
Renin-a in the Subfornical Organ Plays a Critical Role in the Maintenance of Salt-Sensitive Hypertension
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DOCA-Salt Hypertension Model
2.3. Droplet Digital PCR
2.4. RNAscope In Situ Hybridization of Renin mRNA
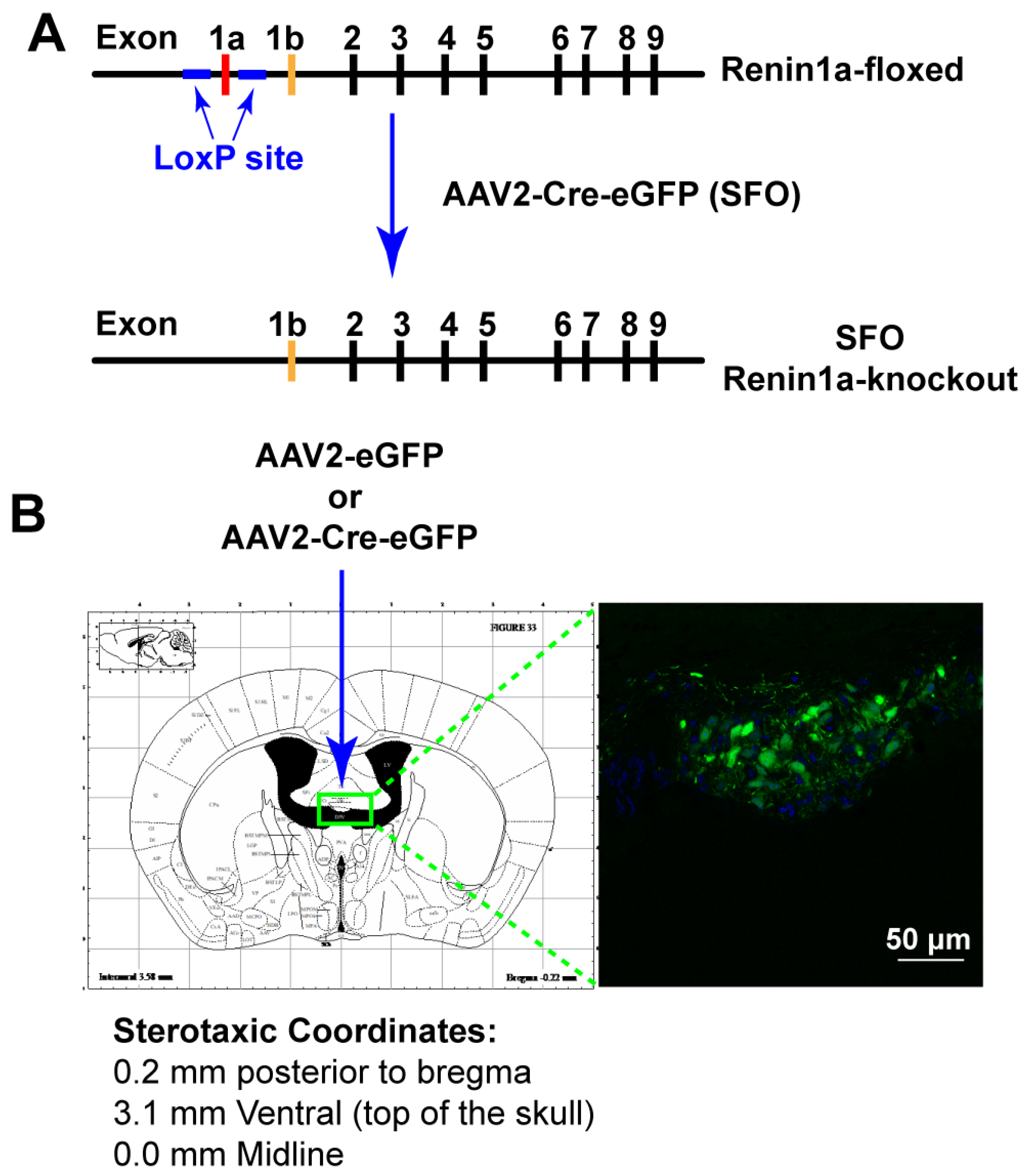
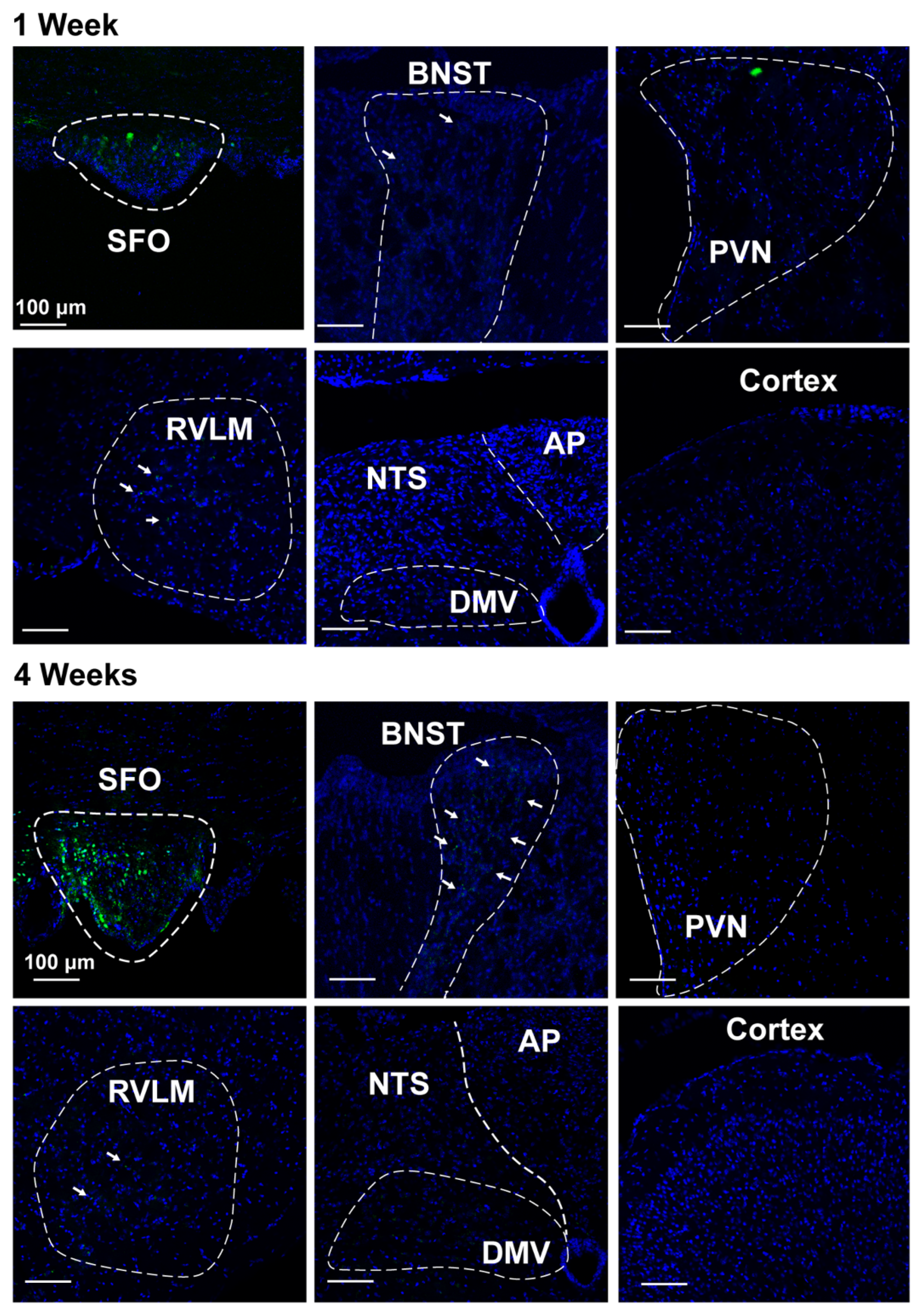
2.5. Nano-Injection of AAVs into the SFO
2.6. Telemetric Measurement of BP, Heart Rate, and Autonomic Function in Conscious, Freely Moving Mice
2.7. Real-Time PCR
2.8. Statistical Analysis
3. Results
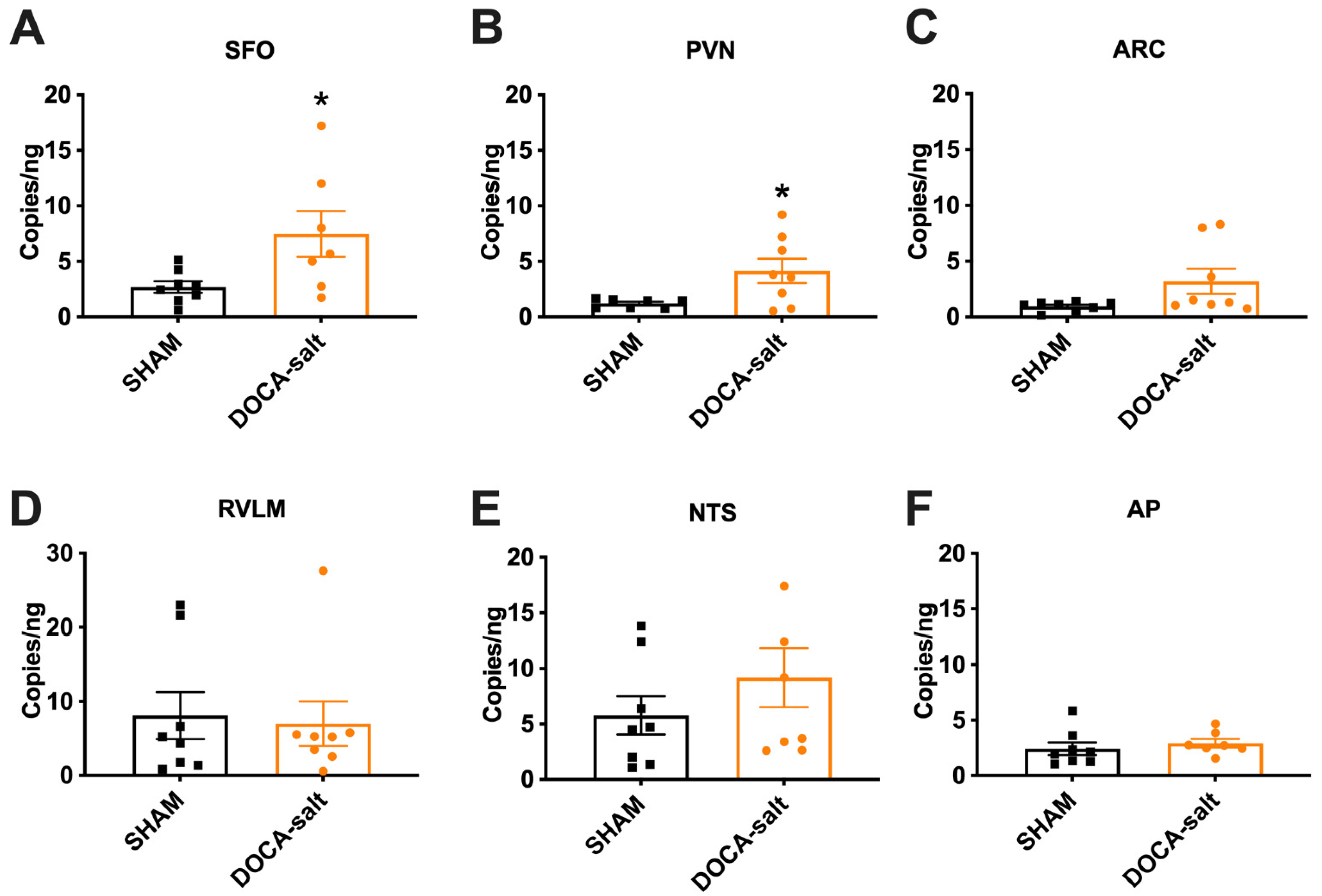
3.1. Elevated Renin mRNA in the SFO and PVN of DOCA-Salt Hypertensive Mice
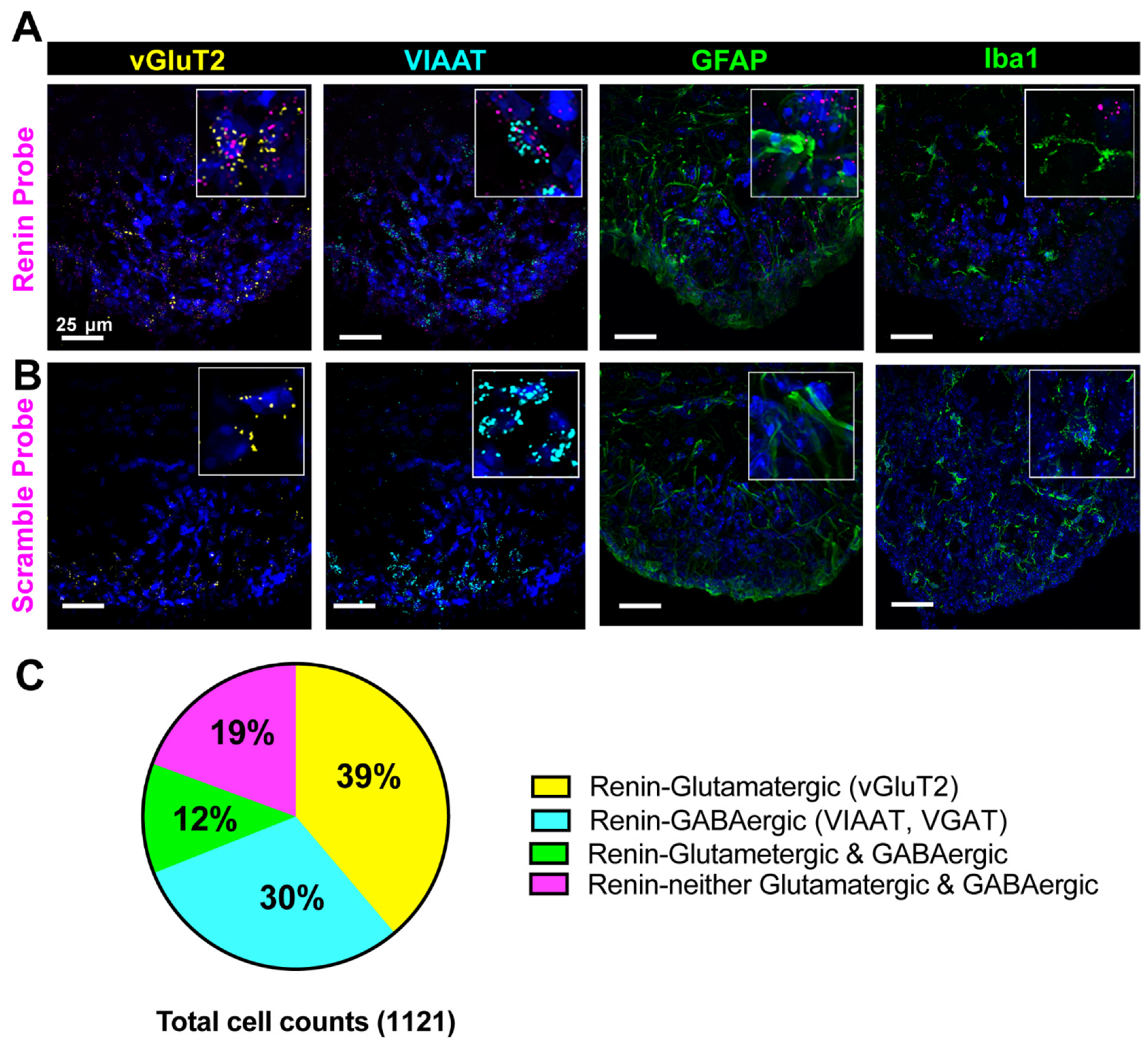
3.2. Cellular Characterization of Renin mRNA in the Brain
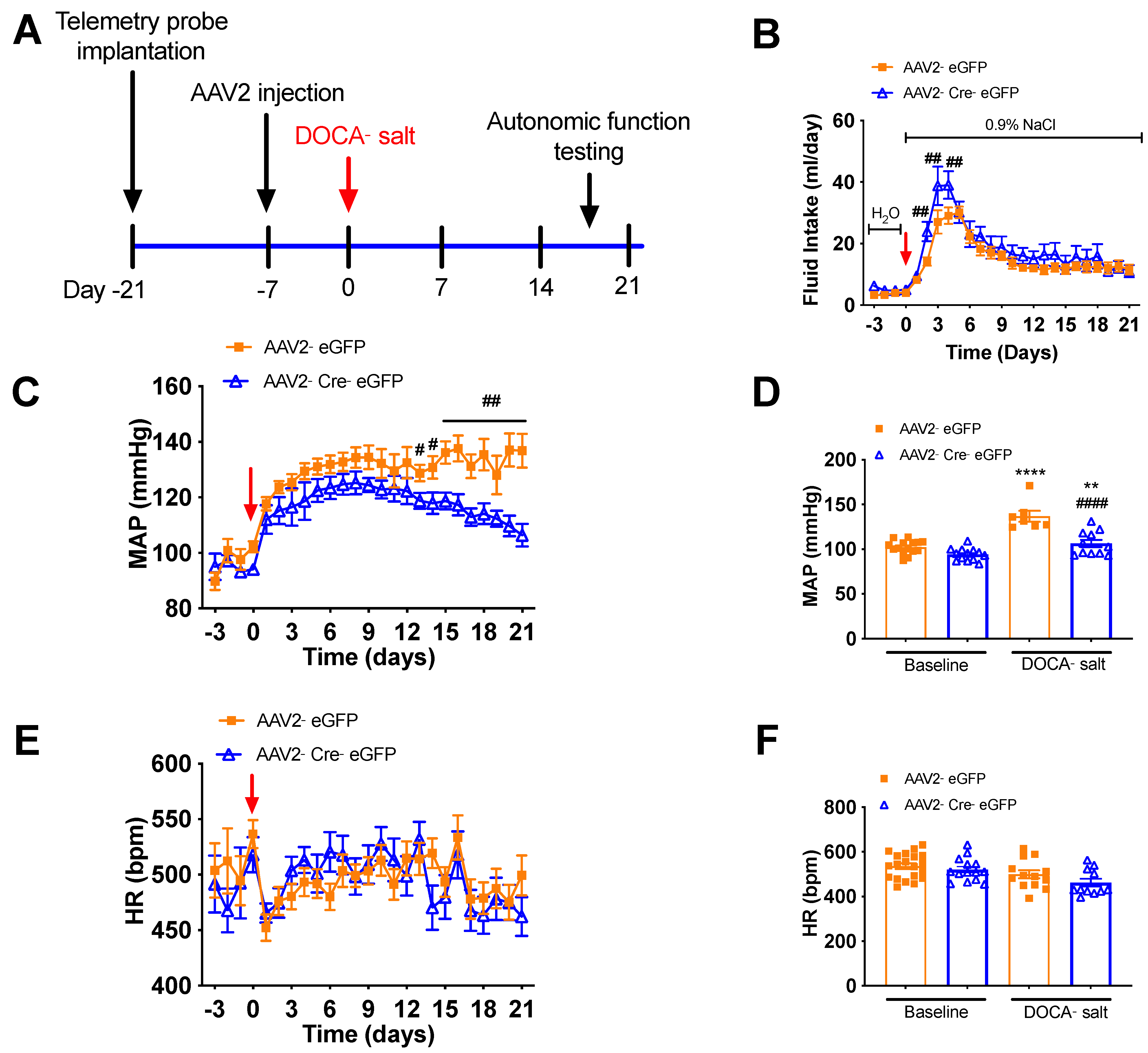
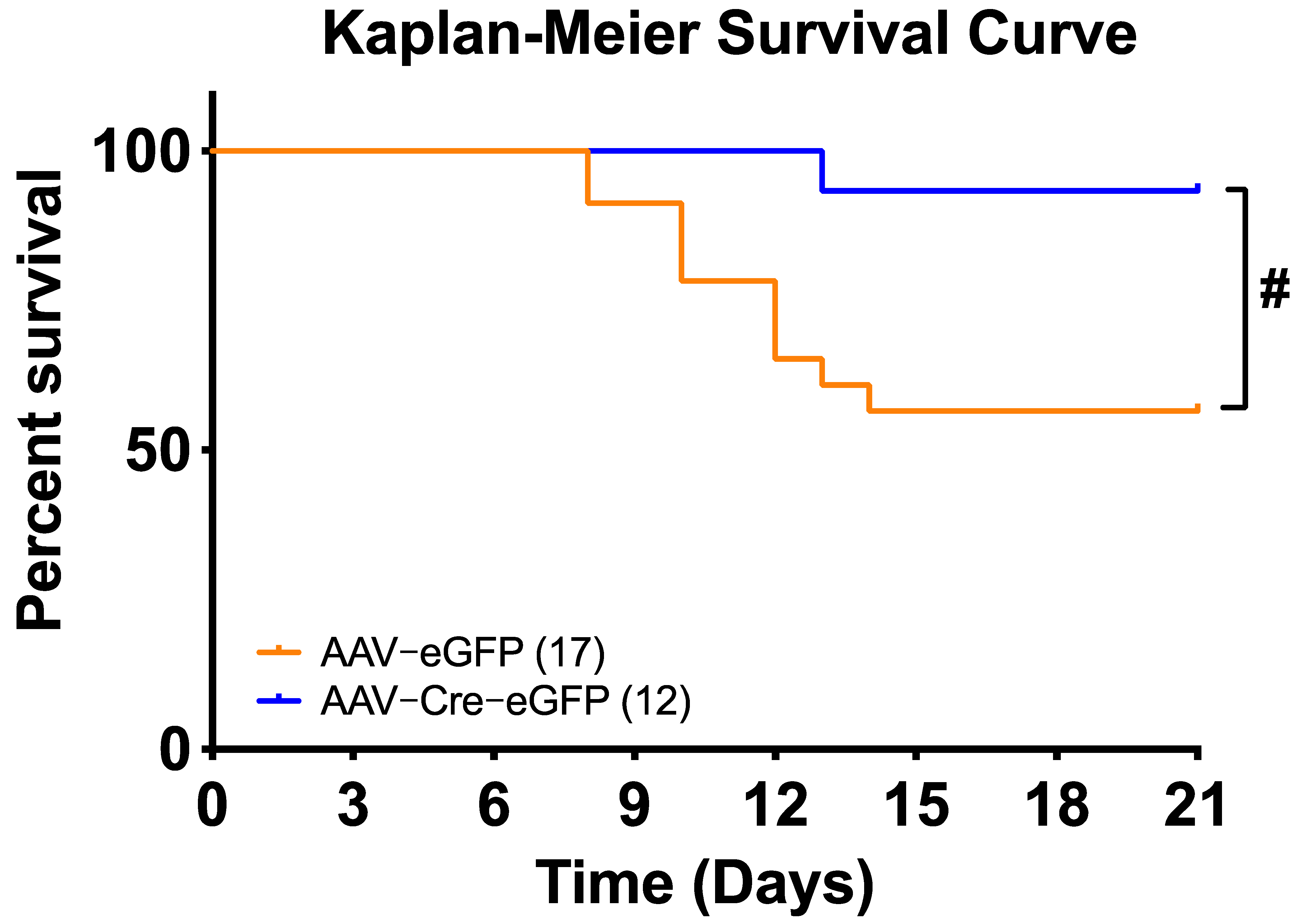
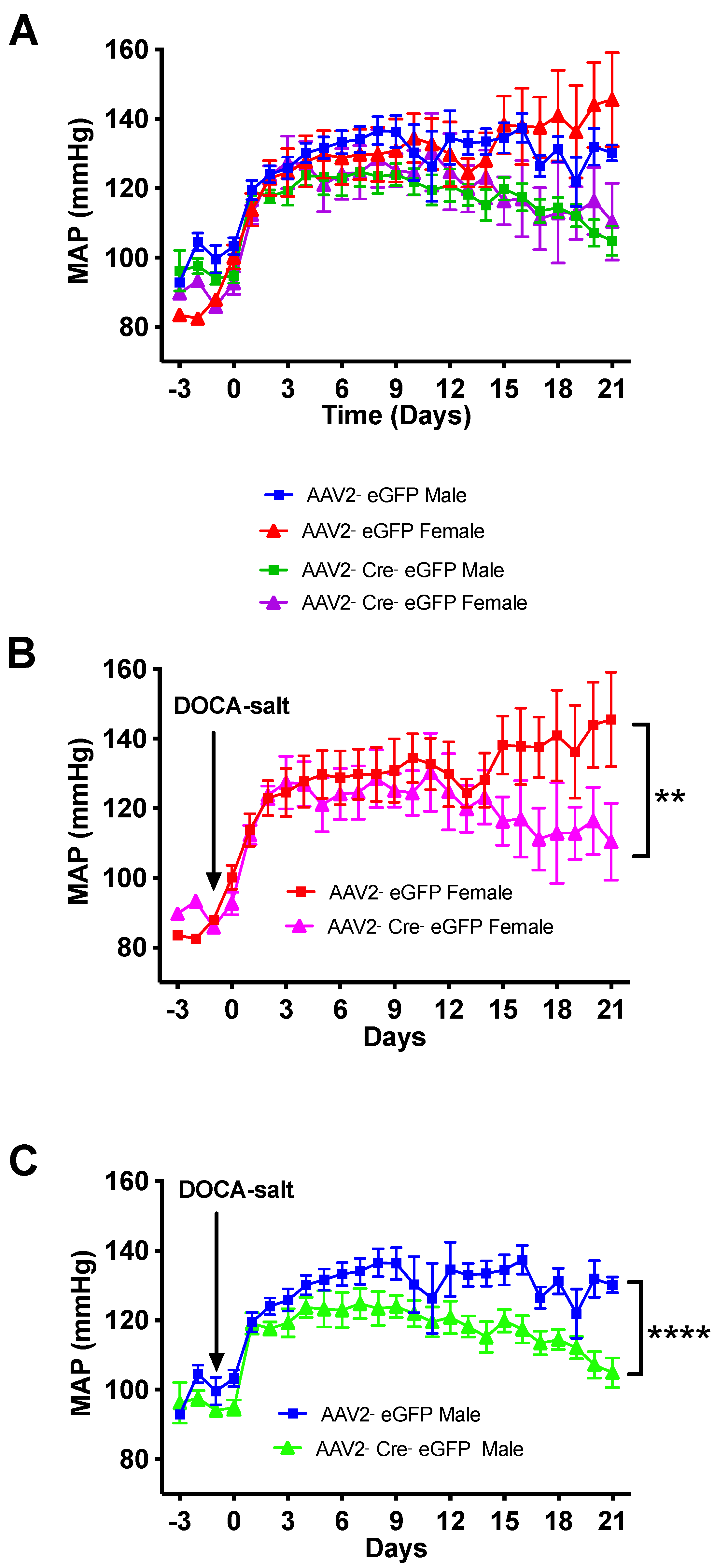
3.3. Deletion of Renin-a in the SFO Attenuates DOCA-Salt–Induced Hypertension
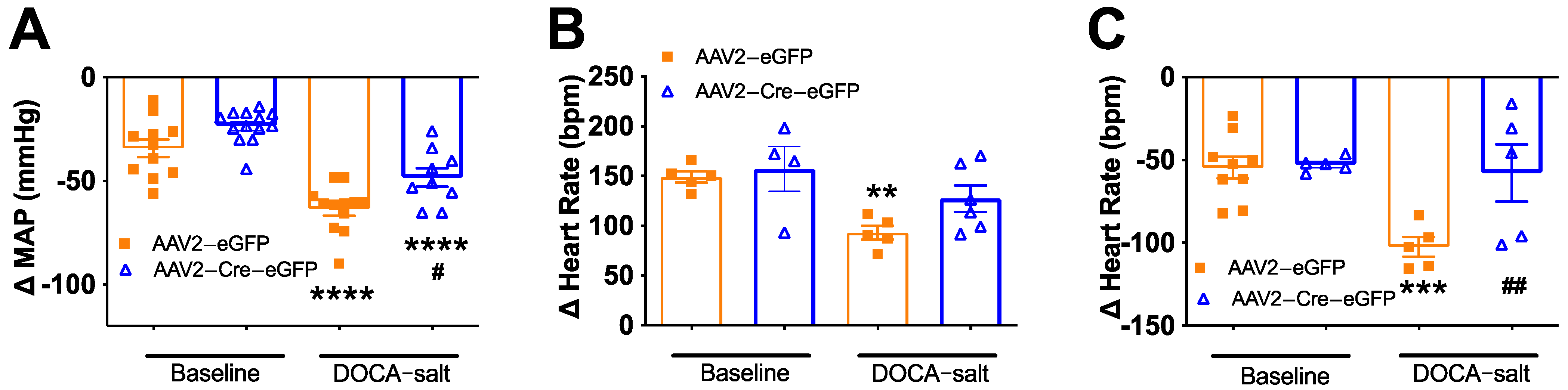
3.4. Deletion of Renin-a in the SFO Improves Autonomic Function in DOCA-Salt Hypertensive Mice
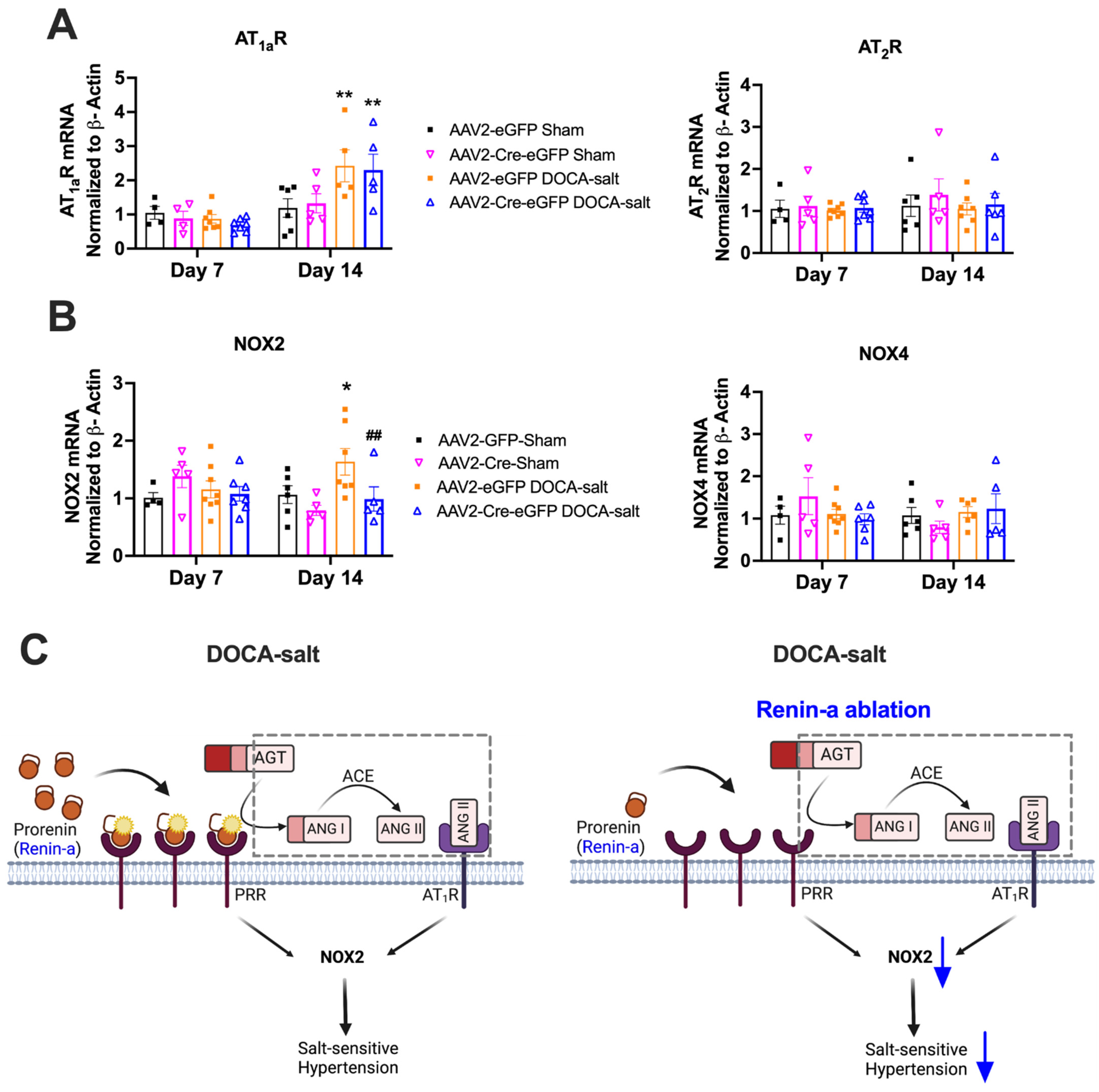
3.5. Renin-a Ablation in the SFO Prevents Upregulation of NOX2 in DOCA-Salt Hypertension
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worker, C.J.; Li, W.; Feng, C.Y.; Souza, L.A.C.; Gayban, A.J.B.; Cooper, S.G.; Afrin, S.; Romanick, S.; Ferguson, B.S.; Feng Earley, Y. The Neuronal (Pro)renin Receptor and Astrocyte Inflammation in the Central Regulation of Blood Pressure and Blood Glucose in Mice Fed a High-Fat Diet. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E765–E778. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.F.; Tofteng, S.S.; Madsen, K.; Jensen, B.L. Role of the renin-angiotensin system in kidney development and programming of adult blood pressure. Clin. Sci. 2020, 134, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, P.; Gomez, J.; Grobe, J.L.; Sigmund, C.D. The Renin-Angiotensin System in the Central Nervous System and Its Role in Blood Pressure Regulation. Curr. Hypertens. Rep. 2020, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Circulating versus local renin-angiotensin system in cardiovascular homeostasis. Circulation 1988, 77, I4–I13. [Google Scholar]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Lavoie, J.L.; Sigmund, C.D. Minireview: Overview of the renin-angiotensin system—An endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef]
- Fleming, I.; Kohlstedt, K.; Busse, R. The tissue renin-angiotensin system and intracellular signalling. Curr. Opin. Nephrol. Hypertens. 2006, 15, 8–13. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Seth, D.M.; Feng, Y. The Prorenin and (Pro)renin Receptor: New Players in the Brain Renin-Angiotensin System? Int. J. Hypertens. 2012, 2012, 290635. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Mehaffey, E.P.; Kimball, C.D.; Grobe, J.L.; van Gool, J.M.; Sullivan, M.N.; Earley, S.; Danser, A.H.; Ichihara, A.; et al. Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension. Hypertension 2014, 63, 316–323. [Google Scholar] [CrossRef]
- Lavoie, J.L.; Cassell, M.D.; Gross, K.W.; Sigmund, C.D. Localization of renin expressing cells in the brain, by use of a REN-eGFP transgenic model. Physiol. Genom. 2004, 16, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.L.; Cassell, M.D.; Gross, K.W.; Sigmund, C.D. Adjacent expression of renin and angiotensinogen in the rostral ventrolateral medulla using a dual-reporter transgenic model. Hypertension 2004, 43, 1116–1119. [Google Scholar] [CrossRef]
- Ganten, D.; Minnich, J.L.; Grenger, P.; Hayduk, K.; Brecht, H.M.; Barbeau, A.; Boucher, R.; Genest, J. Angiotensin-Forming Enzyme in Brain Tissue. Science 1971, 173, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Schelling, P.; Hutchinson, J.S.; Ganten, U.; Sponer, G.; Ganten, D. Impermeability of the blood-cerebrospinal fluid barrier for angiotensin II in rats. Clin. Sci. Mol. Med. Suppl. 1976, 3, 399s–402s. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sullivan, M.N.; Zhang, S.; Worker, C.J.; Xiong, Z.; Speth, R.C.; Feng, Y. Intracerebroventricular infusion of the (Pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension 2015, 65, 352–361. [Google Scholar] [CrossRef]
- Lee-Kirsch, M.A.; Gaudet, F.; Cardoso, M.C.; Lindpaintner, K. Distinct renin isoforms generated by tissue-specific transcription initiation and alternative splicing. Circ. Res. 1999, 84, 240–246. [Google Scholar] [CrossRef]
- Sinn, P.L.; Sigmund, C.D. Identification of three human renin mRNA isoforms from alternative tissue-specific transcriptional initiation. Physiol. Genom. 2000, 3, 25–31. [Google Scholar] [CrossRef]
- Nakagawa, P.; Nair, A.R.; Agbor, L.N.; Gomez, J.; Wu, J.; Zhang, S.Y.; Lu, K.T.; Morgan, D.A.; Rahmouni, K.; Grobe, J.L.; et al. Increased Susceptibility of Mice Lacking Renin-b to Angiotensin II-Induced Organ Damage. Hypertension 2020, 76, 468–477. [Google Scholar] [CrossRef]
- Grobe, J.L.; Xu, D.; Sigmund, C.D. An intracellular renin-angiotensin system in neurons: Fact, hypothesis, or fantasy. Physiology 2008, 23, 187–193. [Google Scholar] [CrossRef]
- Lavoie, J.L.; Liu, X.; Bianco, R.A.; Beltz, T.G.; Johnson, A.K.; Sigmund, C.D. Evidence supporting a functional role for intracellular renin in the brain. Hypertension 2006, 47, 461–466. [Google Scholar] [CrossRef]
- Shinohara, K.; Liu, X.; Morgan, D.A.; Davis, D.R.; Sequeira-Lopez, M.L.; Cassell, M.D.; Grobe, J.L.; Rahmouni, K.; Sigmund, C.D. Selective Deletion of the Brain-Specific Isoform of Renin Causes Neurogenic Hypertension. Hypertension 2016, 68, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Nakagawa, P.; Gomez, J.; Morgan, D.A.; Littlejohn, N.K.; Folchert, M.D.; Weidemann, B.J.; Liu, X.; Walsh, S.A.; Ponto, L.L.; et al. Selective Deletion of Renin-b in the Brain Alters Drinking and Metabolism. Hypertension 2017, 70, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Borges, G.R.; Grobe, J.L.; Pelham, C.J.; Yang, B.; Sigmund, C.D. Preservation of intracellular renin expression is insufficient to compensate for genetic loss of secreted renin. Hypertension 2009, 54, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Borges, G.R.; Davis, D.R.; Agassandian, K.; Sequeira Lopez, M.L.; Gomez, R.A.; Cassell, M.D.; Grobe, J.L.; Sigmund, C.D. Neuron- or glial-specific ablation of secreted renin does not affect renal renin, baseline arterial pressure, or metabolism. Physiol. Genom. 2011, 43, 286–294. [Google Scholar] [CrossRef]
- Souza, L.A.C.; Worker, C.J.; Li, W.; Trebak, F.; Watkins, T.; Gayban, A.J.B.; Yamasaki, E.; Cooper, S.G.; Drumm, B.T.; Feng, Y. (Pro)renin receptor knockdown in the paraventricular nucleus of the hypothalamus attenuates hypertension development and AT1 receptor-mediated calcium events. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1389–H1405. [Google Scholar] [CrossRef]
- Woods, T.C.; Satou, R.; Miyata, K.; Katsurada, A.; Dugas, C.M.; Klingenberg, N.C.; Fonseca, V.A.; Navar, L.G. Canagliflozin Prevents Intrarenal Angiotensinogen Augmentation and Mitigates Kidney Injury and Hypertension in Mouse Model of Type 2 Diabetes Mellitus. Am. J. Nephrol. 2019, 49, 331–342. [Google Scholar] [CrossRef]
- Satou, R.; Cypress, M.W.; Woods, T.C.; Katsurada, A.; Dugas, C.M.; Fonseca, V.A.; Navar, L.G. Blockade of sodium-glucose cotransporter 2 suppresses high glucose-induced angiotensinogen augmentation in renal proximal tubular cells. Am. J. Physiol. Renal. Physiol. 2020, 318, F67–F75. [Google Scholar] [CrossRef]
- De Kloet, A.D.; Wang, L.; Ludin, J.A.; Smith, J.A.; Pioquinto, D.J.; Hiller, H.; Steckelings, U.M.; Scheuer, D.A.; Sumners, C.; Krause, E.G. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct. Funct. 2016, 221, 891–912. [Google Scholar] [CrossRef]
- Peng, H.; Jensen, D.D.; Li, W.; Sullivan, M.N.; Buller, S.A.; Worker, C.J.; Cooper, S.G.; Zheng, S.; Earley, S.; Sigmund, C.D.; et al. Overexpression of the neuronal human (pro)renin receptor mediates angiotensin II-independent blood pressure regulation in the central nervous system. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H580–H592. [Google Scholar] [CrossRef]
- Grobe, J.L.; Rahmouni, K.; Liu, X.; Sigmund, C.D. Metabolic rate regulation by the renin-angiotensin system: Brain vs. body. Pflugers Arch. 2013, 465, 167–175. [Google Scholar] [CrossRef]
- Vivas, L.; Godino, A.; Dalmasso, C.; Caeiro, X.E.; Macchione, A.F.; Cambiasso, M.J. Frontiers in Neuroscience. Neurochemical Circuits Subserving Fluid Balance and Baroreflex: A Role for Serotonin, Oxytocin, and Gonadal Steroids. In Neurobiology of Body Fluid Homeostasis: Transduction and Integration; De Luca, L.A., Menani, J.V., Jr., Johnson, A.K., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2014. [Google Scholar]
- Rossi, N.F.; Zenner, Z.; Rishi, A.K.; Levi, E.; Maliszewska-Scislo, M. AT(1) receptors in the subfornical organ modulate arterial pressure and the baroreflex in two-kidney, one-clip hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R172–R185. [Google Scholar] [CrossRef] [PubMed]
- Zubcevic, J.; Santisteban, M.M.; Perez, P.D.; Arocha, R.; Hiller, H.; Malphurs, W.L.; Colon-Perez, L.M.; Sharma, R.K.; de Kloet, A.; Krause, E.G.; et al. A Single Angiotensin II Hypertensive Stimulus Is Associated with Prolonged Neuronal and Immune System Activation in Wistar-Kyoto Rats. Front. Physiol. 2017, 8, 592. [Google Scholar] [CrossRef]
- Wiinberg, N.; Høegholm, A.; Christensen, H.R.; Bang, L.E.; Mikkelsen, K.L.; Nielsen, P.E.; Svendsen, T.L.; Kampmann, J.P.; Madsen, N.H.; Bentzon, M.W. 24-h ambulatory blood pressure in 352 normal Danish subjects, related to age and gender. Am. J. Hypertens. 1995, 8, 978–986. [Google Scholar] [CrossRef]
- Colafella, K.M.M.; Denton, K.M. Sex-specific differences in hypertension and associated cardiovascular disease. Nat. Rev. Nephrol. 2018, 14, 185–201. [Google Scholar] [CrossRef]
- Pilote, L.; Dasgupta, K.; Guru, V.; Humphries, K.H.; McGrath, J.; Norris, C.; Rabi, D.; Tremblay, J.; Alamian, A.; Barnett, T.; et al. A comprehensive view of sex-specific issues related to cardiovascular disease. Cmaj 2007, 176, S1–S44. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, L.M.; Sampson, A.K.; Brown, R.D.; Denton, K.M. The “his and hers” of the renin-angiotensin system. Curr. Hypertens. Rep. 2013, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Pamidimukkala, J.; Hay, M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2177–H2184. [Google Scholar] [CrossRef]
- Peng, H.; Li, W.; Seth, D.M.; Nair, A.R.; Francis, J.; Feng, Y. (Pro)renin receptor mediates both angiotensin II-dependent and -independent oxidative stress in neuronal cells. PLoS ONE. 2013, 8, e58339. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Cao, T.; Sato, R.; McDaniels, S.J.; Kobori, H.; Navar, L.G.; Feng, Y. Brain-targeted (pro)renin receptor knockdown attenuates angiotensin II-dependent hypertension. Hypertension 2012, 59, 1188–1194. [Google Scholar] [CrossRef]
- Lob, H.E.; Schultz, D.; Marvar, P.J.; Davisson, R.L.; Harrison, D.G. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 2013, 61, 382–387. [Google Scholar] [CrossRef]
- Case, A.J.; Li, S.; Basu, U.; Tian, J.; Zimmerman, M.C. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H19–H28. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Baxter, J.D. Human prorenin. Hypertension 1991, 17, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Sealey, J.E.; Glorioso, N.; Itskovitz, J.; Laragh, J.H. Prorenin as a reproductive hormone. New form of the renin system. Am. J. Med. 1986, 81, 1041–1046. [Google Scholar] [CrossRef]
- Genain, C.P.; Van Loon, G.R.; Kotchen, T.A. Distribution of renin activity and angiotensinogen in rat brain. Effects of dietary sodium chloride intake on brain renin. J. Clin. Investig. 1985, 76, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Van Thiel, B.S.; Góes Martini, A.; Te Riet, L.; Severs, D.; Uijl, E.; Garrelds, I.M.; Leijten, F.P.J.; van der Pluijm, I.; Essers, J.; Qadri, F.; et al. Brain Renin-Angiotensin System: Does It Exist? Hypertension 2017, 69, 1136–1144. [Google Scholar] [CrossRef]
- Sakai, K.; Agassandian, K.; Morimoto, S.; Sinnayah, P.; Cassell, M.D.; Davisson, R.L.; Sigmund, C.D. Local production of angiotensin II in the subfornical organ causes elevated drinking. J. Clin. Investig. 2007, 117, 1088–1095. [Google Scholar] [CrossRef]
- Schinke, M.; Baltatu, O.; Böhm, M.; Peters, J.; Rascher, W.; Bricca, G.; Lippoldt, A.; Ganten, D.; Bader, M. Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc. Natl. Acad. Sci. USA 1999, 96, 3975–3980. [Google Scholar] [CrossRef]
- Baltatu, O.; Silva, J.A.; Ganten, D.; Bader, M. The Brain Renin-Angiotensin System Modulates Angiotensin II–Induced Hypertension and Cardiac Hypertrophy. Hypertension 2000, 35, 409–412. [Google Scholar] [CrossRef]
- Itaya, Y.; Suzuki, H.; Matsukawa, S.; Kondo, K.; Saruta, T. Central renin-angiotensin system and the pathogenesis of DOCA-salt hypertension in rats. Am. J. Physiol. 1986, 251, H261–H268. [Google Scholar] [CrossRef]
- Park, C.G.; Leen, F.H.H. Effects of Centrally Administered Losartan on Deoxycorticosterone-salt Hypertension Rats. J. Korean Med. Sci. 2001, 16, 553–557. [Google Scholar] [CrossRef]
- Nakagawa, P.; Sigmund, C.D. How Is the Brain Renin-Angiotensin System Regulated? Hypertension 2017, 70, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Jensen, D.D.; Peng, H.; Feng, Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharm. Ther. 2016, 164, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Wanka, H.; Lutze, P.; Albers, A.; Golchert, J.; Staar, D.; Peters, J. Overexpression of Transcripts Coding for Renin-b but Not for Renin-a Reduce Oxidative Stress and Increase Cardiomyoblast Survival under Starvation Conditions. Cells 2021, 10, 1024. [Google Scholar] [CrossRef] [PubMed]
- Basting, T.; Lazartigues, E. DOCA-Salt Hypertension: An Update. Curr. Hypertens. Rep. 2017, 19, 32. [Google Scholar] [CrossRef]
- Yemane, H.; Busauskas, M.; Burris, S.K.; Knuepfer, M.M. Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)-salt hypertension in rats. Exp. Physiol. 2010, 95, 51–55. [Google Scholar] [CrossRef]
- De Oliveira-Sales, E.B.; Nishi, E.E.; Boim, M.A.; Dolnikoff, M.S.; Bergamaschi, C.T.; Campos, R.R. Upregulation of AT1R and iNOS in the rostral ventrolateral medulla (RVLM) is essential for the sympathetic hyperactivity and hypertension in the 2K-1C Wistar rat model. Am. J. Hypertens. 2010, 23, 708–715. [Google Scholar] [CrossRef]
- Chen, A.; Huang, B.S.; Wang, H.W.; Ahmad, M.; Leenen, F.H. Knockdown of mineralocorticoid or angiotensin II type 1 receptor gene expression in the paraventricular nucleus prevents angiotensin II hypertension in rats. J. Physiol. 2014, 592, 3523–3536. [Google Scholar] [CrossRef]
- Frazier, C.J.; Harden, S.W.; Alleyne, A.R.; Mohammed, M.; Sheng, W.; Smith, J.A.; Elsaafien, K.; Spector, E.A.; Johnson, D.N.; Scott, K.A.; et al. An Angiotensin-Responsive Connection from the Lamina Terminalis to the Paraventricular Nucleus of the Hypothalamus Evokes Vasopressin Secretion to Increase Blood Pressure in Mice. J. Neurosci. 2021, 41, 1429–1442. [Google Scholar] [CrossRef]
- Jancovski, N.; Carter, D.A.; Connelly, A.A.; Stevens, E.; Bassi, J.K.; Menuet, C.; Allen, A.M. Angiotensin type 1A receptor expression in C1 neurons of the rostral ventrolateral medulla contributes to the development of angiotensin-dependent hypertension. Exp. Physiol. 2014, 99, 1597–1610. [Google Scholar] [CrossRef]
- Wang, H.W.; Huang, B.S.; White, R.A.; Chen, A.; Ahmad, M.; Leenen, F.H. Mineralocorticoid and angiotensin II type 1 receptors in the subfornical organ mediate angiotensin II-induced hypothalamic reactive oxygen species and hypertension. Neuroscience 2016, 329, 112–121. [Google Scholar] [CrossRef]
- Hilzendeger, A.M.; Cassell, M.D.; Davis, D.R.; Stauss, H.M.; Mark, A.L.; Grobe, J.L.; Sigmund, C.D. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension 2013, 61, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.Y.; Peng, W.; Zhang, Y.P.; Li, J.D.; Shen, Y.; Sun, X.F. Brain endogenous angiotensin II receptor type 2 (AT2-R) protects against DOCA/salt-induced hypertension in female rats. J. Neuroinflamm. 2015, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Burmeister, M.A.; Tian, X.; Zhou, Y.; Guruju, M.R.; Stupinski, J.A.; Sharma, R.V.; Davisson, R.L. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension 2009, 54, 1106–1114. [Google Scholar] [CrossRef]
- Callera, G.E.; Tostes, R.C.; Yogi, A.; Montezano, A.C.; Touyz, R.M. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin. Sci. 2006, 110, 243–253. [Google Scholar] [CrossRef]
- De Queiroz, T.M.; Xia, H.; Filipeanu, C.M.; Braga, V.A.; Lazartigues, E. α-Lipoic acid reduces neurogenic bhypertension by blunting oxidative stress-mediated increase in ADAM17. Am J. Physiol. Heart Circ. Physiol. 2015, 309, H926–H934. [Google Scholar] [CrossRef]
- Sriramula, S.; Xia, H.; Xu, P.; Lazartigues, E. Brain-targeted angiotensin-converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase-mediated inflammation. Hypertension 2015, 65, 577–586. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′ to 3′ | ||
|---|---|---|
| AT1aR | Forward | TCACCAGATCAAGTGCATTTTGA |
| Reverse | AGAGTTAAGGGCCATTTTGCTTT | |
| AT2R | Forward | TACCCGTGACCAAGTCCTGA |
| Reverse | TACCCATCCAGGTCAGAGCA | |
| Beta actin | Forward | CCAGCCTTCCTTCTTGGGTA |
| Reverse | AGAGGTCTTTACGGATGTCAACG | |
| NOX 2 | Forward | CCCTTTGGTACAGCCAGTGAAGAT |
| Reverse | CAATCCCGGCTCCCACTAACATCA | |
| NOX 4 | Forward | TGAACTACAGTGAAGATTTCCTTGAAC |
| Reverse | GACACCCGTCAGACCAGGAA | |
| Total Renin | Forward | TGCTTGTGGGATTCACAGCCTCTA |
| Reverse | TGTGTCACAGTGATTCCACCCACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, S.G.; Souza, L.A.C.; Worker, C.J.; Gayban, A.J.B.; Buller, S.; Satou, R.; Feng Earley, Y. Renin-a in the Subfornical Organ Plays a Critical Role in the Maintenance of Salt-Sensitive Hypertension. Biomolecules 2022, 12, 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12091169
Cooper SG, Souza LAC, Worker CJ, Gayban AJB, Buller S, Satou R, Feng Earley Y. Renin-a in the Subfornical Organ Plays a Critical Role in the Maintenance of Salt-Sensitive Hypertension. Biomolecules. 2022; 12(9):1169. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12091169
Chicago/Turabian StyleCooper, Silvana G., Lucas A. C. Souza, Caleb J. Worker, Ariana Julia B. Gayban, Sophie Buller, Ryosuke Satou, and Yumei Feng Earley. 2022. "Renin-a in the Subfornical Organ Plays a Critical Role in the Maintenance of Salt-Sensitive Hypertension" Biomolecules 12, no. 9: 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12091169