Dual Role of the Alternative Reading Frame ARF Protein in Cancer


1
Department of Pharmacology, Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA
2
Division of Hematology and Medical Oncology, Laura and Isaac Perlmutter Cancer Center, NYU Langone Medical Center, New York, NY 10016, USA
3
Department of Biology, University of Naples Federico II, 80126 Naples, Italy
*
Author to whom correspondence should be addressed.
†
Contributed equally to this manuscript.
Biomolecules 2019, 9(3), 87; https://0-doi-org.brum.beds.ac.uk/10.3390/biom9030087
Submission received: 11 December 2018
/
Revised: 20 February 2019
/
Accepted: 22 February 2019
/
Published: 4 March 2019
Abstract
:The CDKN2a/ARF locus expresses two partially overlapping transcripts that encode two distinct proteins, namely p14ARF (p19Arf in mouse) and p16INK4a, which present no sequence identity. Initial data obtained in mice showed that both proteins are potent tumor suppressors. In line with a tumor-suppressive role, ARF-deficient mice develop lymphomas, sarcomas, and adenocarcinomas, with a median survival rate of one year of age. In humans, the importance of ARF inactivation in cancer is less clear whereas a more obvious role has been documented for p16INK4a. Indeed, many alterations in human tumors result in the elimination of the entire locus, while the majority of point mutations affect p16INK4a. Nevertheless, specific mutations of p14ARF have been described in different types of human cancers such as colorectal and gastric carcinomas, melanoma and glioblastoma. The activity of the tumor suppressor ARF has been shown to rely on both p53-dependent and independent functions. However, novel data collected in the last years has challenged the traditional and established role of this protein as a tumor suppressor. In particular, tumors retaining ARF expression evolve to metastatic and invasive phenotypes and in humans are associated with a poor prognosis. In this review, the recent evidence and the molecular mechanisms of a novel role played by ARF will be presented and discussed, both in pathological and physiological contexts.
1. Introduction
The ARF (alternative reading frame) protein is encoded by the Alternative Reading Frame of the CDKN2a locus, one of the most frequently mutated sites in human cancers after the p53 locus [1,2,3]. The locus, located on human chromosome 9p21, encodes two completely unrelated proteins, p16INK4a and p14ARF, both of which are potent tumor suppressors. The mechanism by which the two proteins are produced is quite unusual. Each gene is endowed with its own promoter that guides the transcription of an α- or β-transcript. Each transcript has a specific 5’ exon, E1α or E1β for INK4a and ARF respectively, spliced to a common exon 2 (Figure 1a) in which two overlapped ORFs (Open Reading Frame) are translated into two proteins sharing no amino acid sequence identity at all.
The alpha transcript encodes the p16INK4a protein, a member of the INK4 family of inhibitors of the cyclin-dependent kinases 4 and 6 (Inhibitor of CDK4). In response to specific signals, they block the assembly and/or inhibit the kinase activity of the cyclin D-CDK4/6 complex required for G1 to S cell cycle progression [6,7]. In this way, the retinoblastoma protein pRB is maintained in an active hypo-phosphorylated state and sequesters the transcription factors of the E2F family causing G1-phase cell cycle arrest [7,8] (Figure 1b). The ARF protein instead inhibits the functions of the MDM2 oncoprotein (Mouse Double Minute 2, HDM2 in human) thus inducing p53 stabilization and the activation of p53-dependent pathways (Figure 1b).
In humans, the β transcript results in a polypeptide of 132 amino acids (14 kDa) named p14ARF while, in mice, the transcript is translated into a 169 amino acid polypeptide named p19ARF (19kDa). Human and mouse proteins share only 50% of identity. Interestingly, the exon 1β-encoded N-terminal region, that is necessary and sufficient to fulfil almost all of the known ARF tumor suppressor functions, is only modestly conserved between species, whereas the exon 2-encoded C-terminal region shows a stronger degree of identity between human and mouse (57% of identity) [5]. By comparison, mouse and human INK4a are more conserved, sharing the 65% of identity overall [9]. ARF proteins are highly basic (> 20% arginine content) and hydrophobic molecules. The basic nature of ARF renders this protein highly insoluble and this is likely the reason for which neither NMR (nuclear magnetic resonance) nor crystal structure has been determined, despite its small size. ARF probably needs to form complexes with other molecules to assume specific spatial conformation and to neutralize its charge at physiological pH, thus explaining the incredible number of ARF binding partners [10].
ARF is a potent tumor suppressor, regulating cell cycle arrest and/or apoptosis by both p53-dependent and independent pathways [11]. Interestingly, it represents a link between the pRb and the p53 pathway, the most important tumor suppressor pathways within the cell. It has been demonstrated the ARF triggers a p53-dependent checkpoint when the pRb pathway is compromised. In particular, when inactivated, pRb causes the release of E2F transcription factors, which in turn induce the increase of ARF transcription [5,12].
The classical ARF tumor suppressor function has been challenged by the recent observations suggesting that this protein is overexpressed and plays pro-oncogenic functions in specific types of human cancers. In this review, ARF dependent tumor suppressor mechanisms will be described together with the new evidence outlining its pro-survival functions.
2. ARF Inactivation in Human and Murine Cancer
A contribution of ARF to tumor formation has been documented using genetic analysis of tumors, molecular and cell biology approaches, and animal models [13,14]. In respect to p16, ARF seems to assume a more prominent role as a tumor suppressor in mice rather than in humans, where the mutation frequency of this gene is low. Arf-null mice usually die after one year since they develop tumors, mostly sarcomas (43%), lymphoid malignancies (29%), carcinomas (17%), and tumors of the nervous system (11%). Furthermore, tumors arising in the heterozygous mice undergo further deletion of the remaining allele [15].
In most cases of human cancers, both ARF and p16 are inactivated, making it difficult to determine their individual roles in tumor suppression [16,17,18,19,20]. Alterations of the CDKN2a locus were found in roughly 30% of human tumors such as glioblastoma, melanoma and pancreatic adenocarcinoma [8,21]. However, there are specific examples in which only ARF appears to be affected in human cancer. Promoter methylation in ARF gene has been reported to occur independently of the INK4a promoter methylation, thus suggesting that this is a specific alteration of ARF expression. A CpG island within the promoter region of p14ARF has been found to be hyper-methylated [21] in a wide spectrum of human cancers such as colorectal [22], gastric [23] and prostate carcinomas [24]. Deletions and point mutations of the exon 1β has been reported in familial melanoma syndromes and in glioblastoma [11,17,25,26,27,28]. In breast cancer, several insults such as homozygous deletion, loss of heterozygosity and promoter hyper-methylation, have all been described [29]. Interestingly, in the majority of non-small cell lung cancer (NSCLC), p14ARF protein is expressed at low levels, albeit no genetic mutations or known transcriptional alterations have been reported so far [30].
3. ARF is Involved in the p53 Pathway
One of the most well-defined ARF function is the suppression of aberrant cell growth in response to oncogene activation [12,31,32,33,34]. In particular, in this condition, ARF activates the transcription factor p53 that triggers the expression of many apoptosis inducers and cell cycle inhibitory genes. TP53 mutation is the most frequent genetic alteration in human cancers. In response to a wide variety of cellular stresses, p53 is able to induce the expression of target genes leading to cell cycle arrest, apoptosis or senescence. A transcriptional target of p53 is the MDM2 protein, which plays a central role in regulating p53 functions [35,36,37]. MDM2 is an E3 ubiquitin ligase that ubiquitinates p53 through its RING (Really Interesting New Gene) domain, common to many E3 ubiquitin ligases [38]. The two proteins are part of a negative feedback loop that keeps p53 levels low during normal growth and development. Interestingly, the dynamics of p53 activation appears to be the signal that dictates cell fate. While transient p53 activation induces cell cycle arrest, long-lasting p53 oscillations promote apoptosis [39]. The mechanisms that increase the stability and the activity of p53 point to alleviating the negative effect of MDM2 on p53. By interacting with p53, MDM2 blocks p53-mediated transactivation and targets the p53 protein for ubiquitin-dependent proteasome-mediated degradation [35,37,40,41]. Early studies reported that the p19ARF protein was involved in the p53 pathway via its interaction and inhibition of MDM2 [42,43]. Several studies then clarified that ARF inhibition of cell proliferation also relies on p53 independent pathways through the functional and molecular interaction with a number of different proteins (Figure 2).
ARF Inhibits MDM2 Through Multiple Mechanisms
Several mechanisms have been formulated to explain ARF-mediated p53 stabilization and activation. Initial evidence reported that ARF is able to block the nucleus-cytoplasm shuttling of the MDM2-p53 complex [44]. ARF is predominantly located in the nucleolus in different cell lines and in different experimental conditions [45]. In regard to the human protein, this localization depends on two aminoacidic stretches, one located at the N-terminal region (aa 2–14), while the other resides at the C-terminal and comprises a sequence of basic amino acids (aa 82–101) [45,46]. The co-expression of ARF together with p53 and MDM2 blocks the export of both p53 and MDM2 to the cytoplasm. Furthermore, it was observed a re-localization of ARF from the nucleolus to the nucleus in sub-nuclear structures defined as nuclear bodies, in which p53 is stabilized and activated. Another mechanism reports that ARF induces p53 stabilization and activation by sequestering MDM2 in the nucleolus. This hypothesis is based on the evidence that the interaction between ARF and MDM2 reveals a cryptic nucleolar localization signal present in the C-terminal region of MDM2 [47]. This signal, in synergy with similar sequences within ARF, induces re-localization of the ARF-MDM2 complex in the nucleolus, thus preventing p53 binding and export to the cytoplasm [48,49].
An alternative mechanism, that probably integrates the previously described ones, reports that ARF, by MDM2 binding, inhibits its ubiquitin-ligase activity [50,51]. In order to be exported in the cytoplasm and degraded, p53 needs to be modified through the covalent binding to ubiquitin. Ubiquitination, in addition to targeting p53 to the proteasome for degradation, prevents the formation of p53 tetramers, thus inducing the exposure of the p53 nuclear export signals and its re-localization to the cytoplasm.
The observations that ARF mediated p53 stabilization can occur without relocation of MDM2 in the nucleolus, and that ARF mutants that do not exhibit nucleolar accumulation retain the ability to stabilize p53 [52], suggested that its nucleolar localization could contribute to, but is not essential for, p53 stabilization. As mouse and human homologues are poorly conserved it has been difficult to dissect which are the domains required for ARF localization, MDM2 binding and inhibition, and above all, how these aspects led to p53 stabilization. Moreover, the MDM2 binding and the p53 stabilization domains, as well as the nucleolar driving sequences, show a certain degree of overlap. In mice, for example, the domain mapped between aa 26–37 is not only required to induce p19ARF nucleolar localization but also for p53 accumulation and cell cycle arrest [53]. Another layer of complexity comes from the observation that N-terminally deleted p19ARF mutants, although impaired in p53 stabilization, are still able to induce its transcriptional activation and to inhibit cell growth. Conversely, expression of ARF mutants not able to block cell proliferation through p53 can still induce MDM2 accumulation in the nucleolus. Altogether these data point to the notion that MDM2 subcellular localization and p53 stabilization are not both required to guarantee p53-dependent ARF inhibition of cell growth, as previously supposed [54].
4. ARF is Involved In p53 Independent Pathways of Tumor Suppression
Although ARF is undoubtedly a critical component of the p53 pathway, there are many lines of evidence that ARF restrains cell growth independently of p53 (Figure 2). Analysis of mouse models in which Arf, Trp53 and Mdm2 genes were simultaneously ablated (triple knockout or TKO mice) showed a higher tendency towards tumor development than those lacking only p53 and MDM2 [55]. Furthermore, Arf -/- and Arf +/- mice develop a broader spectrum of tumors than Trp53 null mice and ARF overexpression can induce cell cycle arrest in cells lacking p53. Finally, in cells derived from TKO mice, the reintroduction of ARF prevents S phase entry and/or trigger apoptosis by mechanisms that did not require the expression of p53 protein [55]. Similarly, in human lung tumor cells devoid of p53 expression, p14ARF induces cell cycle arrest accompanied by features of tumor regression [56]. Also, xenograft models of pancreatic cancer showed that ARF expression impedes cell colonization and thus metastasis formation in a p53-independent fashion [13]. The notion that ARF acts independently of the Mdm2–p53 axis in tumor surveillance is in line with the observation that ARF interacts with a multitude of different cellular partners, such as proteins involved in transcriptional control (E2Fs, DP1, p63, c-Myc, Hif1α), nucleolar proteins such as nucleophosmin (NPM/B23), viral proteins (HIV-1Tat), mitochondrial protein (p32) and others [10,57]. This variety of ARF’s interactors suggested that the protein could play a wide role in cell protection from different types of insults. In particular, for some targets, the interaction with ARF causes an alteration of their stability. For example, NPM/B23 and E2F become degraded by the proteasome in a ubiquitin-dependent manner upon ARF interaction. Other targets change their localization upon ARF expression, while others become activated or stabilized [58]. Moreover, it has been demonstrated that the interaction with the mitochondrial protein p32 determines ARF’s mitochondrial recruitment, through its domain in exon 2 (aa stretch 82–101) [59,60]. The ability to localize to the mitochondrion appears to be p53 independent and required for ARF-induced apoptosis. Interestingly, the ARF exon 2 encoded domain appears to have a role in the defense mechanism that protects cells from oxidative stress. In senescent melanocytes, endogenous ARF is constitutively expressed as a cytoplasmic protein recruited to dysfunctional mitochondria as part of a pathway aimed at maintaining low levels of intracellular superoxide [61]. ARF translocation to mitochondria induces dissipation of mitochondrial potential, with the consequent decrease of free radical levels and cell growth inhibition. This mitochondrial activity involves an evolutionarily conserved acidic motif GHDDGQ (residues 65–70 in p14ARF), which mediates the interaction with, and the consequent inhibition of, BCL-xL, a positive regulator of mitochondrial potential. This cell-protective mechanism appears to be targeted in familial melanoma thus explaining why ARF mutations predispose carriers to melanoma. This function appears to be p3-independent. Moreover, although the C-terminal domain is also required for ARF induced autophagy (as we will discuss below), it is also independent of ARF function in autophagy.
Another tumor-suppressive function of ARF relies on its ability to ensure chromosomal stability [1] through its functional interaction with Aurora B [62]. ARF ablation in primary mouse embryonic fibroblasts (MEFs) results in aneuploidy, caused by misaligned and lagging chromosomes and multipolar spindles. Albeit this appears to be a p53-independent ARF function, it has been shown that this mechanism can also follow p53-dependent routes [63].
It has been widely reported that ARF is able to promote the sumoylation of some of its interactors [64,65,66,67]. This modification modulates a variety of phenomena such as protein stability, transport, modulation of gene expression, ubiquitylation, DNA repair, and centromeric chromatid cohesion. ARF induces sumoylation of both MDM2 and NPM/B23 by direct interaction with these proteins and in a p53-independent manner [65,68,69]. In addition, it has been reported that ARF interacts with the Myc-associated zinc finger protein (Miz1) and, by inducing its sumoylation, facilitates the assembly of the Myc-Miz1 complex that assists the switch from G1 arrest to apoptosis [70]. It has been suggested that ARF promotes this process through direct interaction with the Small Ubiquitin Modifier (SUMO)-conjugating enzyme Ubc9 [65]. Other evidence of ARF involvement in sumoylation is derived from the observation that it can inhibit the function of a desumoylating protein, SENP3 [71]. Although ARF involvement in the sumoylation process is well documented, the biological meaning of ARF-mediated sumoylation is mostly still unclear. Upon thermal or oxidative stress, an increase of SUMO 2 and 3 levels has been reported. Cells over-expressing SUMO 2 and 3 exhibit premature senescence [72], and an increase of both p53 and pRb sumoylation [73,74,75]. Given the established role of ARF in senescence, it would be interesting to analyze ARFs role in sumoylation-induced senescence.
Intriguingly, while ARF mediates p53 stabilization, it has the opposite function with another member of the p53 family, the transcription factor p63 that plays a fundamental role in the development of epithelial derivatives [76,77]. ARF interacts with one of the p63 isoforms, namely DNp63α [78], and increase its sumoylation and its subsequent proteasome-mediated degradation in both tumoral and immortalized cell lines [79,80]. ΔNp63α has an established role in oncogenesis being expressed in several cancerous cells [81,82,83] and acting as dominant negative in respect to p53. ARF’s role in p63 degradation suggests that ARF while stabilizing p53 and eliciting p53-dependent pathways, at the same time can inhibit p63 oncogenic functions.
5. Mechanisms Governing ARF Degradation
The study of ARF interacting partners has led to the accumulation of a bulk of knowledge about the mechanisms regulating its turnover within the cell. Both mouse and human ARF are relatively stable proteins in the nucleolus, with a half-life of about 6 h [88]. This suggested, and was lately experimentally demonstrated, that ARF nucleolar localization could be a mechanism to stabilize the protein [89]. Early studies provided evidence that ARF association with MDM2 in the nucleoplasm decreases its half-life [4,10,42] and that it rapidly decreases when the protein is forced to the nucleoplasm. In the nucleolus, nucleophosmin plays an important role in the control of ARF turnover [68,90]. In response to increased levels of NPM, the turnover of p19ARF is retarded, whereas NPM protein levels downregulation accelerates its degradation [88] and cause p19ARF exclusion from nucleoli. Both NPM binding and nucleolar localization are required to promote ARF stabilization [91,92,93,94]. Conversely, ARF causes NPM poly-ubiquitination and degradation [90,95,96].
For the vast majority of proteins, conjugation of ubiquitin to internal lysines is the initial event in their degradation by the ubiquitin-proteasome system (UPS). Although both human and mouse ARF proteins accumulate following treatment with proteasome inhibitors, early studies excluded the possibility that ARF turnover could be regulated by UPS because the human protein is lysine-less and the murine one has a single lysine residue not required for proteasome degradation [89,97,98]. Interestingly, it was observed that ARF is subjected to N-terminal ubiquitination, a rare mechanism of ubiquitin conjugation also displayed by MyoD (Myogenic Differentiation 1) [99] and p21 [100]. While ARF degradation is inhibited in cancer cell due to UPS dysfunction, in normal cell lines its half-life is strongly reduced [101]. Through biochemical purification, an ARF specific E3 ubiquitin ligase named TRIP12/ULF (Thyroid hormone Receptor Interactor Protein 12/Ubiquitin Ligase For ARF) promoting its N-terminal-ubiquitylation and degradation was identified. Oncogenic stress such as c-Myc and NPM overexpression both inhibit ULF-mediated ARF ubiquitylation [102].
ARF can also be degraded through ubiquitin-independent mechanisms. One of these mechanisms reports that the interaction with TBP-1 (Tat-Binding Protein-1) could cause ARF folding thus making this protein a poor substrate for proteasome destruction [98,103]. Another mechanism involves the 20S/REG-γ proteasome system [104] that resides in the nucleus of interphase cells and is involved in the degradation of small unstructured proteins, such as ARF, p21, and p16INK4a. While NPM prevents ARF degradation retaining it in the nucleolus, TBP-1 protects ARF mainly in the nuclear compartment.
6. ARF Plays a Role During Development and Differentiation
It has been observed that INK4-ARF expression increases with age, mirroring a decline in tissue regenerative capacity. In stem cells, the entire INK4-ARF locus is kept in an epigenetically silenced state due to the function of the Polycomb group of proteins [105,106]. It has been proposed that, as cells lose stemness and acquire differentiated features, the INK4-ARF locus is remodeled in order to become responsive to stress and mitogenic signals arising during cell differentiation [107,108]. Physiological expression of ARF during development has been described in a precise time frame during the differentiation of several tissue types, such as the developing eye, spermatogonia, during epithelial differentiation, and in the yolk sac. One of the first pieces of evidence for ARF involvement in development came from the observation that ARF knockout mice developed smaller eyes compared to wild type mice due to defects in the neuroretina and lens, resulting in blindness [109]. A closer examination revealed that the persistence of the platelet-derived growth factor (PDGF) signaling in Arf -/- cells of the hyaloid vascular system (HVS) stimulates aberrant proliferation and survival of vitreal perivascular cells in the eye in a p53 independent manner. In line with this, Pdgfrβ (Platelet-Derived Growth Factor Receptor β) deletion rescues the Arf -/- eye phenotype and restores vision. Analysis of mouse models indicated that the role of ARF in hyaloid vascular regression is p53-independent [110,111]. Further studies showed that ARF induces the expression of several microRNAs (miR) that repress Pdgfrβ synthesis. In particular, the expression of miR-34 is required for both Pdgfrβ repression and cell proliferation arrest elicited by p19ARF [112].
Interestingly, Li and colleagues reported that ARF is physiologically expressed in the fetal yolk sac, a tissue derived from the extra-embryonic endoderm (ExEn) [113]. In in vitro cultured embryoid bodies, ARF expression regulates the late stages of ExEn differentiation through the induction of another microRNA, miR-205, that in turn induces a set of genes required for cell migration and adhesion. Interestingly miR-205 is also regulated by p53 (and by the other p53 family members, p63 and p73) [113].
A role played by ARF has been reported in male germ cell development in mice [111]. ARF is over-expressed in mitotically dividing spermatogonia lining the basement membrane of the seminiferous tubules. Undifferentiated spermatogonia evolve into spermatocytes during a spatio-temporal coordinated process by which cells detach from the basement of the tubules and through a series of mitotic and meiotic cellular divisions, move towards the lumen. In Arf-null progenitors, detachment triggers DNA damage and p53-dependent apoptosis (anoikis) thus resulting in a reduced number of mature sperm and testicular atrophy. These data surprisingly indicate that, in a physiological process such as differentiation of spermatocytes, ARF can actually prevent instead of inducing, p53-dependent apoptosis [114]. The exact mechanism(s) by which ARF contributes to germ cells genomic integrity during their maturation is still lacking. It has been reported that ARF levels increase upon treatment with ATM (ataxia-telangiectasia mutated) inhibitors, underlining its involvement in a defense pathway that protects cells upon DNA damage. Also, ARF involvement in chromosomal stability and its cross-talk with the machinery of homologous recombination during meiosis could be part of the mechanism underlining its role in differentiation. The continuing pursuit of ARF’s functions in development led to the discovery of cross-talk between the DNA damage response and angiogenesis [115,116,117]. An inverse correlation has been observed between the expression levels of the vascular endothelial growth factor (VEGF) and ARF leading to the hypothesis that ARF expression can inhibit angiogenesis both in physiological and pathological conditions. These aspects and the therapeutic approaches potentially deriving from these studies have been clearly reviewed in Kostinas et al., 2014 [116].
As previously mentioned, ARF interacts with and sumoylates p63 in keratinocytes. During differentiation, the proliferative cells of the basal level of epidermis detach, lose the ability to self-renew and start to express molecular markers of differentiating skin. In particular, p63 levels decrease whereas ARF protein levels increase at the onset of keratinocyte differentiation [79]. The increase of differentiation-dependent protein sumoylation machinery [118] led to a scenario in which ARF expression is required for p63 sumoylation and thus inactivation. Interestingly, it has been shown that p63 represses ARF promoter during development, and in line with this, ARF knockout rescues the severe epithelial phenotype of p63 null mice allowing both cell proliferation and their subsequent differentiation in keratinocytes [119]. Figure 3 shows a comprehensive picture of ARF involvement in several biological mechanisms.
7. ARF has Pro-Oncogenic Functions
ARF is found overexpressed in a significant fraction of human tumors, such as Burkitt’s lymphoma [120] and the majority of tumors with mutant p53 [43]. In 74 samples of aggressive B-cell lymphomas, p14ARF is mainly localized in the nucleoplasm and is associated with cancer progression and unfavorable prognosis. Also, at the transcriptional level, ARF expression is increased in patients with haematological malignancies, especially in the early stage of CML (chronic myelogenous leukemia), suggesting that this over-expression could be associated with CML development [121]. Inactivation of the CDKN2a locus is rare in thyroid follicular adenomas and carcinomas, where p14ARF protein levels are strongly increased. Remarkably, ARF delocalization to the cytoplasm has been observed in thyroid aggressive papillary carcinomas, not accompanied by mutations in the ARF gene [122]. Although for several years ARF was considered to be dysfunctional when over-expressed in cancer tissues, surprisingly, ARF silencing was shown to limit the progression of Myc-driven lymphomas in mouse xenograft models [123]. In this study, Humbey and co-authors proposed for the first time that ARF can promote tumor progression through its ability to induce autophagy [60,124,125,126,127]. Autophagy is a lysosome-mediated process of self-digestion occurring during periods of nutrient deprivation. Target proteins and organelles are enveloped by the autophagosome, a double-membrane vesicle, that fuses with the lysosome leading to the degradation of its contents. The released amino acids are thus recycled for the synthesis of essential proteins [124]. ARF silencing in Myc-driven lymphoma cells impaired autophagy and decreased the ability of these cells to form tumors in vivo. In this cellular model, autophagy induction is thus a mechanism that helps cancer cells to survive during periods of metabolic or oxidative stress [123].
Interesting, a dual role in cancer has been proposed for autophagy [128]. The tumor suppressor function of this pathway has been inferred by the analysis of loss of function mouse models. Genetically engineered mouse models in which AuTophaGy essential genes (ATGs) were ablated show increased tissue and DNA damage, chronic inflammation, and chromosomal instability due to the accumulation of non-functional proteins and organelles and free radicals. This has been observed both in the liver and in the pancreas, tissues in which chronic inflammation creates a tumor-promoting environment [129,130,131]. Furthermore, the increased levels of the autophagosome cargo protein p62 upon dysfunctional autophagy promotes cancer survival by activation of the nuclear factor (erythroid-derived-2) like (NRF-2) a master regulator of the antioxidant defense response [132].
On the other hand, several observations showed that, in established tumors, autophagy functions as a survival pathway [133]. The metabolic survival function of autophagy is essential during starvation of all cells. However, some cancer cell lines have increased levels of autophagy also under non-starving conditions. In these cells, aberrant metabolic programmes are present in order to support hyper-proliferation and survival [134], cause the production of waste products, such as non-functional or misfolded proteins and DNA damage [135,136] that need to be discarded through the induction of autophagy. Moreover, activation of the antioxidant defense pathway induced by high levels of free radicals further promotes chemoresistance. During cancer evolution, the possibility to recycle cellular constituents is also an advantage for those cells that escape the primary tumor and disseminate through the body, a process that requires the ability to survive prolonged periods of starvation. In this scenario, it appears clear that while autophagy can contribute to ARF tumor suppression functions [137] the same mechanism can be hijacked to promote cancer evolution and survival [123].
Another evidence of an ARF pro-oncogenic role comes from the analysis of mouse models of prostate cancer. Genetic ablation of p19Arf in prostate epithelium does not accelerate but rather partially inhibits the prostate cancer phenotype of Pten-/- mice [138]. Tissue microarrays analysis performed on human prostate samples showed that p14ARF inactivation is indeed very rare in human prostate cancer and inversely correlated with disease aggressiveness. The authors observed that the double knock-out Pten-/-/p19Arf -/- causes a decrease of prostatic intraepithelial neoplasia incidence in the ventral region of the prostate respect to Pten-/- mice. Interestingly, this effect appears highly cell type specific as mouse embryo fibroblasts (MEFs) derived from these mice show instead increased proliferation rate and present features of cellular transformation.
Using a similar approach, the analysis of p19Arf deletion was analyzed in the context of Pten/Trp53 knockout mice [139]. In this study authors showed that ARF positively regulates prostate tumors growth by increasing the stability of the transcriptional factor Slug. Slug is a zinc-finger protein known to bind and repress the E-cadherin promoter thus mediating the so-called epithelial to mesenchymal transition (EMT), a cellular switch that provides cancer cells with the ability to disseminate through the body. In this study authors observed that in the nucleus of PC3 cells, ARF induces Slug stabilization by increasing its sumoylation. This results in the down-regulation of E-cadherin and, consequently, in the repression of the cell-cell junctions thus favoring cell migration and cancer metastasis. Both mathematical and experimental evidence suggested that the negative effect of Slug on E-cadherin can be counteracted by the expression of NRF2 [140]. In addition to a role in oxidative stress, it has been shown that NRF2 is required to allow collective cells migration, a hallmark of hybrid epithelial/mesenchymal phenotype (E/M). The hybrid E/M phenotype, a status characterized by the simultaneous presence of both epithelial and mesenchymal traits at morphological and molecular levels, has been suggested to be potentially more aggressive than a complete EMT, correlating with the acquisition of cancer stem-like traits, enhanced drug resistance and poor prognosis. Given the oscillation of NRF2 upon autophagy as previously discussed [132], and the regulative circuit between ARF and E-cadherin, it would be interesting to analyze how the interplay between these proteins functionally interact with the progression/establishment of the E/M phenotype. In a recent study, it was demonstrated that ARF is able to induce EMT in prostate cancer through the upregulation of metallopeptidase 7 (MMP-7), a secreted member of the peptidase family of matrix metalloproteinase (MMPs) [141,142]. The degradation of the extra cellular matrix (ECM) by matrix metalloproteinases favors cell detachment and promotes cell migration. Interestingly, the MMP-7 protein, shows ARF-dependent nuclear localization in Pten/Trp53 deficient tumors, a feature of an aggressive cancer phenotype [143].
Recent findings in bladder cancer also unveiled a novel role of ARF in drug resistance. The majority of bladder cancers are no–muscle-invasive tumors with favorable survival outcomes. Although ARF expression is known to be elevated in bladder cancer, as a consequence of p53 loss-of-function, the INK4a/ARF locus has been found to be amplified and associated with poor survival in several cases of muscle-invasive bladder cancer (MIBC) [144]. An analysis of The Cancer Genome Atlas [145] showed that, in addition to TP53 mutations, deletion or mutations of the CDKN2A locus has been found in 42% of cases [146]. Interestingly, a substantial percentage (35%) of MIBCs, displays mutant TP53 without corresponding alterations of INK4a/ARF locus. In particular, ARF and not p16 appears significantly overexpressed in MIBC, and mainly localized in the nucleolus of patients-derived bladder cancerous cells [147], a feature that correlates with the tendency to develop chemoresistance. Using a mouse model in which the ARF coding region was conditionally ablated in the bladder tissue of Trp53-/-/Pten-/- mice, the authors showed that Arf wild-type tumors were markedly resistant to treatment with cisplatin compared with the Arf-null counterpart. Similarly, the overexpression of the human ARF protein in human bladder cancer cells and their use in orthotopic assays showed that it promotes cancer formation in vivo.
Additional support to ARF pro-proliferative role is provided by the observation that ARF interacts with the focal adhesion kinase (FAK) [67]. During cytoskeleton remodeling induced by cell spreading, ARF is recruited to cell periphery at points of focal adhesions, where it co-localizes and interacts with the active focal adhesion kinase (pFAKY397). Accordingly, ARF silencing induces cytoskeleton disorganization causing anoikis through a mechanism dependent on the death-associated protein kinase (DAPK). The DAPK is a pro-apoptotic serine/threonine kinase, activated following cytoskeleton–matrix disengagement [148]. The presented data suggest that p14ARF blocks DAPK functions avoiding DAPK-mediated cell death by both p53-dependent and -independent pathways [67].
Collectively this experimental evidence suggests that ARF has pro-survival functions, thus implying that it may function in both tumor suppressive and oncogenic pathways in cancers, depending on the genetic context.
8. ARF and Autophagy
Given the dual role of autophagy in cancer, the involvement of ARF in autophagy could be the key to interpret ARF role in cancer as well. Early evidence of ARF role in autophagy led to the identification of a shorter isoform of the protein translated by an internal methionine residue (Met 45 in mice and Met 48 in humans), which is not conserved in other ARF orthologs. Translation from this residue produces the so-called smARF (short mitochondrial ARF), which lacks the well-conserved N-terminus end, displaying most of the known p53-dependent functions of the protein. In response to viral and cellular oncogenes, smARF is upregulated and localizes to mitochondria, where it is stabilized by the interaction with the mitochondrial protein p32 [149]. Here, smARF induces the dissipation of mitochondrial membrane potential not accompanied by cytochrome c release to the cytoplasm, suggesting that there is no apoptotic cell death. In line with this, overexpression of smARF in 293T cells causes caspase-independent cell death characterized by the accumulation of autophagic vacuoles. Interestingly, the knockdown of autophagy essential genes such as Atg5 or Beclin-1 attenuates cell death in smARF-expressing cells, leading to the hypothesis that autophagy contributed to the observed process [60,150]. However, the smARF localization to mitochondria has been related to its ability to induce a selective form of autophagy called mitophagy, a cellular process that specifically removes damaged or excessive mitochondria [125,137,151,152]. Although mitophagy shares many features with autophagy, the selectivity of the autophagosome for mitochondrial cargo is due to a set of proteins that are activated under specific stimuli. Indeed, smARF recruitment to mitochondria induces membrane depolarization that in turn, provokes the stabilization of the mitochondrial PINK1 kinase and the PINK1-mediated recruitment of the Parkin ubiquitin ligase on the outer membrane of the mitochondrion. Parkin, by ubiquitinating several mitochondrial proteins on the outer membrane, primes the mitochondrion for subsequent degradation through the autophagosome [151]. Further complexity has been added following the observation that smARF can have a negative role on stress-induced mitophagy, a process mediated by the JNK2 kinase [153]. Authors observed that JNK2 induces the ubiquitination and the proteasome-mediated degradation of smARF upon stress. In this condition, basal autophagy (smARF dependent) is impaired and the protein levels of p62 increase. This protein, recruited to the mitochondrial membrane, mediates the formation of the autophagosomal vesicles thus promoting the selective degradation of mitochondria. Interestingly, smARF re-expression in ARF null mice rescues the ocular defects and the reduction of sperm production, thus suggesting that these developmental functions depend on smARF dependent functions [149].
It has been now also extensively shown that full-length ARF proteins (both human and mouse) can promote autophagy in a p53-dependent and independent manner [137]. Mitochondrial localized full-length ARF physically interacts with BCL-xL, a member of the Bcl2 family, that plays an important role in autophagy. The autophagy-inducing activity of ARF was shown to involve the displacement of Beclin-1 from BCL-xL complexes through direct binding of ARF to BCL-xL [124,154,155,156,157,158,159,160,161,162] thus promoting the activation of the key mediator of autophagy, the Beclin-1/Vps34 complex [124,154,155].
Analysis of ARF mutants showed that the ARF C-terminal domain, comprising amino acids 100 to 120, is necessary for autophagy induction by both human and murine protein [125]. Natural melanoma mutations in exon 2 (R98L, L104I and R115G), affecting the coding potential of ARF, but not of p16INK4a, impair ARF ability to induce autophagy [125]. Interestingly, a domain coded by exon 2, within the 100–132 aminoacidic stretch, has been shown to promote both FAK activation and cell survival thus suggesting the speculation that ARF-mediated autophagy could be, at least in part, the mechanism underlining its role in cell spreading [67]. In support of this hypothesis, in cells that detach from the substrate, or in cells with low level of activated FAK, autophagy inhibition induces anoikis [156]. Indeed autophagy, by protecting matrix-detached epithelial cells from anoikis could promote metastatic processes [157]. In support of the cross-talk between cell morphology and autophagy-mediated survival, ATG proteins have been shown to co-localize with FAK at focal adhesions [156]. Moreover, autophagy regulates cell spreading, especially cell protrusions extensions [158], and is required for focal adhesion turnover and cell migration [159] further supporting the view that ARF could promote cell spreading through its ability to induce autophagy. It should be underlined, that while the expression of the N-terminal ARF domain is impaired in mediating cell spreading, the C-terminal region, in which the autophagy-promoting domain has been mapped, completely rescues the morphology defect of ARF silenced HeLa cells [67]. Further experiments will be necessary to validate this hypothesis and clarify the possible involvement of autophagy in this ARF function and/or in tumor progression. A summary of ARF dependent functions is provided in Table 1.
9. Conclusions
Mammalian cells sustaining oncogenic insults invoke defensive programs resulting either in cell growth arrest or apoptosis. In non-pathological conditions, ARF protein level is almost undetectable within the cell. Upon oncogenic stimuli or developmental cues, its levels increase, eliciting p53-dependent or independent pathways to restrain cell proliferation. ARF protein interacts with a cohort of different partners involved in different cellular processes.
Recent experimental evidence, also pursued by the observation of high ARF protein levels in specific tumors, suggested that this protein might have pro-survival functions. The possibility that ARF might promote or improve the survival of a subset of tumors has been at least in part correlated with ARFs ability to induce autophagy. Experimental data suggest that within a tumor cell, ARF protein can be stabilized by decreased proteasome-mediated degradation [101] or by protein kinase C (PKC) activation [160]. In this latter case, ARF phosphorylation mediates its accumulation in the cytoplasm where it cannot efficiently exert its classical role in the control cell proliferation [160,161]. Although this can be a way to escape ARF surveillance in tumorigenesis, it might likewise mean that phosphorylation can confer pro-survival properties to the protein favoring cancer progression. In line with this last hypothesis, the over-expression of an ARF mutant mimicking the phosphorylated status of the protein promotes proliferation in HeLa cells, induces FAK stabilization and allows cell spreading [162]. Adhesion/spreading interplay has a well-documented role not only in malignancy but also in physiological processes such as embryogenesis [113]. Interestingly ARF involvement in cytoskeleton organization relies on an evolutionarily conserved mechanism, as p19ARF is able to rescue spread morphology defects caused by p14ARF silencing. The ARF exon 2-encoded region (aa 65–132) which, compared to the exon 1β shows a stronger degree of identity between human and its mouse homologue, is required for FAK binding, activation and cell spreading. These observations add a crucial element in the understanding of the functions of the exon 2 encoded domain, in which the bulk of ARF mutations in human cancers resides. The data presented thus implies that the cellular environment, both within and outside the cell, can dictate the function of ARF as a tumor suppressor or as a pro-metastatic player in cancer. Unravelling the molecular mechanisms underlying ARF duality in cancer progression will be of great interest for the future design of cancer therapy.
Author Contributions
M.V. wrote the paper with the help of R.F., M.R. All the authors revised the manuscript.
Funding
This research received funding from Grant GLAMANTIA 2018 assigned to GLM from University of Naples Federico II.
Acknowledgments
We thank Samuel John Taylor for the editing support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Di Tommaso, A.; Hagen, J.; Tompkins, V.; Muniz, V.; Dudakovic, A.; Kitzis, A.; Ladeveze, V.; Quelle, D.E. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Exp. Cell Res. 2009, 315, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Soussi, T.; Shomer, B.; Hollstein, M.; Greenblatt, M.; Hovig, E.; Harris, C.C.; Montesano, R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997, 25, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and CDK inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2a locus, p14(Arf), participates in a regulatory feedback loop with p53 and mdm2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase CDK6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R.A. The INK4a/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the ink4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [PubMed]
- Sherr, C.J. Divorcing arf and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The Arf tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- De Stanchina, E.; McCurrach, M.E.; Zindy, F.; Shieh, S.Y.; Ferbeyre, G.; Samuelson, A.V.; Prives, C.; Roussel, M.F.; Sherr, C.J.; Lowe, S.W. E1a signaling to p53 involves the p19(Arf) tumor suppressor. Genes Dev. 1998, 12, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.P.; Barnes, J.M.; Paliwal, S.; Zhang, X.; Tang, X.; Chen, S.; Zamba, K.D.; Cullen, J.J.; Meyerholz, D.K.; Meyers, S.; et al. The ARF tumor suppressor inhibits tumor cell colonization independent of p53 in a novel mouse model of pancreatic ductal adenocarcinoma metastasis. Mol. Cancer Res. 2011, 9, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ishikawa, T.; Sugihara, E.; Kuninaka, S.; Miyamoto, T.; Mabuchi, Y.; Matsuzaki, Y.; Tsunoda, T.; Miya, F.; Morioka, H.; et al. c-Myc overexpression with loss of INK4a/ARF transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene 2010, 29, 5687–5699. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Bodner, S.; van de Kamp, E.; Randle, D.H.; Sherr, C.J. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999, 59, 2217–2222. [Google Scholar] [PubMed]
- Del Arroyo, A.G.; Peters, G. The INK4a/ARF network—Cell cycle checkpoint or emergency brake? Adv. Exp. Med. Biol. 2005, 570, 227–247. [Google Scholar] [PubMed]
- Rizos, H.; Darmanian, A.P.; Holland, E.A.; Mann, G.J.; Kefford, R.F. Mutations in the INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J. Biol. Chem. 2001, 276, 41424–41434. [Google Scholar] [CrossRef] [PubMed]
- Gardie, B.; Cayuela, J.M.; Martini, S.; Sigaux, F. Genomic alterations of the p19ARF encoding exons in T-cell acute lymphoblastic leukemia. Blood 1998, 91, 1016–1020. [Google Scholar] [PubMed]
- Rutter, J.L.; Goldstein, A.M.; Davila, M.R.; Tucker, M.A.; Struewing, J.P. CDKN2a point mutations d153spl(c.457g > t) and ivs 2 + 1g > t result in aberrant splice products affecting both p16ink4a and p14ARF. Oncogene 2003, 22, 4444–4448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Maggi, L.B., Jr.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar] [PubMed]
- Tsujimoto, H.; Hagiwara, A.; Sugihara, H.; Hattori, T.; Yamagishi, H. Promoter methylations of p16INK4a and p14Arf genes in early and advanced gastric cancer. Correlations of the modes of their occurrence with histologic type. Pathol. Res. Pract. 2002, 198, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Konishi, N.; Nakamura, M.; Kishi, M.; Nishimine, M.; Ishida, E.; Shimada, K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am. J. Pathol. 2002, 160, 1207–1214. [Google Scholar] [CrossRef]
- Hewitt, C.; Lee Wu, C.; Evans, G.; Howell, A.; Elles, R.G.; Jordan, R.; Sloan, P.; Read, A.P.; Thakker, N. Germline mutation of ARF in a melanoma kindred. Hum. Mol. Genet. 2002, 11, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Puig, S.; Badenas, C.; Malvehy, J.; Darmanian, A.P.; Jimenez, L.; Mila, M.; Kefford, R.F. A melanoma-associated germline mutation in exon 1β inactivates p14ARF. Oncogene 2001, 20, 5543–5547. [Google Scholar] [CrossRef] [PubMed]
- Randerson-Moor, J.A.; Harland, M.; Williams, S.; Cuthbert-Heavens, D.; Sheridan, E.; Aveyard, J.; Sibley, K.; Whitaker, L.; Knowles, M.; Bishop, J.N.; et al. A germline deletion of p14ARF but not cdkn2a in a melanoma-neural system tumour syndrome family. Hum. Mol. Genet. 2001, 10, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sakaki, T.; Hashimoto, H.; Nakase, H.; Ishida, E.; Shimada, K.; Konishi, N. Frequent alterations of the p14arf and p16ink4a genes in primary central nervous system lymphomas. Cancer Res. 2001, 61, 6335–6339. [Google Scholar] [PubMed]
- Silva, J.; Dominguez, G.; Silva, J.M.; Garcia, J.M.; Gallego, I.; Corbacho, C.; Provencio, M.; Espana, P.; Bonilla, F. Analysis of genetic and epigenetic processes that influence p14ARF expression in breast cancer. Oncogene 2001, 20, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Vonlanthen, S.; Heighway, J.; Tschan, M.P.; Borner, M.M.; Altermatt, H.J.; Kappeler, A.; Tobler, A.; Fey, M.F.; Thatcher, N.; Yarbrough, W.G.; et al. Expression of p16ink4a/p16alpha and p19ARF/p16β is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene 1998, 17, 2779–2785. [Google Scholar] [CrossRef] [PubMed]
- Palmero, I.; Pantoja, C.; Serrano, M. P19arf links the tumour suppressor p53 to Ras. Nature 1998, 395, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, E.; Lamed, Y.; Ginsberg, D. E2F and Ras synergize in transcriptionally activating p14arf expression. Cell Cycle 2003, 2, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Parisi, T.; Pollice, A.; Di Cristofano, A.; Calabro, V.; La Mantia, G. Transcriptional regulation of the human tumor suppressor p14ARF by e2f1, e2f2, e2f3, and sp1-like factors. Biochem. Biophys. Res. Commun. 2002, 291, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. MDM2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase e3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by MDM2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Tsai, K.Y.; Jacks, T. An intact HDM2 ring-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2000, 2, 563–568. [Google Scholar] [CrossRef] [PubMed]
- MarikoOkada-Hatakeyama, S.M.K.I. Current status of mathematical modeling of cancer—From the viewpoint of cancer hallmarks. Curr. Opin. Syst. Biol. 2017, 2, 39–48. [Google Scholar]
- Chen, J.; Wu, X.; Lin, J.; Levine, A.J. MDM-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol. Cell. Biol. 1996, 16, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The INK4a tumor suppressor gene product, p19ARf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and MDM2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Levine, A.J. P19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of MDM2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell 1999, 3, 579–591. [Google Scholar] [CrossRef]
- Rizos, H.; Darmanian, A.P.; Mann, G.J.; Kefford, R.F. Two arginine rich domains in the p14ARF tumour suppressor mediate nucleolar localization. Oncogene 2000, 19, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Ashcroft, M.; Kubbutat, M.H.; Vousden, K.H. Contribution of two independent MDM2-binding domains in p14ARF to p53 stabilization. Curr. Biol. 2000, 10, 539–542. [Google Scholar] [CrossRef]
- Weber, J.D.; Kuo, M.L.; Bothner, B.; DiGiammarino, E.L.; Kriwacki, R.W.; Roussel, M.F.; Sherr, C.J. Cooperative signals governing ARF-MDM2 interaction and nucleolar localization of the complex. Mol. Cell. Biol. 2000, 20, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar ARF sequesters MDM2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19ARF with MDM2 inhibits ubiquitin ligase activity of MDM2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.A.; Desterro, J.M.; Saville, M.K.; Howard, S.; Sparks, A.; Hay, R.T.; Lane, D.P. An N-terminal p14ARF peptide blocks MDM2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 2000, 19, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Llanos, S.; Peters, G. Multiple interacting domains contribute to p14ARF mediated inhibition of MDM2. Oncogene 2002, 21, 4498–4507. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, C.; Zhao, L.; Modestou, M.; Quelle, D.E. ARF function does not require p53 stabilization or MDM2 relocalization. Mol. Cell. Biol. 2002, 22, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. P53-independent functions of the p19ARF tumor suppressor. Genes Dev. 2000, 14, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Leduc, C.; Coll, J.L.; Brambilla, E.; Gazzeri, S. P14arf induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Vivo, M.; La Mantia, G. The promiscuity of ARF interactions with the proteasome. FEBS Lett. 2008, 582, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.; Bartkova, J.; Mistrik, M.; Hall, A.; Lange, M.K.; Ralfkiaer, U.; Bartek, J.; Guldberg, P. A short acidic motif in ARF guards against mitochondrial dysfunction and melanoma susceptibility. Nat. Commun. 2014, 5, 5348. [Google Scholar] [CrossRef] [PubMed]
- Britigan, E.M.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol. Biol. Cell 2014, 25, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, V.S.; Hagen, J.; Frazier, A.A.; Lushnikova, T.; Fitzgerald, M.P.; di Tommaso, A.; Ladeveze, V.; Domann, F.E.; Eischen, C.M.; Quelle, D.E. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J. Biol. Chem. 2007, 282, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; den Besten, W.; Thomas, M.C.; Sherr, C.J. ARF-induced turnover of the nucleolar nucleophosmin-associated sumo-2/3 protease senp3. Cell Cycle 2008, 7, 3378–3387. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the ARF tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Chisholm, J.; Desterro, J.M.; Lane, D.P.; Hay, R.T. P14ARF promotes accumulation of sumo-1 conjugated (h)MDM2. FEBS Lett. 2002, 528, 207–211. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La Mantia, G. P14arf interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed]
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the ARF tumor suppressor protein with nucleophosmin/b23. Mol. Cell. Biol. 2004, 24, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Bertwistle, D.; Den Besten, W.; Kuo, M.L.; Sugimoto, M.; Tago, K.; Williams, R.T.; Zindy, F.; Roussel, M.F. p53-dependent and -independent functions of the ARF tumor suppressor. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Dwertmann, A.; Herold, S.; Abed, M.; Naud, J.F.; Finkernagel, F.; Harms, G.S.; Orian, A.; Wanzel, M.; Eilers, M. The ARF tumor suppressor protein inhibits miz1 to suppress cell adhesion and induce apoptosis. J. Cell Biol. 2010, 188, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses sumo modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Santockyte, R.; Shen, R.F.; Tekle, E.; Wang, G.; Yang, D.C.; Chock, P.B. Expression of sumo-2/3 induced senescence through p53- and pRb-mediated pathways. J. Biol. Chem. 2006, 281, 36221–36227. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J. MDM2-ARF complex regulates p53 sumoylation. Oncogene 2003, 22, 5348–5357. [Google Scholar] [CrossRef] [PubMed]
- Damalas, A.; Velimezi, G.; Kalaitzakis, A.; Liontos, M.; Papavassiliou, A.G.; Gorgoulis, V.; Angelidis, C. Loss of p14ARF confers resistance to heat shock- and oxidative stress-mediated cell death by upregulating beta-catenin. Int. J. Cancer 2011, 128, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Fatyol, K.; Szalay, A.A. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1α (hif-1α) and inhibits hif-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Festa, L.; Duverger, O.; Vivo, M.; Guerrini, L.; La Mantia, G.; Morasso, M.I.; Calabro, V. Homeodomain protein dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle 2009, 8, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Festa, L.; Roscigno, G.; Vivo, M.; Pollice, A.; Morasso, M.; La Mantia, G.; Calabro, V. A dominant mutation etiologic for human tricho-dento-osseous syndrome impairs the ability of DLX3 to downregulate ΔNp63α. J. Cell. Physiol. 2011, 226, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Calabro, V.; Mansueto, G.; Santoro, R.; Gentilella, A.; Pollice, A.; Ghioni, P.; Guerrini, L.; La Mantia, G. Inhibition of p63 transcriptional activity by p14ÂRF: Functional and physical link between human ARF tumor suppressor and a member of the p53 family. Mol. Cell. Biol. 2004, 24, 8529–8540. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Di Costanzo, A.; Fortugno, P.; Pollice, A.; Calabro, V.; La Mantia, G. Downregulation of ΔNp63α in keratinocytes by p14ARF-mediated sumo-conjugation and degradation. Cell Cycle 2009, 8, 3545–3551. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Vivo, M.; De Simone, M.; Guerrini, L.; Pollice, A.; La Mantia, G.; Calabro, V. Sumoylation and ubiquitylation crosstalk in the control of Δnp63α protein stability. Gene 2018, 645, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Guarino, A.M.; Troiano, A.; Pizzo, E.; Bosso, A.; Vivo, M.; Pinto, G.; Amoresano, A.; Pollice, A.; La Mantia, G.; Calabro, V. Oxidative stress causes enhanced secretion of YB-1 protein that restrains proliferation of receiving cells. Genes 2018, 9, 513. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, O.; Troiano, A.; Guarino, A.M.; Pollice, A.; Vivo, M.; La Mantia, G.; Calabro, V. Δnp63α controls YB-1 protein stability: Evidence on YB-1 as a new player in keratinocyte differentiation. Genes Cells 2016, 21, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Troiano, A.; Lomoriello, I.S.; di Martino, O.; Fusco, S.; Pollice, A.; Vivo, M.; La Mantia, G.; Calabro, V. Y-box binding protein-1 is part of a complex molecular network linking Δnp63α to the PI3K/AKT pathway in cutaneous squamous cell carcinoma. J. Cell. Physiol. 2015, 230, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Apicelli, A.J.; Maggi, L.B., Jr.; Hirbe, A.C.; Miceli, A.P.; Olanich, M.E.; Schulte-Winkeler, C.L.; Saporita, A.J.; Kuchenreuther, M.; Sanchez, J.; Weilbaecher, K.; et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol. Cell. Biol. 2008, 28, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Saporita, A.J.; Maggi, L.B., Jr.; Apicelli, A.J.; Weber, J.D. Therapeutic targets in the ARF tumor suppressor pathway. Curr. Med. Chem. 2007, 14, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar ARF tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Ayrault, O.; Andrique, L.; Larsen, C.J.; Seite, P. Human ARF tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene 2004, 23, 8097–8104. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the ARF tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Rodway, H.; Llanos, S.; Rowe, J.; Peters, G. Stability of nucleolar versus non-nucleolar forms of human p14ARF. Oncogene 2004, 23, 6186–6192. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor suppressor ARF degrades b23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (b23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Bonetti, P.; Lazzerini Denchi, E.; Martinelli, P.; Zamponi, R.; Marine, J.C.; Helin, K.; Falini, B.; Pelicci, P.G. Nucleophosmin is required for DNA integrity and p19ARF protein stability. Mol. Cell. Biol. 2005, 25, 8874–8886. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Grisendi, S.; Clohessy, J.G.; Majid, S.; Bernardi, R.; Sportoletti, P.; Pandolfi, P.P. The leukemia-associated cytoplasmic nucleophosmin mutant is an oncogene with paradoxical functions: ARF inactivation and induction of cellular senescence. Oncogene 2007, 26, 7391–7400. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Chiarella, S.; Franceschini, M.; Di Matteo, A.; Brunori, M.; Banci, L.; Federici, L. Identification of a novel nucleophosmin-interaction motif in the tumor suppressor p14ARF. FEBS J. 2018, 285, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Martinelli, P.; Zamponi, R.; Shing, D.C.; Bonetti, P.; Luzi, L.; Volorio, S.; Bernard, L.; Pruneri, G.; Alcalay, M.; et al. Delocalization and destabilization of the ARF tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006, 66, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, W.; Kuo, M.L.; Tago, K.; Williams, R.T.; Sherr, C.J. Ubiquitination of, and sumoylation by, the ARF tumor suppressor. ISR Med. Assoc. J. 2006, 8, 249–251. [Google Scholar] [PubMed]
- Kuo, M.L.; den Besten, W.; Sherr, C.J. N-terminal polyubiquitination of the ARF tumor suppressor, a natural lysine-less protein. Cell Cycle 2004, 3, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Nasti, V.; Ronca, R.; Vivo, M.; Lo Iacono, M.; Calogero, R.; Calabro, V.; La Mantia, G. Functional and physical interaction of the human ARF tumor suppressor with TAT-binding protein-1. J. Biol. Chem. 2004, 279, 6345–6353. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K.; Bengal, E.; Ziv, T.; Admon, A.; Ciechanover, A. A novel site for ubiquitination: The N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998, 17, 5964–5973. [Google Scholar] [CrossRef] [PubMed]
- Sheaff, R.J.; Singer, J.D.; Swanger, J.; Smitherman, M.; Roberts, J.M.; Clurman, B.E. Proteasomal turnover of p21cip1 does not require p21cip1 ubiquitination. Mol. Cell 2000, 5, 403–410. [Google Scholar] [CrossRef]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The trip from ULF to ARF. Cancer Cell 2010, 17, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Sepe, M.; Villella, V.R.; Tolino, F.; Vivo, M.; Calabro, V.; La Mantia, G. Tbp-1 protects the human oncosuppressor p14ARF from proteasomal degradation. Oncogene 2007, 26, 5154–5162. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGγ proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the INK4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16INK4a and p19ARF senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. INK4-ARF locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Williams, R.T.; Baudino, T.A.; Rehg, J.E.; Skapek, S.X.; Cleveland, J.L.; Roussel, M.F.; Sherr, C.J. ARF tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 15930–15935. [Google Scholar] [CrossRef] [PubMed]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.L.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of PDGF signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the ARF tumor suppressor during male germ cell and eye development in ARF-CRE reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Mei, J.; Liu, J.; Skapek, S.X. Mir-34a is essential for p19ARF-driven cell cycle arrest. Cell Cycle 2014, 13, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Finkelstein, D.; Sherr, C.J. ARF tumor suppressor and Mir-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1112–E1121. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Roig, I.; Jasin, M.; Keeney, S.; Sherr, C.J. Expression of ARF tumor suppressor in spermatogonia facilitates meiotic progression in male germ cells. PLoS Genet. 2011, 7, e1002157. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Kotsinas, A.; Papanagnou, P.; Evangelou, K.; Trigas, G.C.; Kostourou, V.; Townsend, P.; Gorgoulis, V.G. ARF: A versatile DNA damage response ally at the crossroads of development and tumorigenesis. Front. Genet. 2014, 5, 236. [Google Scholar] [CrossRef] [PubMed]
- Bieging-Rolett, K.T.; Johnson, T.M.; Brady, C.A.; Beaudry, V.G.; Pathak, N.; Han, S.; Attardi, L.D. p19ARF is required for the cellular response to chronic DNA damage. Oncogene 2016, 35, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Deyrieux, A.F.; Rosas-Acosta, G.; Ozbun, M.A.; Wilson, V.G. Sumoylation dynamics during keratinocyte differentiation. J. Cell Sci. 2007, 120, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Cho, M.S.; Gi, Y.J.; Ayanga, B.A.; Sherr, C.J.; Flores, E.R. Rescue of key features of the p63-null epithelial phenotype by inactivation of INK4a and ARF. EMBO J. 2009, 28, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aguilera, A.; Sanchez-Beato, M.; Garcia, J.F.; Prieto, I.; Pollan, M.; Piris, M.A. p14ARF nuclear overexpression in aggressive B-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Park, J.Y.; Kang, H.J.; Cho, H.C. Overexpression of p16INK4a and p14ARF in haematological malignancies. Clin. Lab. Haematol. 2003, 25, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of CDKN2a α (p16INK4a) and β (p14ARF) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Humbey, O.; Pimkina, J.; Zilfou, J.T.; Jarnik, M.; Dominguez-Brauer, C.; Burgess, D.J.; Eischen, C.M.; Murphy, M.E. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008, 68, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Vivo, M. Dynamics of p14ARF and focal adhesion kinase-mediated autophagy in cancer. Cancers 2018, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14ARF tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kimmelman, A.C. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, M.; Burigo, L.; Durante, M.; Herrlitz, M.; Kramer, M.; Mishustin, I.; Muller, I.; Natale, F.; Pshenichnov, I.; Schramm, S.; et al. Nanolesions induced by heavy ions in human tissues: Experimental and theoretical studies. Beilstein J. Nanotechnol. 2012, 3, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. NRF2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Horl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Scholl, A.; Rapp, A.; Yu, W.; Rausch, C.; Cardoso, M.C. DNA replication and repair kinetics of ALU, LINE1 and satellite III genomic repetitive elements. Epigenet. Chromatin 2018, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Gu, W. p53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Carracedo, A.; Lin, H.K.; Koutcher, J.A.; Behrendt, N.; Egia, A.; Alimonti, A.; Carver, B.S.; Gerald, W.; Teruya-Feldstein, J.; et al. Differential p53-independent outcomes of p19ARF loss in oncogenesis. Sci. Signal. 2009, 2, ra44. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19ARF in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Federico Bocci, S.C.T.; Mercedes, S.V.; George, J.T.; Casabar, J.; Wong, P.K.; Hanash, S.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Nrf2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. bioRxiv 2018. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. MMP7 interacts with ARF in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Wang, S.C.; Morris, J.P.T.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.C.; Cheung, S.T.; Fan, S.T. Atypical localization of membrane type 1-matrix metalloproteinase in the nucleus is associated with aggressive features of hepatocellular carcinoma. Mol. Carcinog. 2007, 46, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Berggren, P.; Kumar, R.; Sakano, S.; Hemminki, L.; Wada, T.; Steineck, G.; Adolfsson, J.; Larsson, P.; Norming, U.; Wijkstrom, H.; et al. Detecting homozygous deletions in the cdkn2a(p16(ink4a))/arf(p14(arf)) gene in urinary bladder cancer using real-time quantitative PCR. Clin. Cancer Res. 2003, 9, 235–242. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, J.P.; Cha, E.K.; Iyer, G.; Scott, S.N.; Zabor, E.C.; Shah, R.H.; Ren, Q.; Bagrodia, A.; Kim, P.H.; Hakimi, A.A.; et al. Genomic characterization of upper tract urothelial carcinoma. Eur. Urol. 2015, 68, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, T.B.; Kobayashi, T.; Ramirez, R.; Rong, L.; Puzio-Kuter, A.M.; Iyer, G.; Teo, M.Y.; Sanchez-Vega, F.; Wang, J.; Schultz, N.; et al. ARF confers a context-dependent response to chemotherapy in muscle-invasive bladder cancer. Cancer Res. 2017, 77, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.C.; Wang, W.J.; Yao, C.C.; Wu, P.R.; Chen, R.H. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J. Cell Biol. 2006, 172, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Kimchi, A. A smarf way to die: A novel short isoform of p19ARF is linked to autophagic cell death. Autophagy 2006, 2, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; Kontogiannea, M.; Fon, E.A. Short mitochondrial ARF triggers parkin/pink1-dependent mitophagy. J. Biol. Chem. 2014, 289, 29519–29530. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase JNK2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor arf for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.S.B.; McEwan, D.G.; Morton, J.P.; Macagno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; Sansom, O.J.; et al. Autophagic targeting of SRC promotes cancer cell survival following reduced fak signalling. Nat. Cell Biol. 2011, 14, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kadandale, P.; Kiger, A.A. Role of selective autophagy in cellular remodeling: “Self-eating” into shape. Autophagy 2010, 6, 1194–1195. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Novel insights into how autophagy regulates tumor cell motility. Autophagy 2016, 12, 1679–1680. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Ranieri, M.; Sansone, F.; Santoriello, C.; Calogero, R.A.; Calabro, V.; Pollice, A.; La Mantia, G. Mimicking p14ARF phosphorylation influences its ability to restrain cell proliferation. PLoS ONE 2013, 8, e53631. [Google Scholar] [CrossRef]
- Vivo, M.; Matarese, M.; Sepe, M.; Di Martino, R.; Festa, L.; Calabro, V.; La Mantia, G.; Pollice, A. Mdm2-mediated degradation of p14arf: A novel mechanism to control ARF levels in cancer cells. PLoS ONE 2015, 10, e0117252. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Guidone, D.; Sangermano, F.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. PKC dependent p14ARF phosphorylation on threonine 8 drives cell proliferation. Sci. Rep. 2018, 8, 7056. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genomic structure of the CDKN2a locus and produced transcripts. (a) Arrows above each exon 1 indicate promoters, continuous and dashed lines above and below the genomic structure indicate p16 and ARF splicing patterns respectively. Transcription of exon 1β, and its splicing to exons 2 and 3 results in the α-transcript, encoding p16INK4a, whereas transcription starting upstream of exon 1β produces the β-transcript in which the exon1β, and the common exons 2 and 3 encode ARF (p14ARF in human, p19Arf in mouse). In yellow and in red are indicated the open reading frames (ORFs) of p16 and ARF respectively, with exon 2 displaying two overlapped ORFs. White boxes represent untranslated regions at the 3’ and 5’ ends while asterisks (*) indicate stop codons (b) Pathways regulated by the two proteins: while p14ARF inhibits Mdm2 (Mouse Double Minute-2) functions with consequential p53 stabilization [4,5], p16INK4a inhibits the cyclinD-CDK4/6 complex thus maintaining the retinoblastoma protein pRb in its growth-suppressive mode [4].
Figure 1.
Genomic structure of the CDKN2a locus and produced transcripts. (a) Arrows above each exon 1 indicate promoters, continuous and dashed lines above and below the genomic structure indicate p16 and ARF splicing patterns respectively. Transcription of exon 1β, and its splicing to exons 2 and 3 results in the α-transcript, encoding p16INK4a, whereas transcription starting upstream of exon 1β produces the β-transcript in which the exon1β, and the common exons 2 and 3 encode ARF (p14ARF in human, p19Arf in mouse). In yellow and in red are indicated the open reading frames (ORFs) of p16 and ARF respectively, with exon 2 displaying two overlapped ORFs. White boxes represent untranslated regions at the 3’ and 5’ ends while asterisks (*) indicate stop codons (b) Pathways regulated by the two proteins: while p14ARF inhibits Mdm2 (Mouse Double Minute-2) functions with consequential p53 stabilization [4,5], p16INK4a inhibits the cyclinD-CDK4/6 complex thus maintaining the retinoblastoma protein pRb in its growth-suppressive mode [4].

Figure 2.
ARF involvement in tumor suppression relies on p53-dependent and independent pathways. Upon over-expression of oncogenes (such as E2F, Ras, E1A and Myc), an increase of ARF intracellular levels are promptly observed (ARF checkpoint) in the cell. By inhibiting MDM2 functions, ARF interferes with the p53/MDM2 circuit (highlighted in grey), leading to p53 stabilization followed by cell cycle arrest and/or apoptosis [10,32,33,34,35,39]. ARF is able to block cell growth also in a p53-independent manner, through the functional interaction with several molecular players as indicated.
Figure 2.
ARF involvement in tumor suppression relies on p53-dependent and independent pathways. Upon over-expression of oncogenes (such as E2F, Ras, E1A and Myc), an increase of ARF intracellular levels are promptly observed (ARF checkpoint) in the cell. By inhibiting MDM2 functions, ARF interferes with the p53/MDM2 circuit (highlighted in grey), leading to p53 stabilization followed by cell cycle arrest and/or apoptosis [10,32,33,34,35,39]. ARF is able to block cell growth also in a p53-independent manner, through the functional interaction with several molecular players as indicated.

Figure 3.
Synopsis of the effect of ARF in cellular processes. Note that the ARF negative effect on cell growth includes its ability to block cell proliferation and to induce apoptosis or senescence.
Figure 3.
Synopsis of the effect of ARF in cellular processes. Note that the ARF negative effect on cell growth includes its ability to block cell proliferation and to induce apoptosis or senescence.


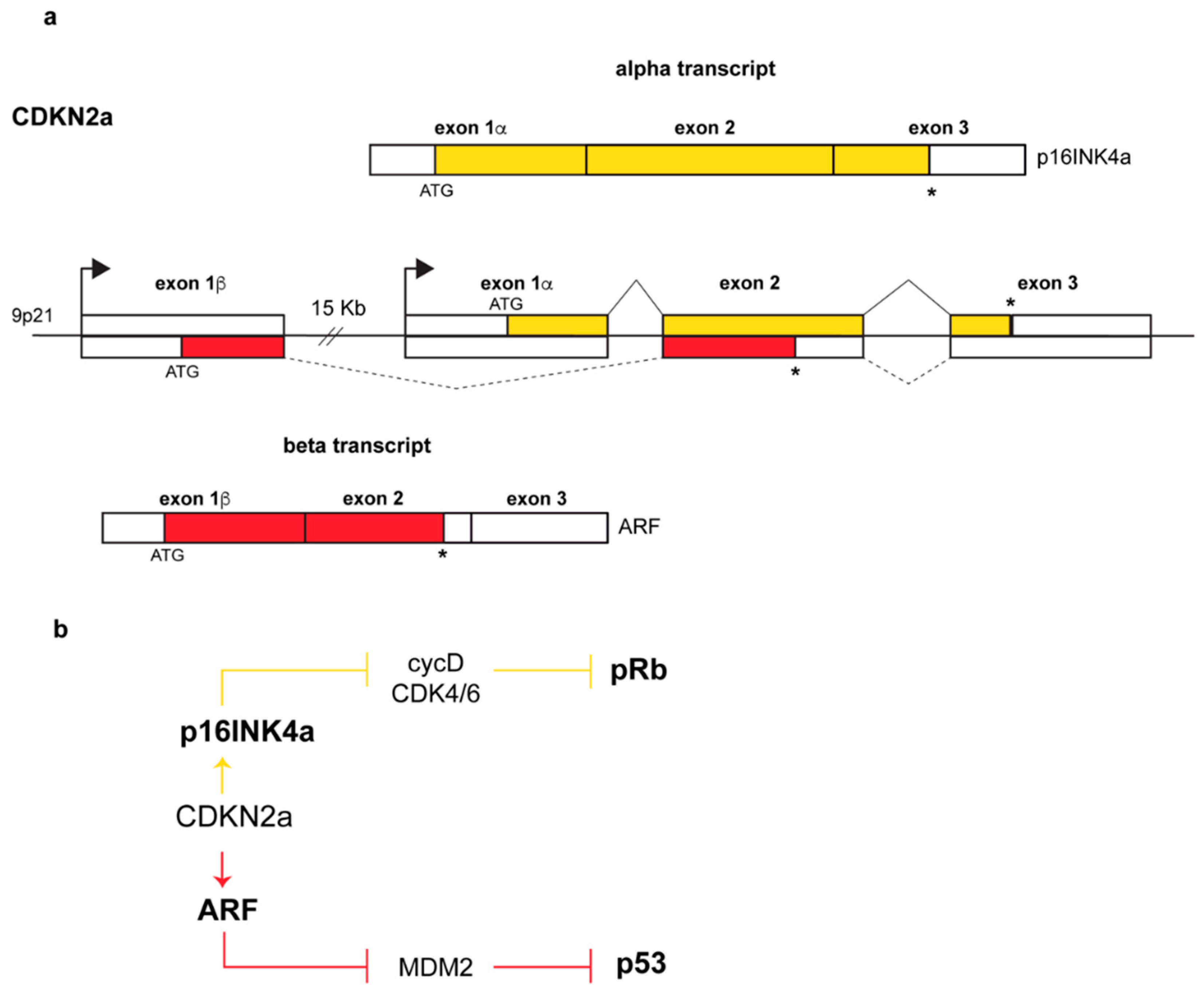{kind=link}
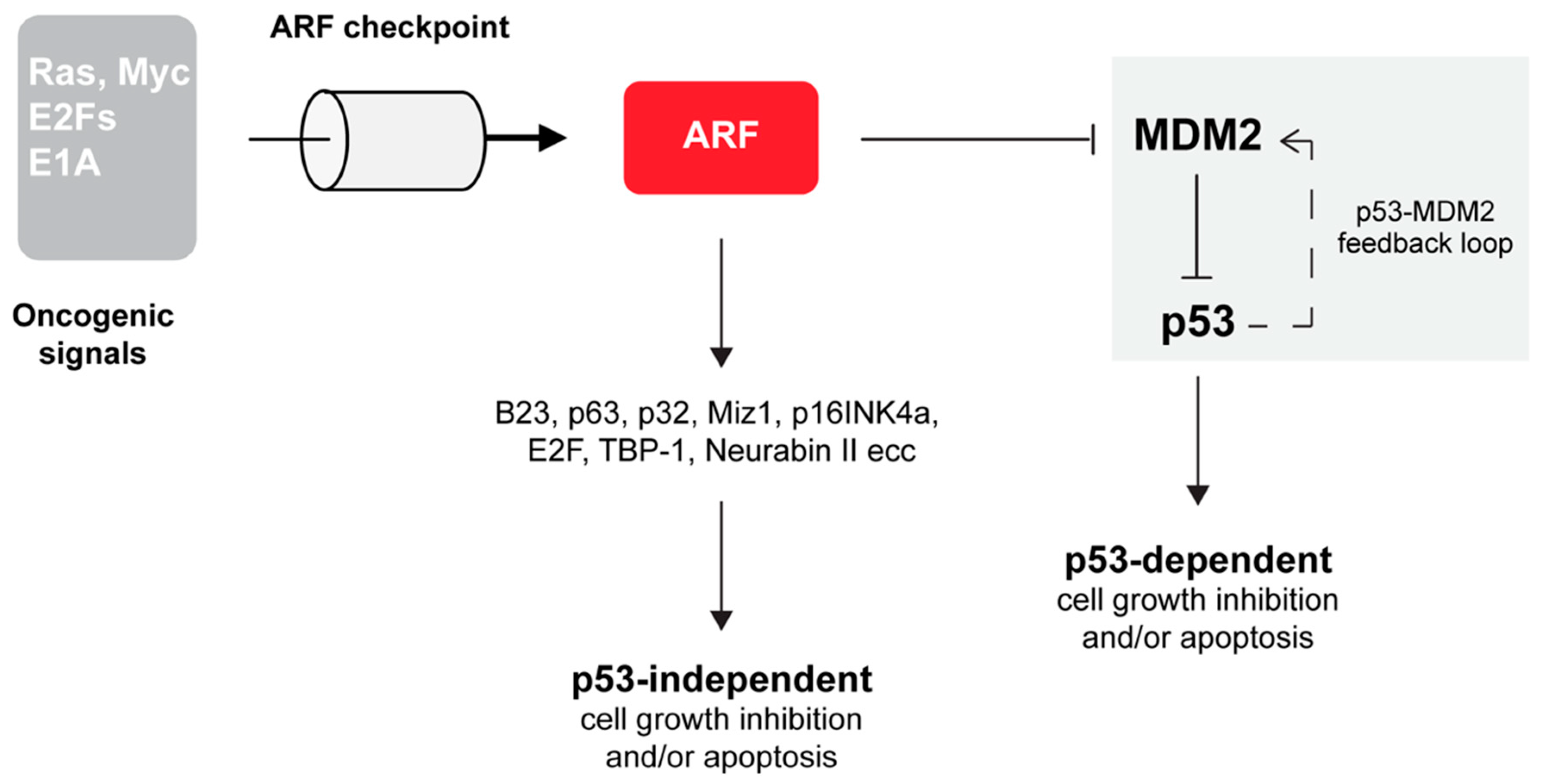{kind=link}
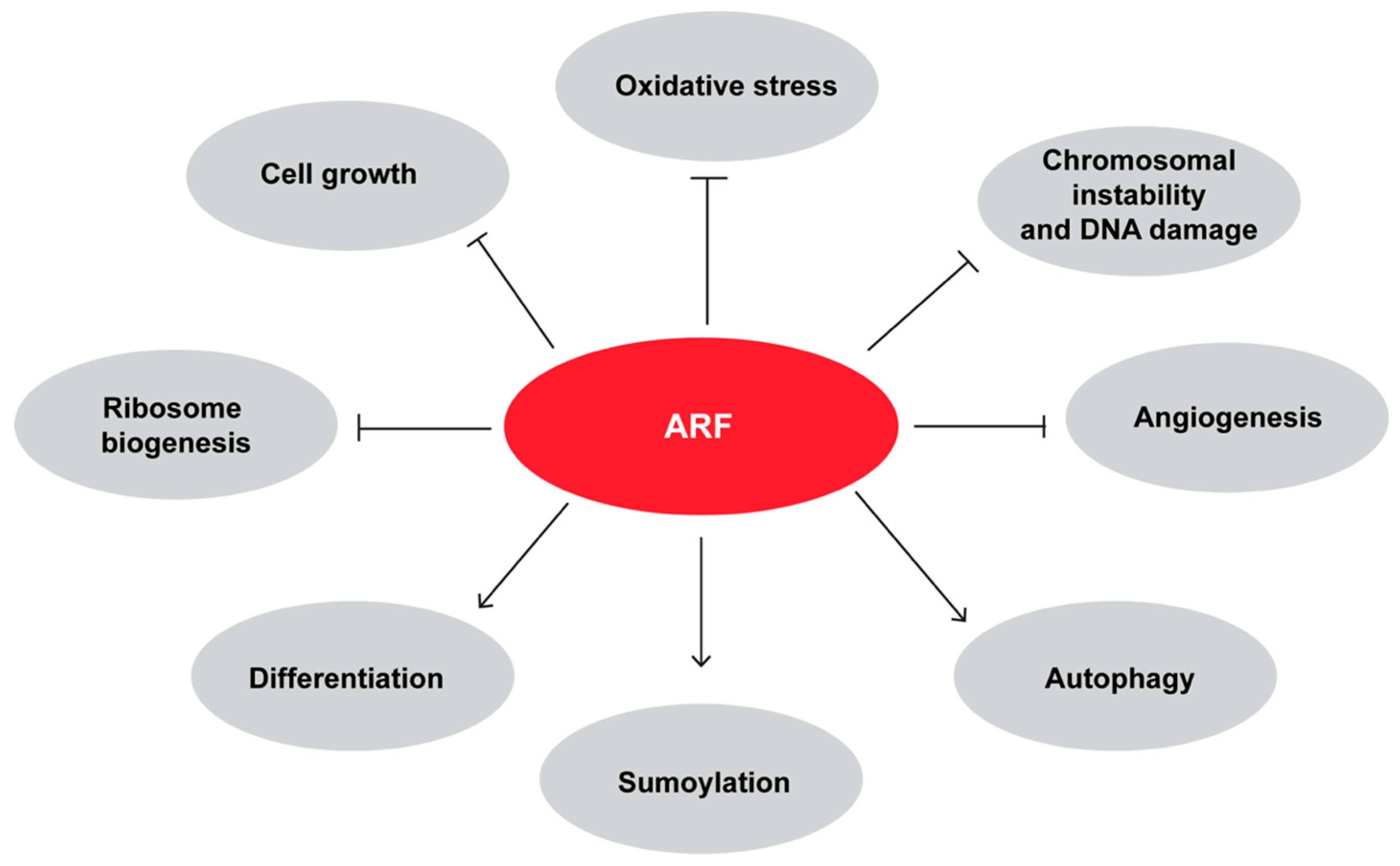{kind=link}
Table 1.
ARF roles in cell proliferation and molecular mechanisms of the observed function.
| Functions | Mechanisms |
|---|---|
| Tumor Suppression | |
| Growth control | p53 dependent and independent call cycle arrest and apoptosis. ΔNp63 inhibition [4,5,11,12,13,31,32,33,34,42,43,55,79,80] |
| Chromosomal stability | Stabilization of miotic spindle, prevents aneuploidy [62] |
| Ribosomal biogenesis | Inhibition or rRNA processing and transcription [84,85,86,87] |
| Oxidative stress | Cellular protection from dysfunctional mitochondria [61] |
| DNA damage | Activation of ATM/ATR/CHK pathway, p53-dependent pathways of DNA repair [115,116] |
| Contest Dependent | |
| Autophagy | Beclin-1 activation, dissipation of mitochondrion potential [60,123,124,125,126,127] |
| Differentiation | Inhibits angiogenesis in developing eye, protects from apoptosis in spermatogonia, allows extraembryonic endoderm migration [79,110,111,112,113,114,115,116,117] |
| Tumor Promoter | |
| Epithelial to mesechimal transition | Slug stabilization [139] |
| Modulation of tumor microenvironment | Metallopeptidase-1 interaction [141] |
| Survival of lymphoma, protease and bladder tumor cells | Promotes prostate cancer in Pten mouse model and autophagy in lymphomas; promotes chemoresistance in bladder cancer [120,121,122,123,124,125,126,127,138,141,147] |
| Anoikis protection, increased proliferation | Inhibition of DAPK mediated cell death; activation focal adhesion kinase [67,160,161,162] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9030087
AMA Style
Fontana R, Ranieri M, La Mantia G, Vivo M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules. 2019; 9(3):87. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9030087
Chicago/Turabian StyleFontana, Rosa, Michela Ranieri, Girolama La Mantia, and Maria Vivo. 2019. "Dual Role of the Alternative Reading Frame ARF Protein in Cancer" Biomolecules 9, no. 3: 87. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9030087
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.