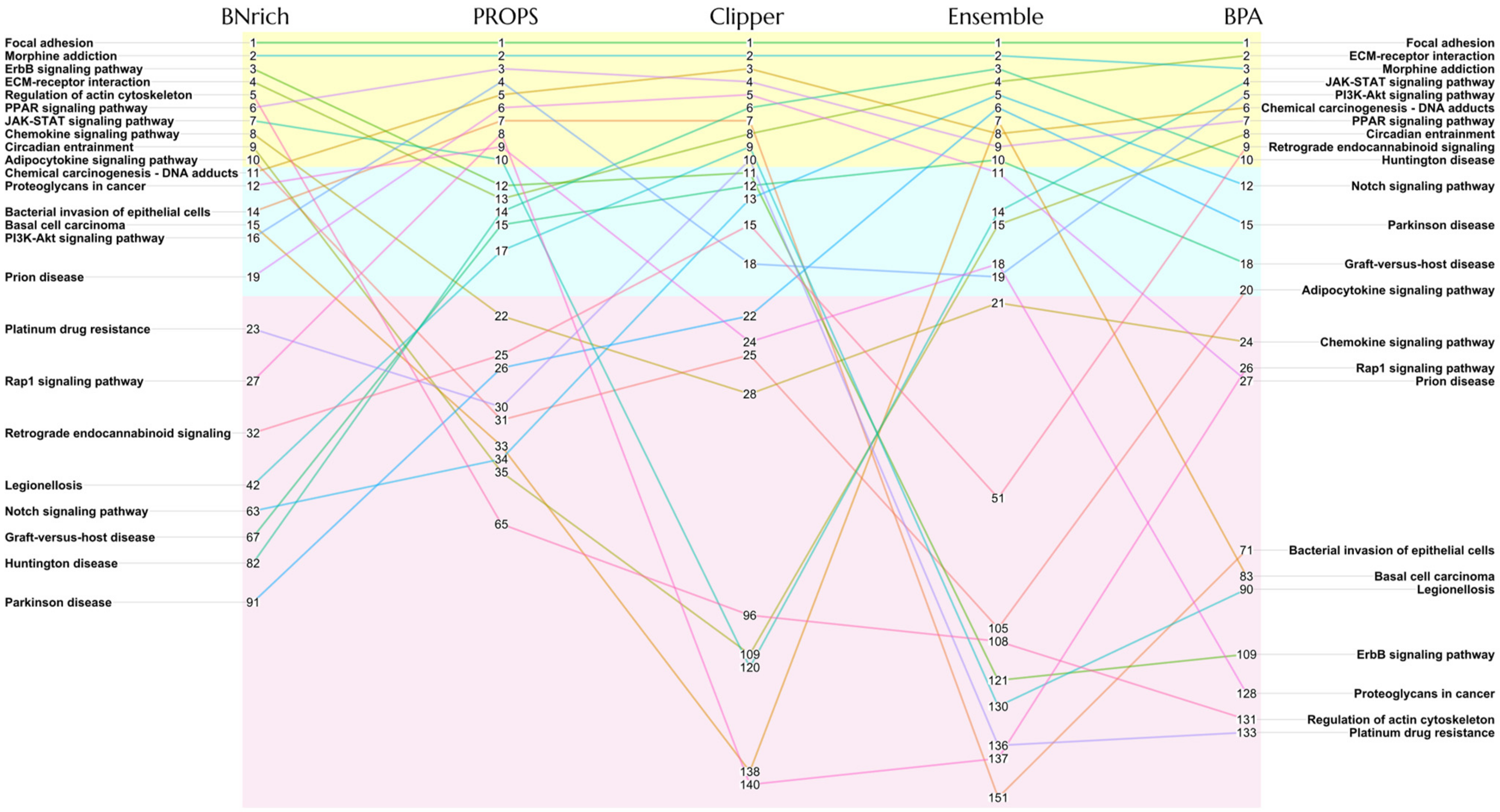
3.1. Different BN Reconstruction Strategies Lead to Different PEA Results
3.2. Different Strategies Generate Different BN Structures
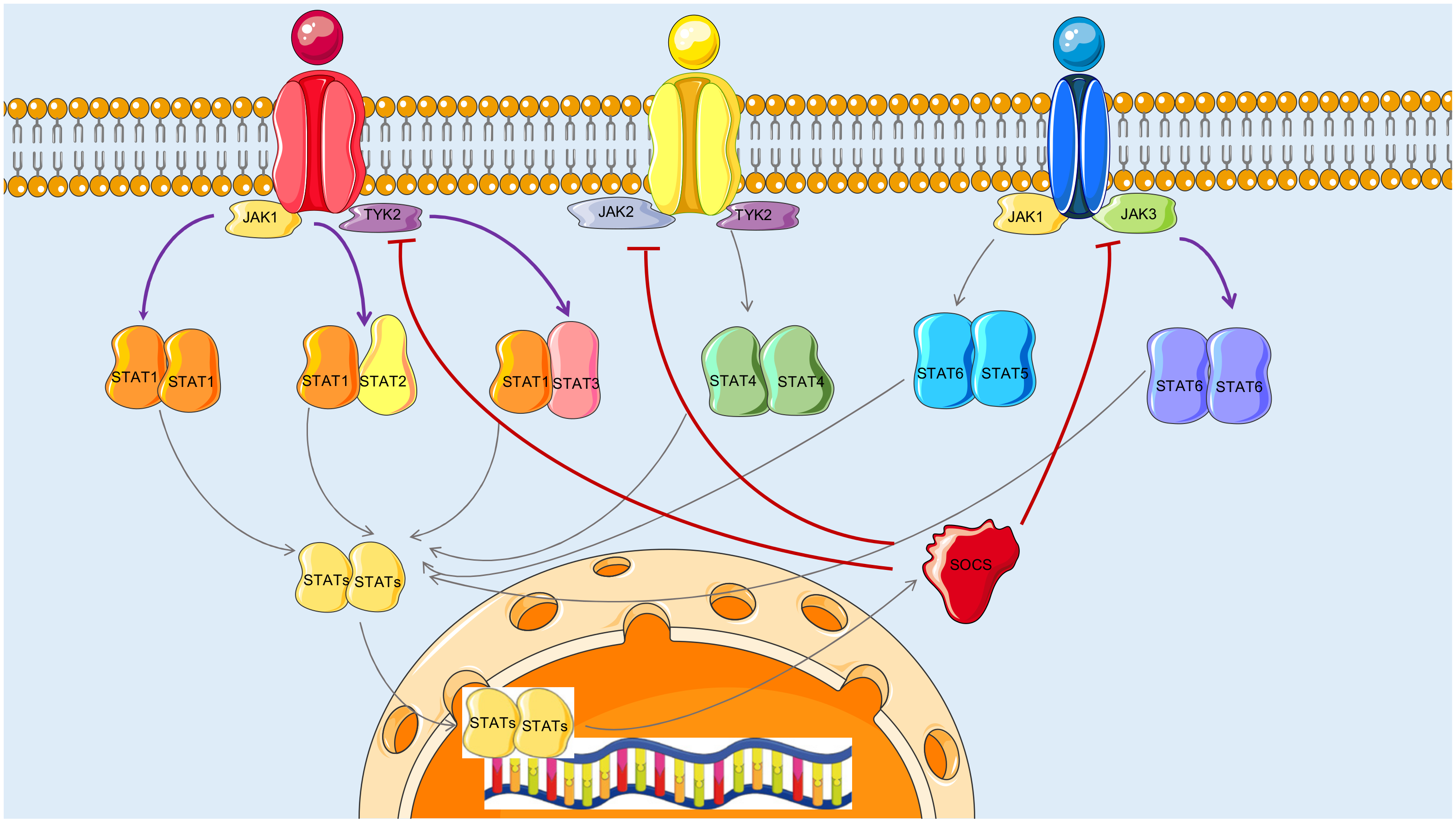
3.3. Reconstructed JAK-STAT Networks and Biological Relevance
4. Discussion
4.1. Concluding Remarks
4.2. Suggestions for Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, P.; Draghici, S.; Ostermeier, G.C.; Krawetz, S.A. Profiling gene expression using onto-express. Genomics 2002, 79, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeeberg, B.R.; Feng, W.; Wang, G.; Wang, M.D.; Fojo, A.T.; Sunshine, M.; Narasimhan, S.; Kane, D.W.; Reinhold, W.C.; Lababidi, S.; et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003, 4, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Greenberg, S.A.; Kong, S.W.; Altschuler, J.; Kohane, I.S.; Park, P.J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl. Acad. Sci. USA 2005, 102, 13544–13549. [Google Scholar] [CrossRef] [Green Version]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim J-S Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2008, 25, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Chen, X.; Wang, C. Systematic elucidation of the pharmacological mechanisms of Rhynchophylline for treating epilepsy via network pharmacology. BMC Complementary Med. Ther. 2021, 21, 9. [Google Scholar] [CrossRef]
- Maleknia, S.; Sharifi-Zarchi, A.; Tabar, V.R.; Namazi, M.; Kavousi, K. BNrich: A Bayesian network approach to the pathway enrichment analysis. bioRxiv 2020. Biorxiv:2020.01.13.905448. [Google Scholar]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: An empirical gene set approach exploiting pathway topology. Nucleic Acids Res. 2012, 41, e19. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Maciejewski, M.; Brockel, C.; Gordon, W.; Snapper, S.B.; Korzenik, J.R.; Afzelius, L.; Altman, R.B. A probabilistic pathway score (PROPS) for classification with applications to inflammatory bowel disease. Bioinformatics 2017, 34, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.S.; Chiogna, M.; Romualdi, C. Gene set analysis exploiting the topology of a pathway. BMC Syst. Biol. 2010, 4, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isci, S.; Ozturk, C.; Jones, J.; Otu, H.H. Pathway analysis of high-throughput biological data within a Bayesian network framework. Bioinformatics 2011, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chen, M.-H.; Pei, B.; Rowe, D.; Shin, D.-G.; Xie, W.; Yu, F.; Kuo, L. A Bayesian Approach to Pathway Analysis by Integrating Gene-Gene Functional Directions and Microarray Data. Stat. Biosci. 2012, 4, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ajwani, D.; Nicholson, P.K.; Sala, A.; Parthasarathy, S. Breaking Cycles in Noisy Hierarchies. In Proceedings of the 2017 ACM on Web Science Conference, Troy, NY, USA, 25–28 June 2017; Association for Computing Machinery: New York, NY, USA, 2017; pp. 151–160. [Google Scholar]
- Mitrea, C.; Taghavi, Z.; Bokanizad, B.; Hanoudi, S.; Tagett, R.; Donato, M.; Voichiţa, C.; Drăghici, S. Methods and approaches in the topology-based analysis of biological pathways. Front. Physiol. 2013, 4, 278. [Google Scholar] [CrossRef] [Green Version]
- Ihnatova, I.; Popovici, V.; Budinska, E. A critical comparison of topology-based pathway analysis methods. PLoS ONE 2018, 13, e0191154. [Google Scholar]
- Rahnenführer, J.; Domingues, F.S.; Maydt, J.; Lengauer, T. Calculating the Statistical Significance of Changes in Pathway Activity From Gene Expression Data. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article16. [Google Scholar] [CrossRef]
- Agrahari, R.; Foroushani, A.; Docking, T.R.; Chang, L.; Duns, G.; Hudoba, M.; Karsan, A.; Zare, H. Applications of Bayesian network models in predicting types of hematological malignancies. Sci. Rep. 2018, 8, 6951. [Google Scholar] [CrossRef]
- Spirtes, P.; Meek, C. Learning Bayesian Networks with Discrete Variables from Data; Morgan Kaufmann: Montreal, QC, Canada, 1995. [Google Scholar]
- Chanumolu, S.K.; Albahrani, M.; Can, H.; Otu, H.H. KEGG2Net: Deducing gene interaction networks and acyclic graphs from KEGG pathways. EMBnet J. 2021, 26, e949. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Aittomäki, S.; Pesu, M. Therapeutic Targeting of the JAK/STAT Pathway. Basic Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezeonwumelu, I.J.; Garcia-Vidal, E.; Ballana, E. JAK-STAT Pathway: A Novel Target to Tackle Viral Infections. Viruses 2021, 13, 2379. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.L.; Zou, X.; Nie, Q.; Bardwell, L. A theoretical framework for specificity in cell signaling. Mol. Syst. Biol. 2005, 1, 2005.0023. [Google Scholar] [CrossRef]
- Nassiri, I.; Masoudi-Nejad, A.; Jalili, M.; Moeini, A. Discovering dominant pathways and signal-response relationships in signaling networks through nonparametric approaches. Genomics 2013, 102, 195–201. [Google Scholar] [CrossRef]
- Palotai, R.; Szalay, M.S.; Csermely, P. Chaperones as integrators of cellular networks: Changes of cellular integrity in stress and diseases. IUBMB Life 2008, 60, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Planes, F.J.; Beasley, J.E. A critical examination of stoichiometric and path-finding approaches to metabolic pathways. Brief. Bioinform. 2008, 9, 422–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubarev, R.A.; Nielsen, M.L.; Fung, E.M.; Savitski, M.M.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Identification of dominant signaling pathways from proteomics expression data. J. Proteom. 2008, 71, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Zhu, X.; Wang, G.; Li, S.; Ao, P. Cancer as robust intrinsic state shaped by evolution: A key issues review. Rep. Prog. Phys. Phys. Soc. 2017, 80, 042701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang-Yen, H.F.; Chambard, J.C.; Sun, Y.L.; Smeal, T.; Schmidt, T.J.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar] [CrossRef]
- Rockne, R.C.; Branciamore, S.; Qi, J.; Frankhouser, D.E.; O’Meally, D.; Hua, W.K.; Cook, G.; Carnahan, E.; Zhang, L.; Marom, A.; et al. State-Transition Analysis of Time-Sequential Gene Expression Identifies Critical Points That Predict Development of Acute Myeloid Leukemia. Cancer Res. 2020, 80, 3157–3169. [Google Scholar] [CrossRef]
- Chen, Y.A.; Almeida, J.S.; Richards, A.J.; Mller, P.; Carroll, R.J.; Rohrer, B. A Nonparametric Approach to Detect Nonlinear Correlation in Gene Expression. J. Comput. Graph. Stat. 2010, 19, 552–568. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Type | Example Application | Data Type | Year | Ref. |
|---|---|---|---|---|---|
| GoMiner | ORA | Compare gene expression profiles in a prostate cancer cell line and a subline selected from it for resistance to the topoisomerase 1-inhibitor 9-nitro-camptothecin. | Microarray | 2003 | [3] |
| WebGestalt | ORA | Disease association analysis and drug association analysis in colorectal cancer; Elucidation of the pharmacological mechanisms of Rhynchophylline for treating epilepsy. | Microarray; RNA-seq | 2013 | [4,8] |
| GSEA | FCS | Searched for significantly associated gene sets from: male vs. female lymphoblastoid cells; p53 status in cancer cell lines; acute lymphoid leukemia (ALL) vs. acute myeloid leukemia (AML); comparing two studies of lung cancer. | Microarray; RNA-seq | 2005 | [5] |
| BNrich | TPA | Discriminate among specific experimental states in: tumor cell line skin, tumor cell line large intestine, breast cancer, Alzheimer, colorectal cancer. | Microarray | 2020 | [9] |
| Clipper | TPA | Peak calling from ChIP-seq data, peptide identification from MS data, DEG identification from bulk or single-cell RNA-seq data, and DIR identification from Hi-C data. | ChIP-seq; microarray; RNA-seq; mass spectrometry | 2012 | [10] |
| PROPS | TPA | Classification of inflammatory bowel disease. | Microarray | 2017 | [11] |
| TopologyGSE | TPA | Identify key regulatory elements in gene expression data on (i) acute lymphocytic leukemia (ALL) with and without BCR/ABL gene rearrangement and (ii) lung adenocarcinoma with and without EGFR mutation. | Microarray | 2010 | [12] |
| BPA | TPA | Investigated the active pathways in NCBI’s GEO database regarding bladder, brain, breast, colon, liver, lung, ovarian, and thyroid cancers. | Microarray | 2011 | [13] |
| BAPA-IGGFD | TPA | Understand the gene expression profile of osteoblast lineage at defined stages of differentiation. | Microarray | 2012 | [14] |
| Ensemble | TPA | Breaking cycles while preserving the logical structure of the directed graph. | Directed graph | 2017 | [15] |
| Dataset | Method | Accuracy | Recall | Precision | F1-Score | AUC |
|---|---|---|---|---|---|---|
| HCC | BNrich | 0.95 ± 0.04 | 0.95 ± 0.03 | 0.95 ± 0.06 | 0.95 ± 0.03 | 0.96 ± 0.04 |
| PROPS | 0.93 ± 0.05 | 0.93 ± 0.06 | 0.94 ± 0.07 | 0.93 ± 0.04 | 0.95 ± 0.04 | |
| Clipper | 0.93 ± 0.04 | 0.92 ± 0.06 | 0.94 ± 0.04 | 0.93 ± 0.04 | 0.95 ± 0.04 | |
| BPA | 0.92 ± 0.06 | 0.93 ± 0.09 | 0.91 ± 0.07 | 0.92 ± 0.06 | 0.95 ± 0.05 | |
| Ensemble | 0.93 ± 0.05 | 0.90 ± 0.08 | 0.96 ± 0.06 | 0.93 ± 0.05 | 0.96 ± 0.05 | |
| HNSC | BNrich | 0.93 ± 0.05 | 0.91 ± 0.12 | 0.96 ± 0.07 | 0.93 ± 0.06 | 0.97 ± 0.03 |
| PROPS | 0.94 ±0.05 | 0.96 ± 0.09 | 0.92 ± 0.08 | 0.94 ± 0.05 | 0.97 ± 0.03 | |
| Clipper | 0.93 ± 0.06 | 0.94 ± 0.12 | 0.93 ± 0.08 | 0.93 ± 0.07 | 0.95 ± 0.04 | |
| BPA | 0.95 ± 0.04 | 0.98 ± 0.04 | 0.93 ± 0.07 | 0.95 ± 0.04 | 0.96 ± 0.03 | |
| Ensemble | 0.95 ± 0.04 | 0.97 ± 0.04 | 0.95 ± 0.07 | 0.96 ± 0.03 | 0.96 ± 0.04 | |
| KIRC | BNrich | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 |
| PROPS | 0.98 ± 0.01 | 0.98 ± 0.03 | 0.99 ± 0.00 | 0.98 ± 0.02 | 0.99 ± 0.00 | |
| Clipper | 0.98 ± 0.01 | 0.98 ± 0.03 | 0.99 ± 0.00 | 0.98 ± 0.02 | 0.99 ± 0.00 | |
| BPA | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | |
| Ensemble | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | |
| LUAD | BNrich | 0.99 ± 0.00 | 0.98 ± 0.04 | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 |
| PROPS | 0.99 ± 0.00 | 0.98 ± 0.04 | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | |
| Clipper | 0.99 ± 0.00 | 0.98 ± 0.04 | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | |
| BPA | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | |
| Ensemble | 0.99 ± 0.00 | 0.98 ± 0.03 | 1.00 ± 0.00 | 0.99 ± 0.00 | 1.00 ± 0.00 | |
| THCA | BNrich | 0.96 ± 0.04 | 0.98 ± 0.04 | 0.95 ± 0.07 | 0.97 ± 0.03 | 0.98 ± 0.02 |
| PROPS | 0.96 ± 0.04 | 0.98 ± 0.04 | 0.95 ± 0.07 | 0.96 ± 0.04 | 0.98 ± 0.03 | |
| Clipper | 0.96 ± 0.04 | 0.99 ± 0.00 | 0.94 ± 0.07 | 0.96 ± 0.04 | 0.98 ± 0.03 | |
| BPA | 0.96 ± 0.04 | 0.99 ± 0.00 | 0.94 ± 0.06 | 0.97 ± 0.03 | 0.97 ± 0.03 | |
| Ensemble | 0.96 ± 0.04 | 0.99 ± 0.00 | 0.94 ± 0.07 | 0.96 ± 0.04 | 0.97 ± 0.03 |
| Method | Averaged Ranks of BIC Scores | ||||
|---|---|---|---|---|---|
| HCC | HNSC | THCA | KIRC | LUAD | |
| BPA | 2.03 | 3.32 | 2.34 | 2.36 | 2.74 |
| Clipper | 3.03 | 3.20 | 2.59 | 2.59 | 2.65 |
| PROPS | 3.45 | 3.84 | 3.77 | 3.61 | 3.56 |
| BNrich | 2.78 | 2.95 | 2.76 | 2.74 | 2.51 |
| Ensemble | 3.70 | 1.69 | 3.54 | 3.69 | 3.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, J.; Huang, D.; Hao, Y.; Li, B.; Wang, K.; Chen, B.; Li, T.; Liu, X. Comparing Bayesian-Based Reconstruction Strategies in Topology-Based Pathway Enrichment Analysis. Biomolecules 2022, 12, 906. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12070906
Wang Y, Li J, Huang D, Hao Y, Li B, Wang K, Chen B, Li T, Liu X. Comparing Bayesian-Based Reconstruction Strategies in Topology-Based Pathway Enrichment Analysis. Biomolecules. 2022; 12(7):906. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12070906
Chicago/Turabian StyleWang, Yajunzi, Jing Li, Daiyun Huang, Yang Hao, Bo Li, Kai Wang, Boya Chen, Ting Li, and Xin Liu. 2022. "Comparing Bayesian-Based Reconstruction Strategies in Topology-Based Pathway Enrichment Analysis" Biomolecules 12, no. 7: 906. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12070906