Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor


Abstract
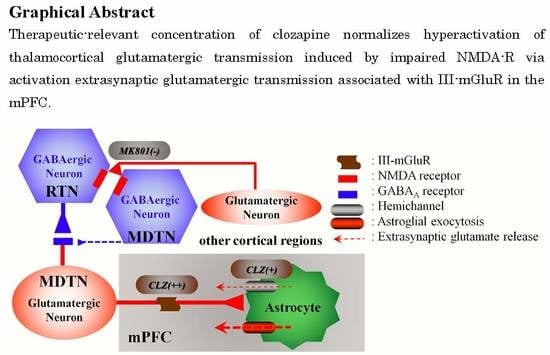
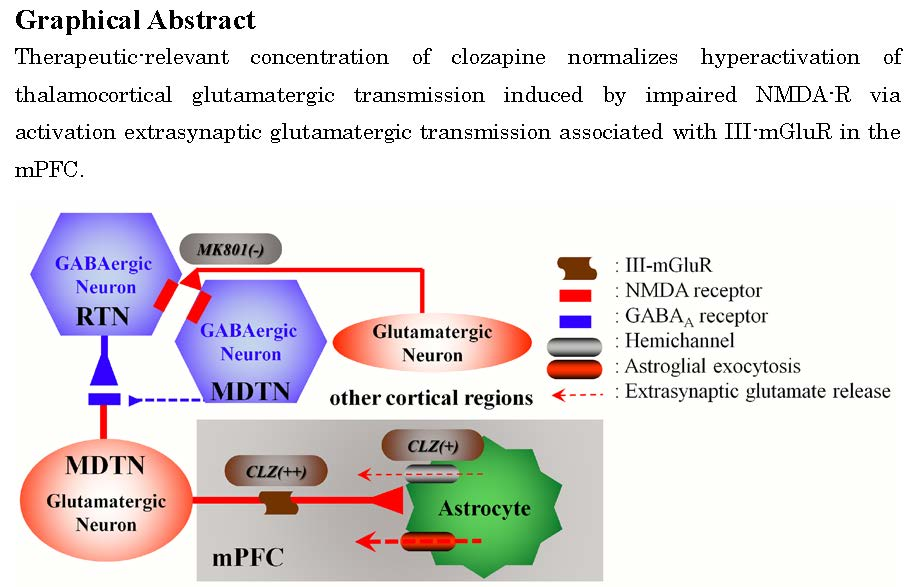
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemical Agents
2.2. Preparation of the Microdialysis System
2.3. Determination of Levels of l-Glutamate and GABA
2.4. Determination of Diffusion Rates of CLZ and MK801
2.5. Statistical Analysis
3. Results
3.1. Microdialysis Study
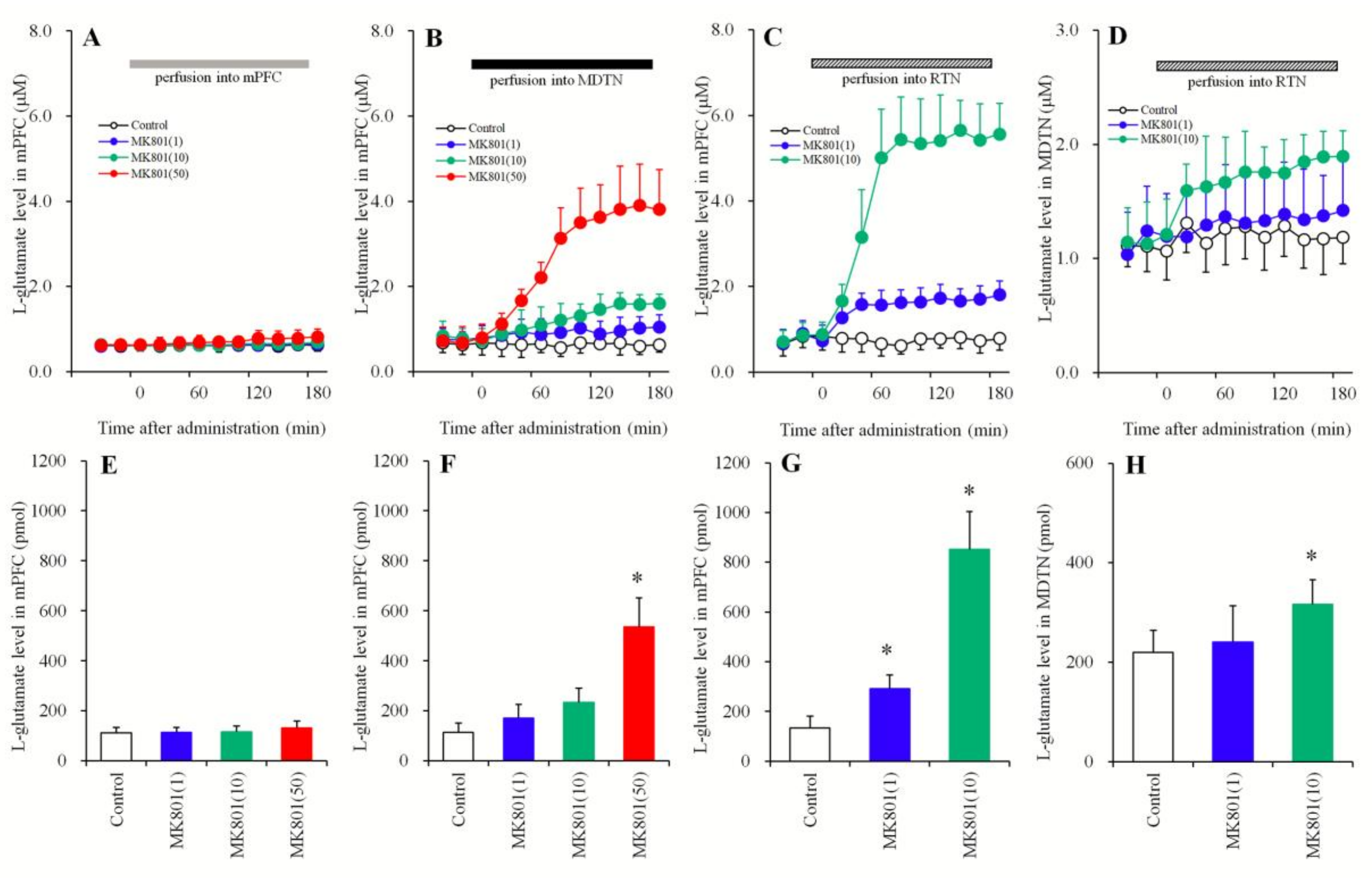
3.1.1. Concentration-Dependent Effects of the Local Administration of MK801 into the mPFC, MDTN, and RTN on Extracellular l-Glutamate Levels in the mPFC and the MDTN
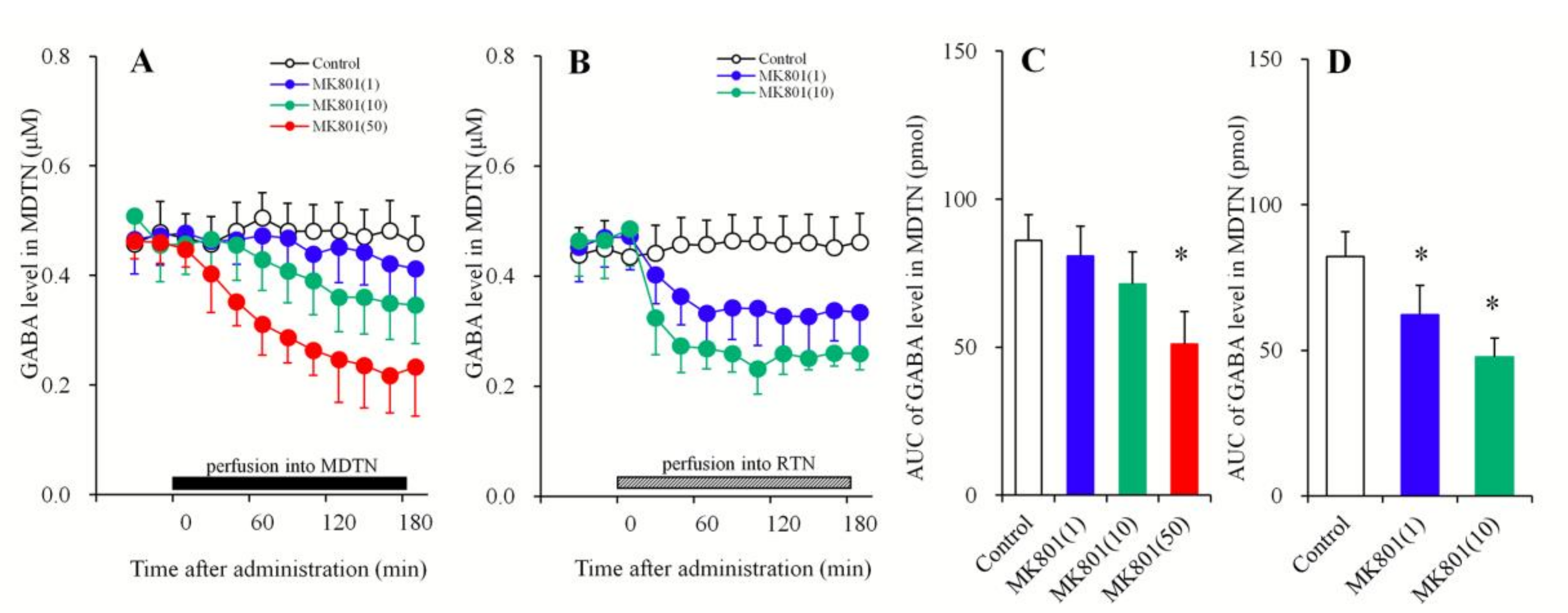
3.1.2. Concentration-Dependent Effects of the Local Administration of MK801 into the MDTN and the RTN on Extracellular GABA Levels in the MDTN
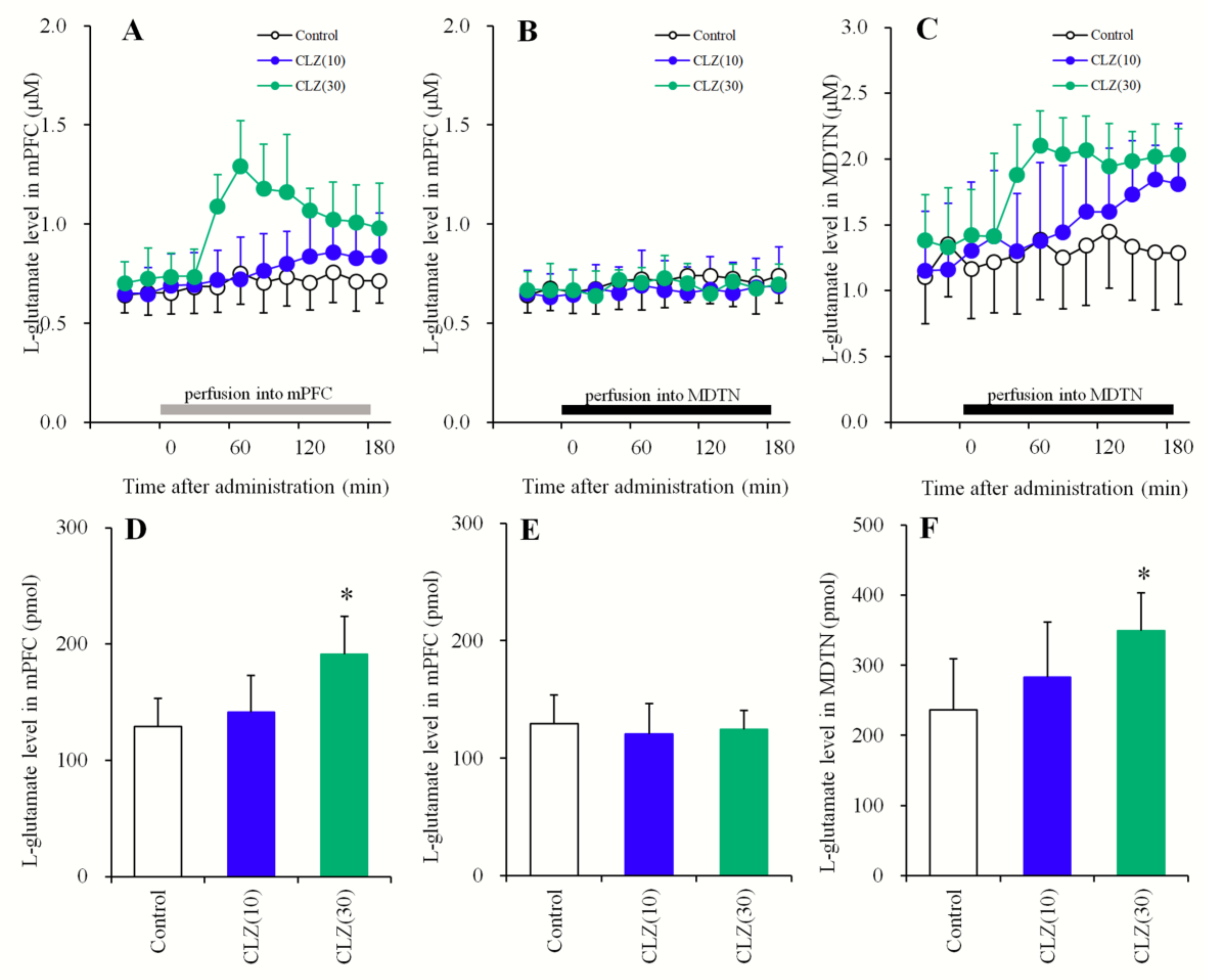
3.1.3. Concentration-Dependent Effects of the Local Administration of CLZ into the mPFC and the MDTN on Extracellular l-Glutamate Levels in the mPFC and MDTN
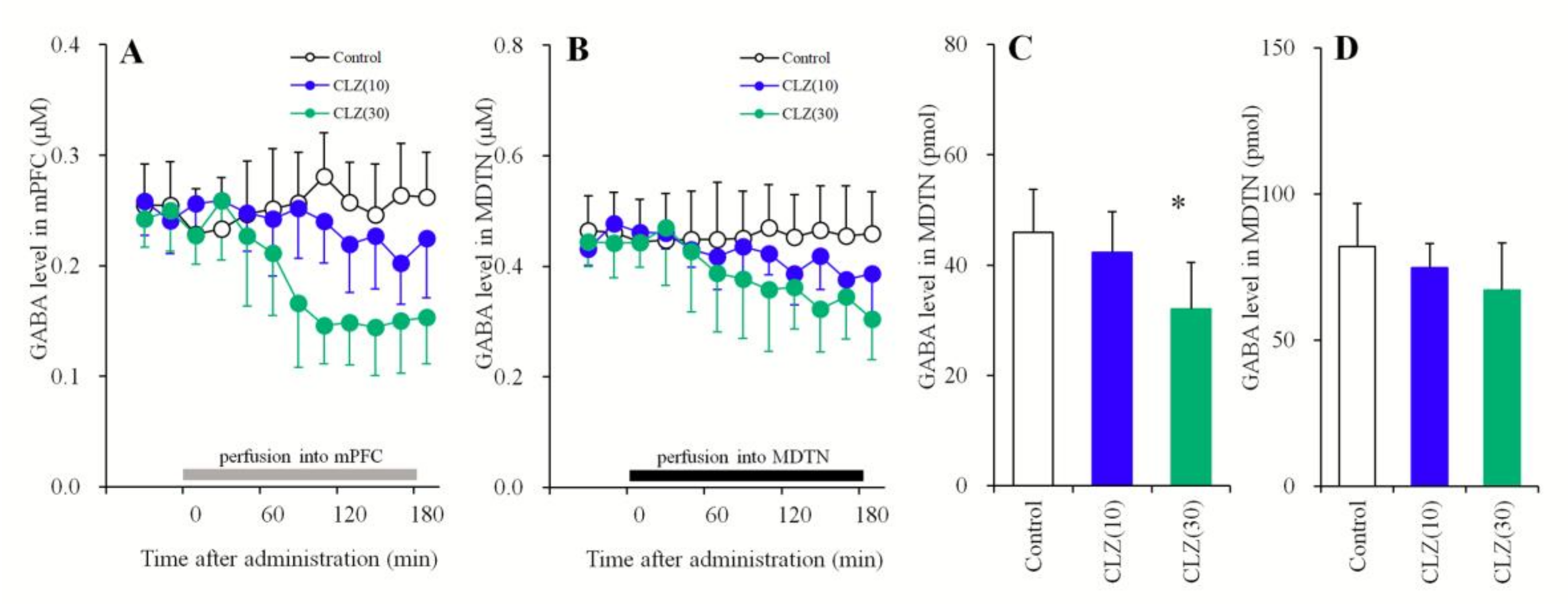
3.1.4. Concentration-Dependent Effects of the Local Administration of CLZ into the mPFC and MDTN on Extracellular GABA Levels in the mPFC and MDTN
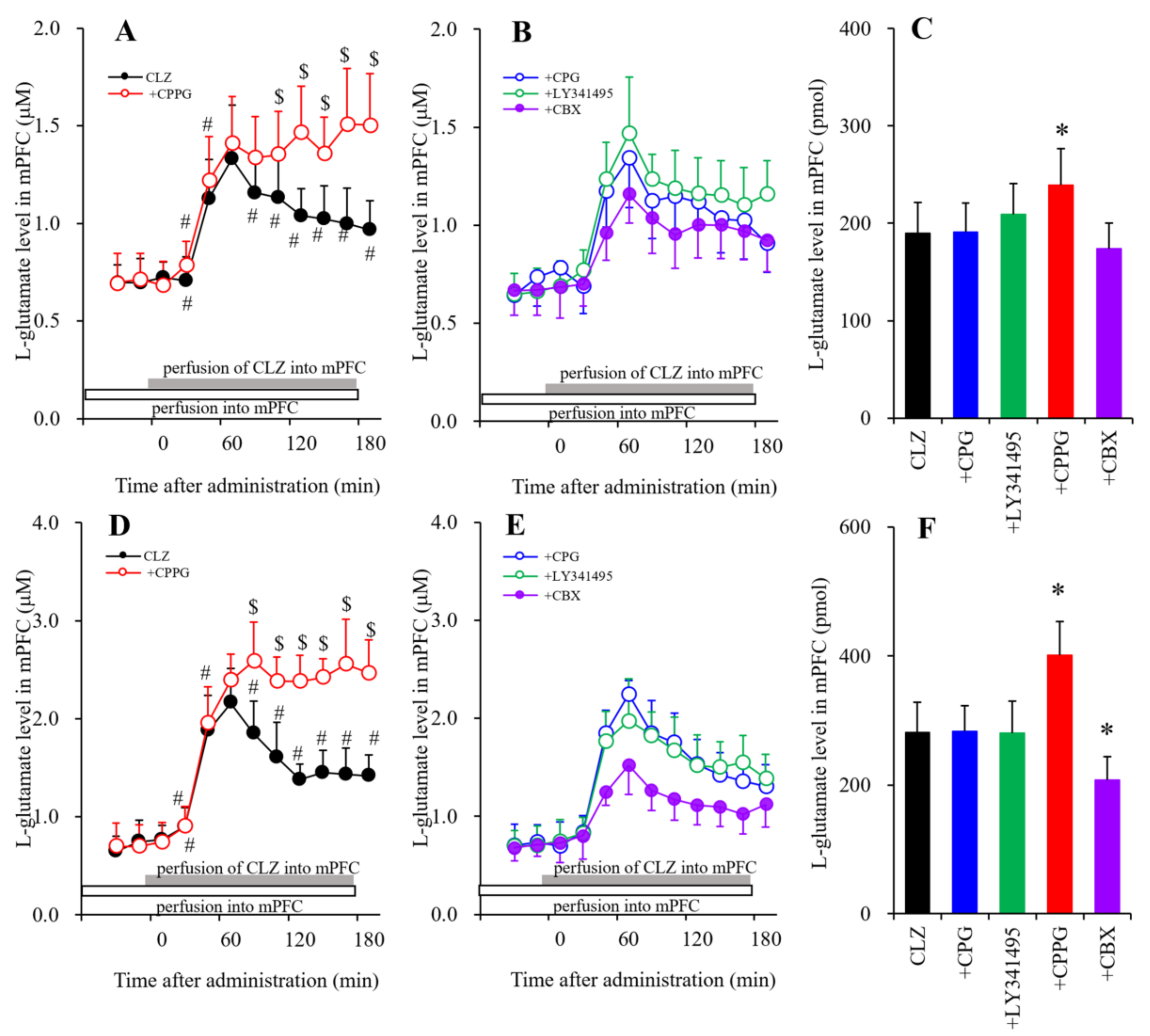
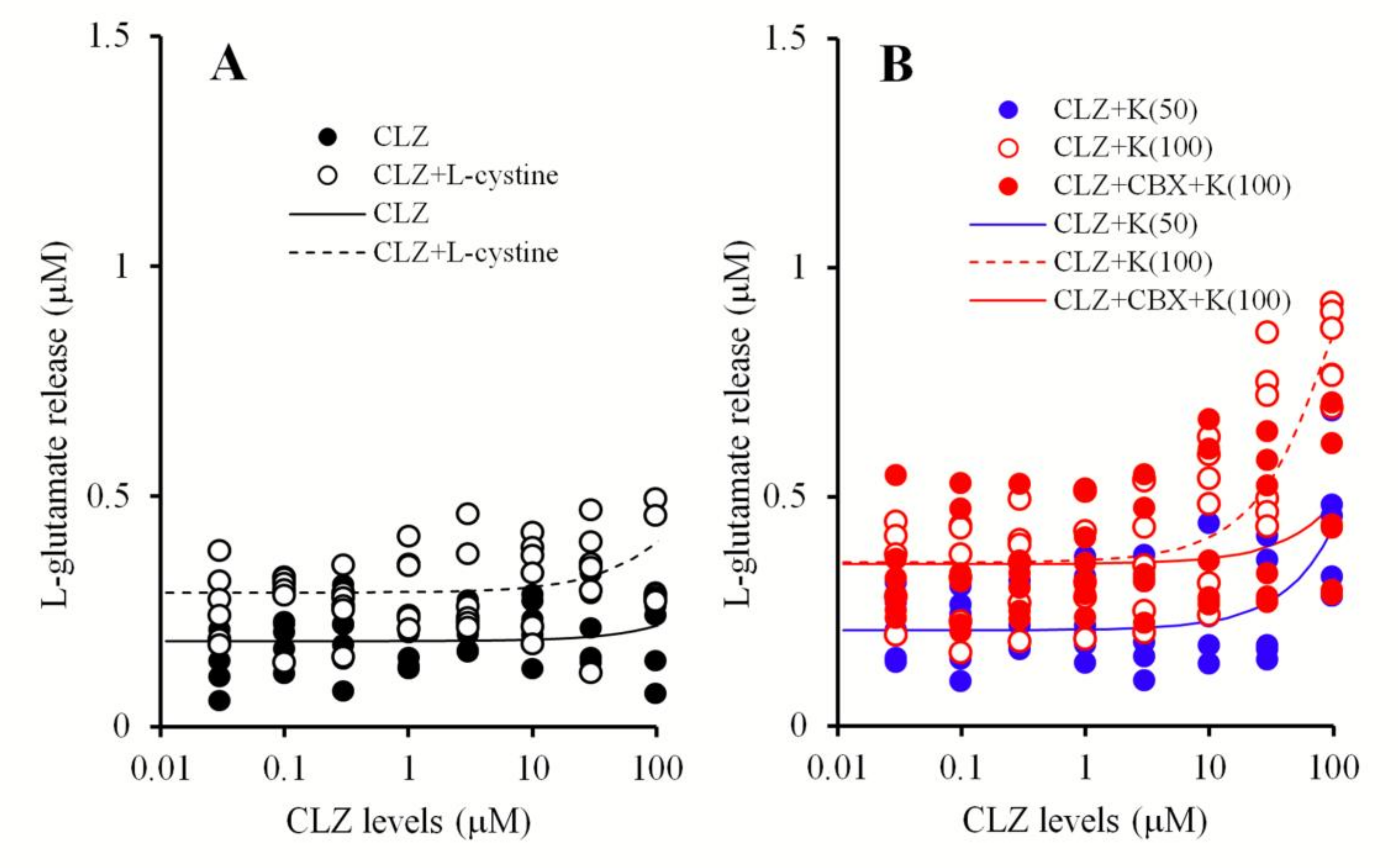
3.1.5. The Interaction of the Perfusion with CLZ and the Modulator of mGluRs and Hemichannels into the mPFC on Extracellular l-Glutamate Levels in the mPFC
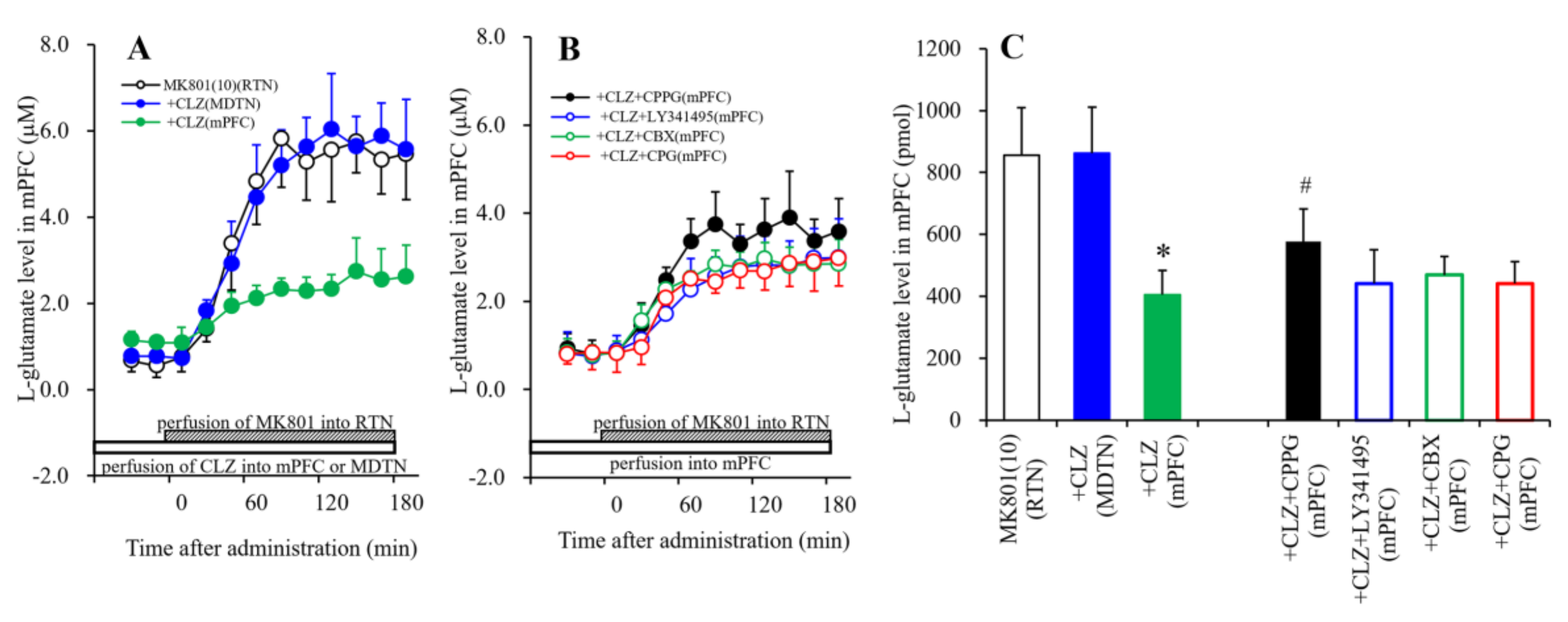
3.1.6. Interaction between MK801 and CLZ and its Impact on Extracellular l-Glutamate Levels in the mPFC
3.2. Primary Cultured Astrocyte Study
Effects of Concentration-Dependent Effects of CLZ on Hemichannel and Sxc Activities of Primary Cultured Astrocytes
4. Discussion
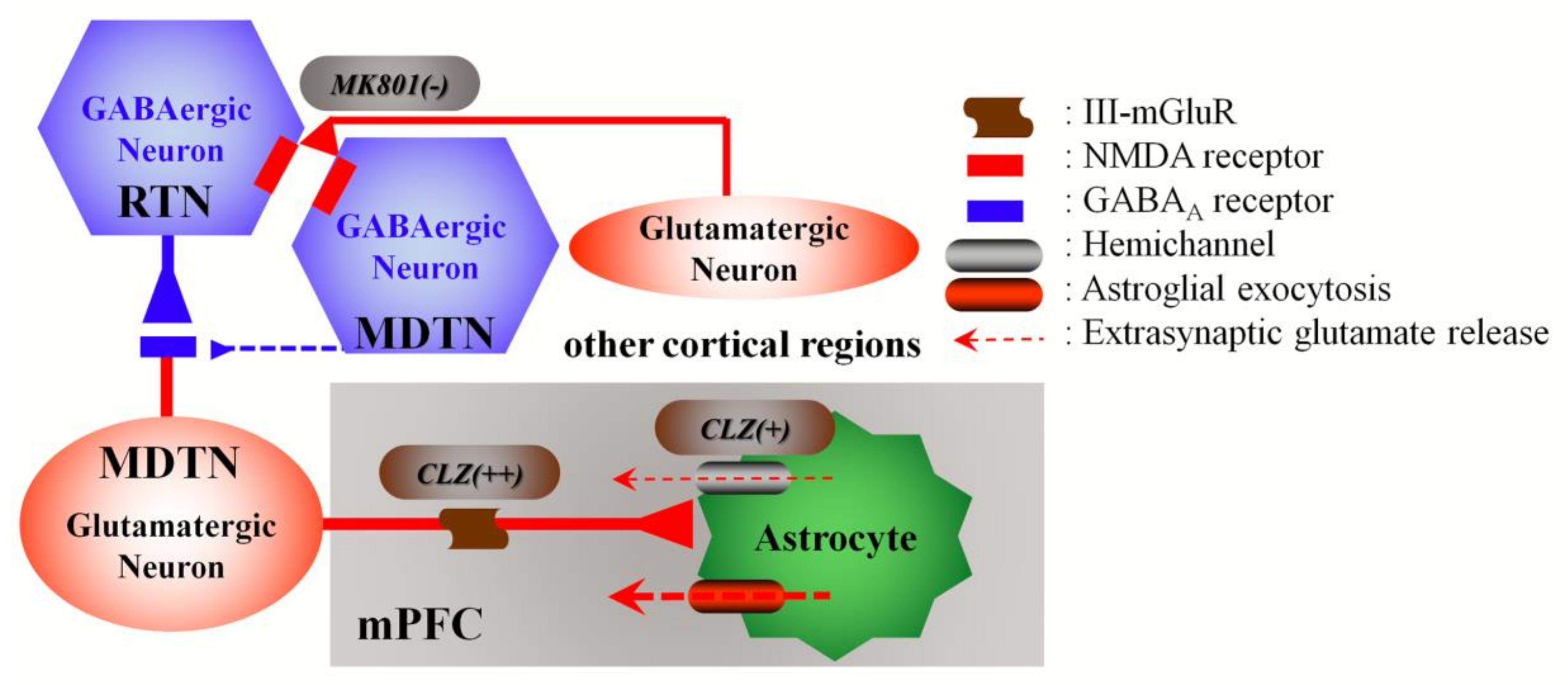
4.1. Candidate Mechanisms of MK801-Induced l-Glutamate Release in the mPFC
4.2. CLZ Regulates Thalamocortical Glutamatergic Transmission
4.3. Candidate Double-Edge Sword Mechanisms of the Action of CLZ
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mitterauer, B. Loss of function of glial gap junctions may cause severe cognitive impairments in schizophrenia. Med. Hypotheses 2009, 73, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Bymaster, F.P.; Meltzer, H.Y.; Deutch, A.Y.; Duncan, G.E.; Marx, C.E.; Aprille, J.R.; Dwyer, D.S.; Li, X.M.; Mahadik, S.P.; et al. Antipsychotic drugs: Comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008, 60, 358–403. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Huang, M. In Vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog. Brain Res. 2008, 172, 177–197. [Google Scholar] [PubMed]
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, n-methyl-d-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [PubMed]
- Labrie, V.; Roder, J.C. The involvement of the nmda receptor d-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 2010, 34, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. Nmda receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996, 14, 301–307. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Krystal, J.H.; D’Souza, D.C.; Mathalon, D.; Perry, E.; Belger, A.; Hoffman, R. Nmda receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology 2003, 169, 215–233. [Google Scholar] [CrossRef]
- Baker, D.A.; Madayag, A.; Kristiansen, L.V.; Meador-Woodruff, J.H.; Haroutunian, V.; Raju, I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 2008, 33, 1760–1772. [Google Scholar] [CrossRef]
- Lorrain, D.S.; Baccei, C.S.; Bristow, L.J.; Anderson, J.J.; Varney, M.A. Effects of ketamine and n-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group ii selective metabotropic glutamate receptor agonist ly379268. Neuroscience 2003, 117, 697–706. [Google Scholar] [CrossRef]
- Lopez-Gil, X.; Babot, Z.; Amargos-Bosch, M.; Sunol, C.; Artigas, F.; Adell, A. Clozapine and haloperidol differently suppress the mk-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 2007, 32, 2087–2097. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/glutamate antiporter and aripiprazole compensate nmda antagonist-induced dysfunction of thalamocortical l-glutamatergic transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by nmda receptor antagonism via activation of system xc(−). Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [PubMed]
- Lorrain, D.S.; Schaffhauser, H.; Campbell, U.C.; Baccei, C.S.; Correa, L.D.; Rowe, B.; Rodriguez, D.E.; Anderson, J.J.; Varney, M.A.; Pinkerton, A.B.; et al. Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 2003, 28, 1622–1632. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.W. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef]
- Ohoyama, K.; Yamamura, S.; Hamaguchi, T.; Nakagawa, M.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Effect of novel atypical antipsychotic, blonanserin, on extracellular neurotransmitter level in rat prefrontal cortex. Eur. J. Pharmacol. 2011, 653, 47–57. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Kashimoto, K.; Nakagawa, M.; Kanehara, S.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Shiroyama, T.; et al. Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology 2009, 206, 243–258. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. Br. J. Pharmacol. 2009, 157, 656–665. [Google Scholar] [CrossRef]
- Leeson, P.D.; Carling, R.W.; James, K.; Smith, J.D.; Moore, K.W.; Wong, E.H.; Baker, R. Role of hydrogen bonding in ligand interaction with the N-methyl-d-aspartate receptor ion channel. J. Med. Chem. 1990, 33, 1296–1305. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology. [CrossRef]
- Asanuma, C. Noradrenergic innervation of the thalamic reticular nucleus: A light and electron microscopic immunohistochemical study in rats. J. Comp. Neurol. 1992, 319, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Acsady, L. Thalamic inhibition: Diverse sources, diverse scales. Trends Neurosci. 2016, 39, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Xu, X.X.; He, S.; He, J. Change detection by thalamic reticular neurons. Nat. Neurosci. 2009, 12, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Choudry, A.; Cohen, D.; Naeem, F.; Ayub, M. Barriers to using clozapine in treatment-resistant schizophrenia: Systematic review. BJPsych Bull. 2019, 43, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Taylor, M. Clozapine: Dangerous orphan or neglected friend? Br. J. Psychiatry 2011, 198, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Siskind, D.; McCartney, L.; Goldschlager, R.; Kisely, S. Clozapine v. First- and second-generation antipsychotics in treatment-refractory schizophrenia: Systematic review and meta-analysis. Br. J. Psychiatry 2016, 209, 385–392. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Adler, C.M.; Kennison, S.D.; Elman, I.; Pickar, D.; Breier, A. Clozapine blunts N-methyl-d-aspartate antagonist-induced psychosis: A study with ketamine. Biol. Psychiatry 1997, 42, 664–668. [Google Scholar] [CrossRef]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine combines astroglial system xc(-) activation with glutamate/NMDA receptor inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef]
- Fell, M.J.; Svensson, K.A.; Johnson, B.G.; Schoepp, D.D. Evidence for the role of metabotropic glutamate (mglu)2 not mglu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (-)-(1r,4s,5s,6s)-4-amino-2-sulfonylbicyclo [3.1.0]hexane-4,6-dicarboxylic acid (ly404039). J. Pharmacol. Exp. Ther. 2008, 326, 209–217. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente Revenga, M.; Ibi, D.; Cuddy, T.; Toneatti, R.; Kurita, M.; Ijaz, M.K.; Miles, M.F.; Wolstenholme, J.T.; Gonzalez-Maeso, J. Chronic clozapine treatment restrains via hdac2 the performance of mglu2 receptor agonism in a rodent model of antipsychotic activity. Neuropsychopharmacology 2019, 44, 443–454. [Google Scholar] [CrossRef]
- Jiang, L.; Wu, X.; Wang, S.; Chen, S.H.; Zhou, H.; Wilson, B.; Jin, C.Y.; Lu, R.B.; Xie, K.; Wang, Q.; et al. Clozapine metabolites protect dopaminergic neurons through inhibition of microglial NADPH oxidase. J. Neuroinflamm. 2016, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wang, X.; Bhardwaj, S.K.; Zhou, X.; Little, P.J.; Quirion, R.; Srivastava, L.K.; Zheng, W. The atypical antipsychotic agent, clozapine, protects against corticosterone-induced death of pc12 cells by regulating the akt/foxo3a signaling pathway. Mol. Neurobiol. 2017, 54, 3395–3406. [Google Scholar] [CrossRef] [PubMed]
- Asenjo Lobos, C.; Komossa, K.; Rummel-Kluge, C.; Hunger, H.; Schmid, F.; Schwarz, S.; Leucht, S. Clozapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst. Rev. 2010, CD006633. [Google Scholar] [CrossRef] [PubMed]
- Lapato, A.S.; Tiwari-Woodruff, S.K. Connexins and pannexins: At the junction of neuro-glial homeostasis & disease. J. Neurosci. Res. 2018, 96, 31–44. [Google Scholar]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Wu, X.M.; Wang, G.L.; Miao, J.; Feng, J.C. Effect of connexin 36 blockers on the neuronal cytoskeleton and synaptic plasticity in kainic acid-kindled rats. Transl. Neurosci. 2015, 6, 252–258. [Google Scholar] [CrossRef]
- Jin, M.; Dai, Y.; Xu, C.; Wang, Y.; Wang, S.; Chen, Z. Effects of meclofenamic acid on limbic epileptogenesis in mice kindling models. Neurosci. Lett. 2013, 543, 110–114. [Google Scholar] [CrossRef]
- Alexander, S.P.; Peters, J.A.; Kelly, E.; Marrion, N.V.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. The concise guide to pharmacology 2017/18: Ligand-gated ion channels. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S130–S159. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S17–S129. [Google Scholar] [CrossRef]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): An obligate exchanger of l-glutamate and l-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. The concise guide to pharmacology 2017/18: Other ion channels. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S195–S207. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Kato, D.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. Ono-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The arrive guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotoxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76 Pt A, 137–145. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-ht1a receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef]
- Yoshitake, M.; Nohta, H.; Sejima, N.; Todoroki, K.; Yoshida, H.; Yamaguchi, M. Selective determination of cysteines through precolumn double-labeling and liquid chromatography followed by detection of intramolecular fret. Anal. Bioanal. Chem. 2011, 399, 1665–1675. [Google Scholar] [CrossRef]
- Yoshitake, M.; Nohta, H.; Ogata, S.; Todoroki, K.; Yoshida, H.; Yoshitake, T.; Yamaguchi, M. Liquid chromatography method for detecting native fluorescent bioamines in urine using post-column derivatization and intramolecular fret detection. J. Chromatogr. B 2007, 858, 307–312. [Google Scholar] [CrossRef]
- Le Quellec, A.; Dupin, S.; Genissel, P.; Saivin, S.; Marchand, B.; Houin, G. Microdialysis probes calibration: Gradient and tissue dependent changes in no net flux and reverse dialysis methods. J. Pharmacol. Toxicol. Methods 1995, 33, 11–16. [Google Scholar] [CrossRef]
- Okada, M.; Yoshida, S.; Zhu, G.; Hirose, S.; Kaneko, S. Biphasic actions of topiramate on monoamine exocytosis associated with both soluble n-ethylmaleimide-sensitive factor attachment protein receptors and Ca2+-induced Ca2+-releasing systems. Neuroscience 2005, 134, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.Q.; Zhang, Z.J.; Weng, Y.Z.; Zhai, Y.M.; Li, W.B.; Cai, Z.J.; Tan, Q.R.; Wang, C.Y. Serum concentrations of clozapine and norclozapine in the prediction of relapse of patients with schizophrenia. Schizophr. Res. 2006, 83, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Bishara, D.; Besag, F.M.; Taylor, D. Clozapine-related EEG changes and seizures: Dose and plasma-level relationships. Ther. Adv. Psychopharmacol. 2011, 1, 47–66. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Bourdelais, A.J.; Deutch, A.Y. The effects of haloperidol and clozapine on extracellular GABA levels in the prefrontal cortex of the rat: An in vivo microdialysis study. Cereb. Cortex 1994, 4, 69–77. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Varga, V.; Jenei, Z.; Janaky, R.; Saransaari, P.; Oja, S.S. Glutathione is an endogenous ligand of rat brain n-methyl-d-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem. Res. 1997, 22, 1165–1171. [Google Scholar] [CrossRef]
- Janaky, R.; Ogita, K.; Pasqualotto, B.A.; Bains, J.S.; Oja, S.S.; Yoneda, Y.; Shaw, C.A. Glutathione and signal transduction in the mammalian CNS. J. Neurochem. 1999, 73, 889–902. [Google Scholar] [CrossRef]
- Evonuk, K.S.; Baker, B.J.; Doyle, R.E.; Moseley, C.E.; Sestero, C.M.; Johnston, B.P.; De Sarno, P.; Tang, A.; Gembitsky, I.; Hewett, S.J.; et al. Inhibition of system xc(-) transporter attenuates autoimmune inflammatory demyelination. J. Immunol. 2015, 195, 450–463. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060234
Fukuyama K, Kato R, Murata M, Shiroyama T, Okada M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules. 2019; 9(6):234. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060234
Chicago/Turabian StyleFukuyama, Kouji, Ryo Kato, Masahiko Murata, Takashi Shiroyama, and Motohiro Okada. 2019. "Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor" Biomolecules 9, no. 6: 234. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060234