Tuning Structure and Dynamics of Blue Copper Azurin Junctions via Single Amino-Acid Mutations

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Atomic-Level Models and Force Fields
2.2. Molecular Dynamic (MD) Simulation Details
2.3. Simulation Protocol for Azurin in Water
2.4. Simulation Protocol for Azurin Adsorption
2.5. Trajectory Analysis
3. Results and Discussion
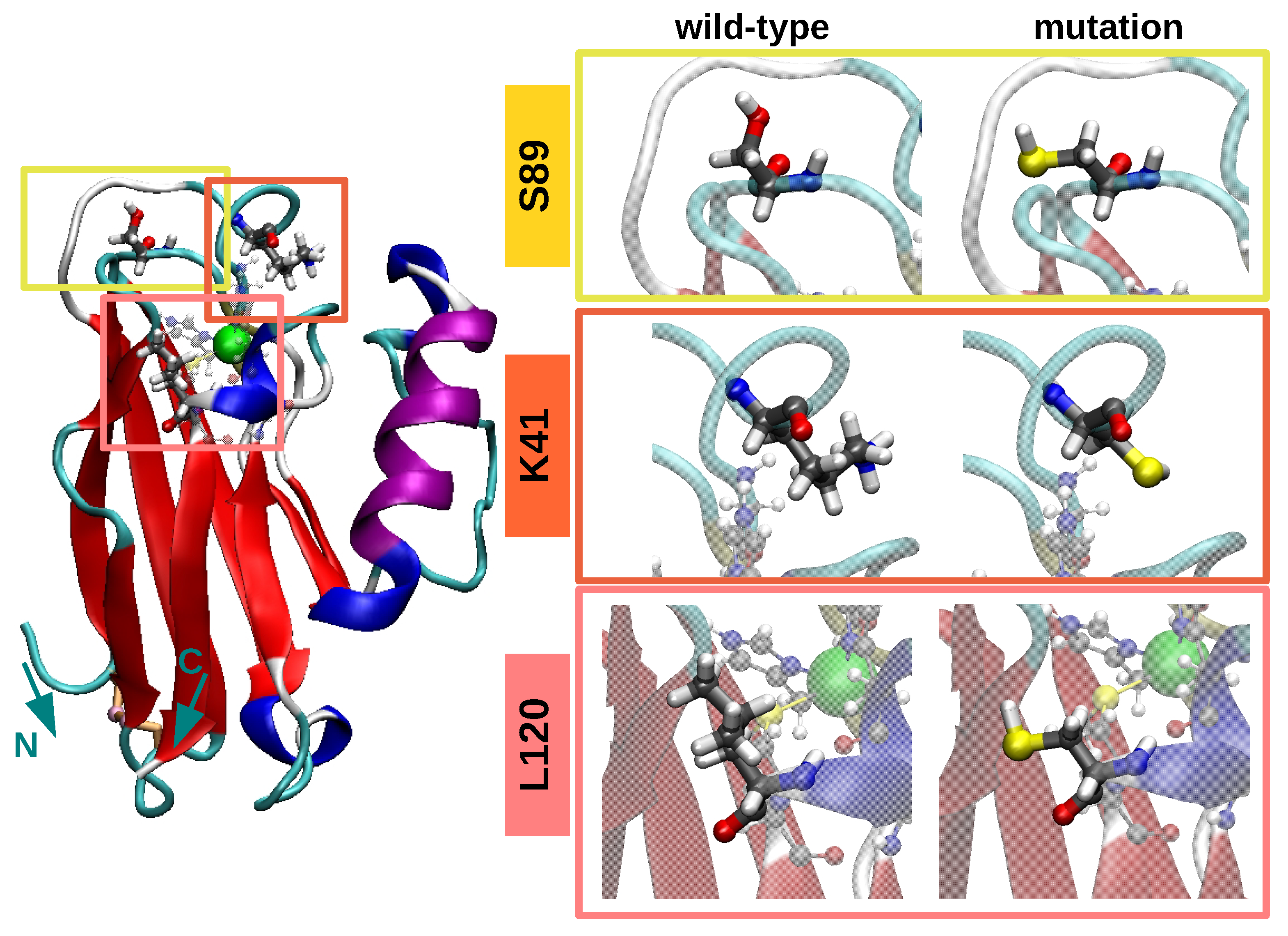
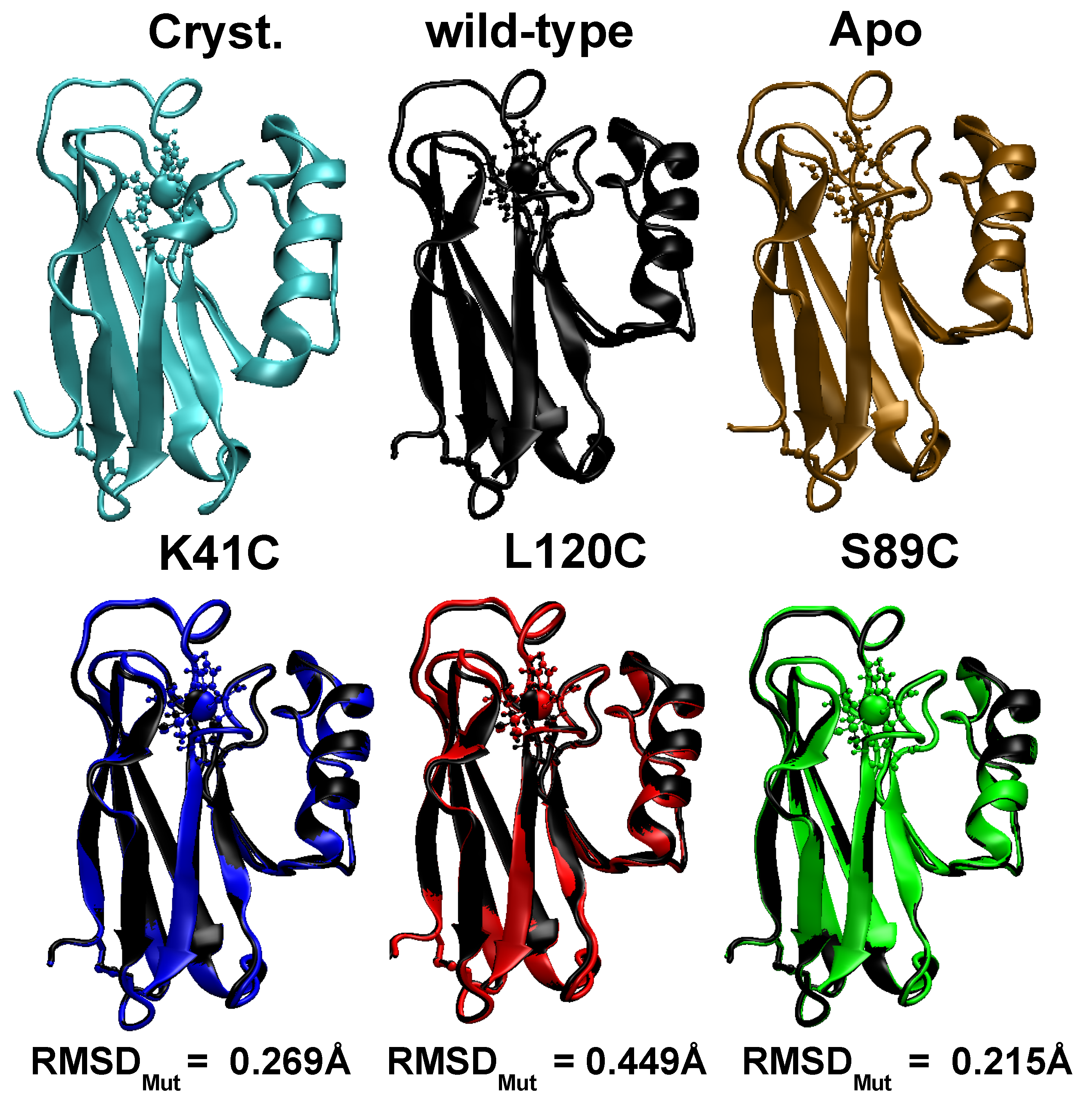
3.1. Equilibrium Structure of Unrestrained Wild-Type and Its Mutants in Water
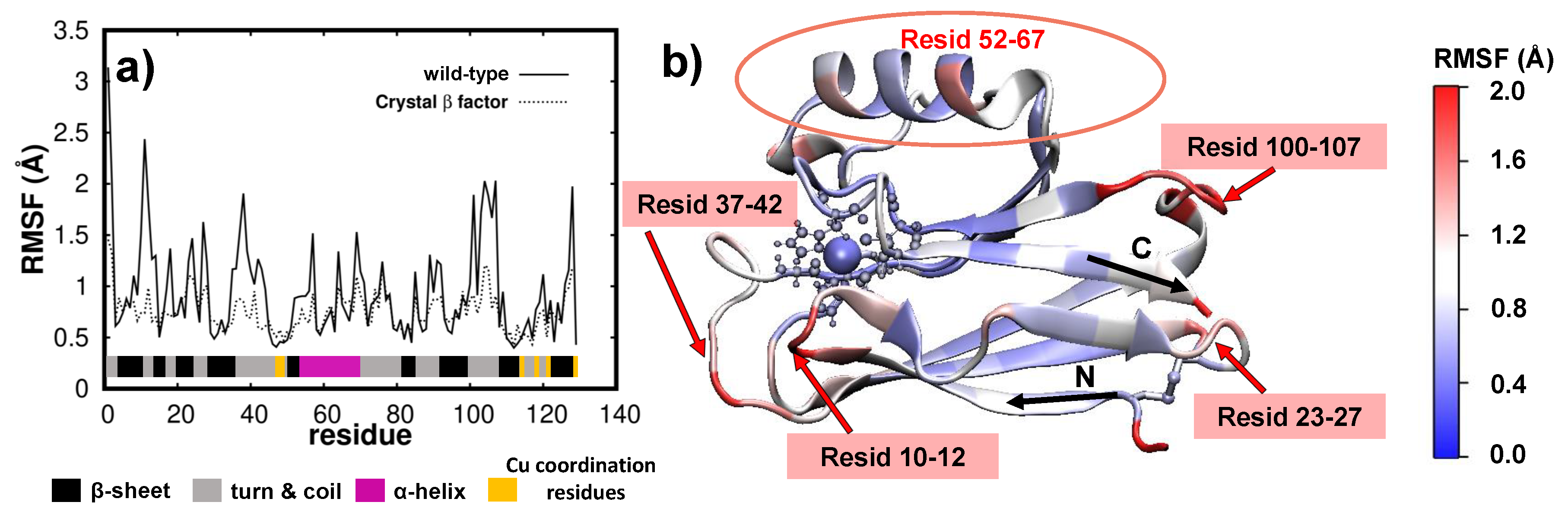
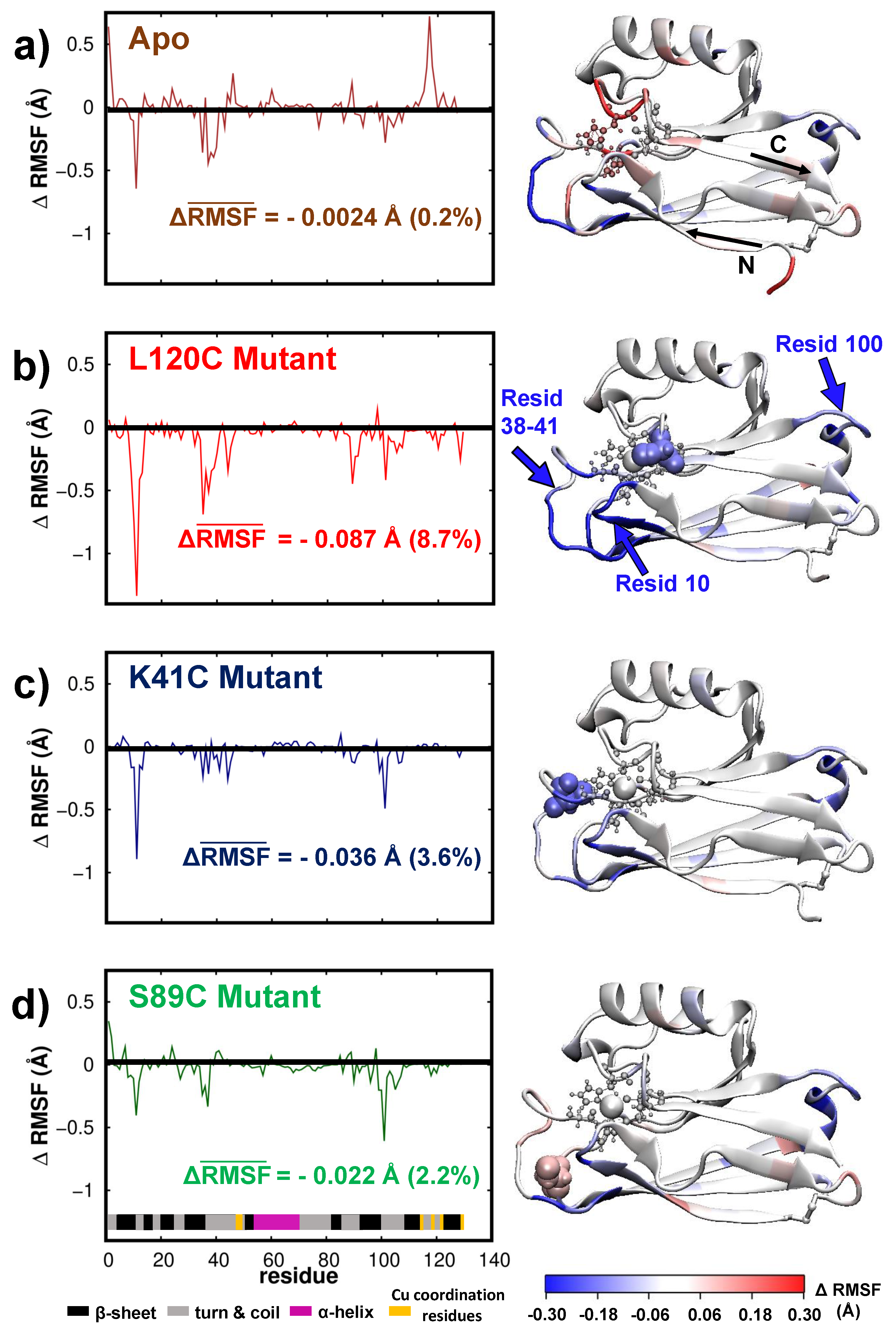
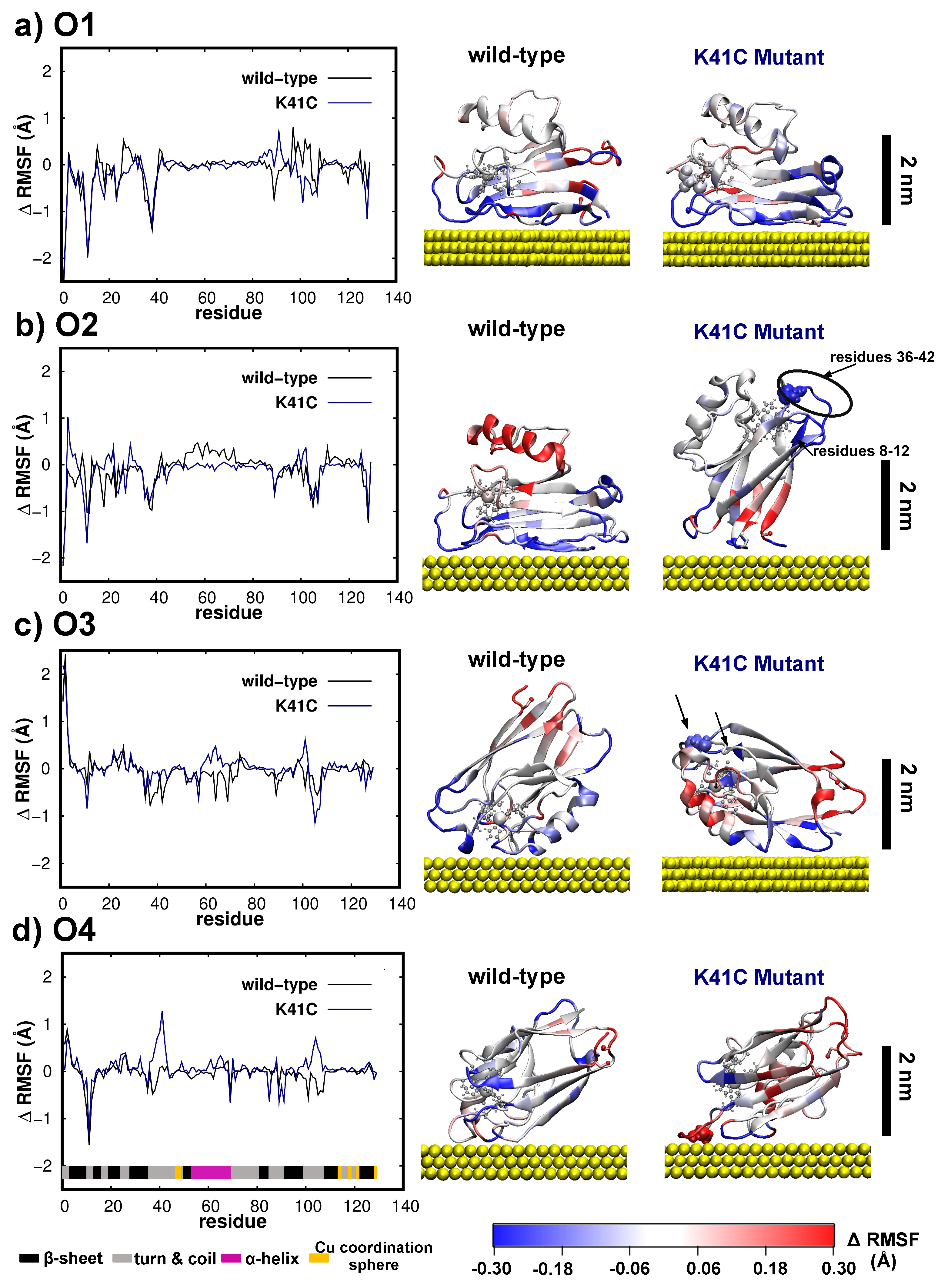
3.2. Dynamics/Fluctuations of Unrestrained Wild-Type and Its Mutants in Water
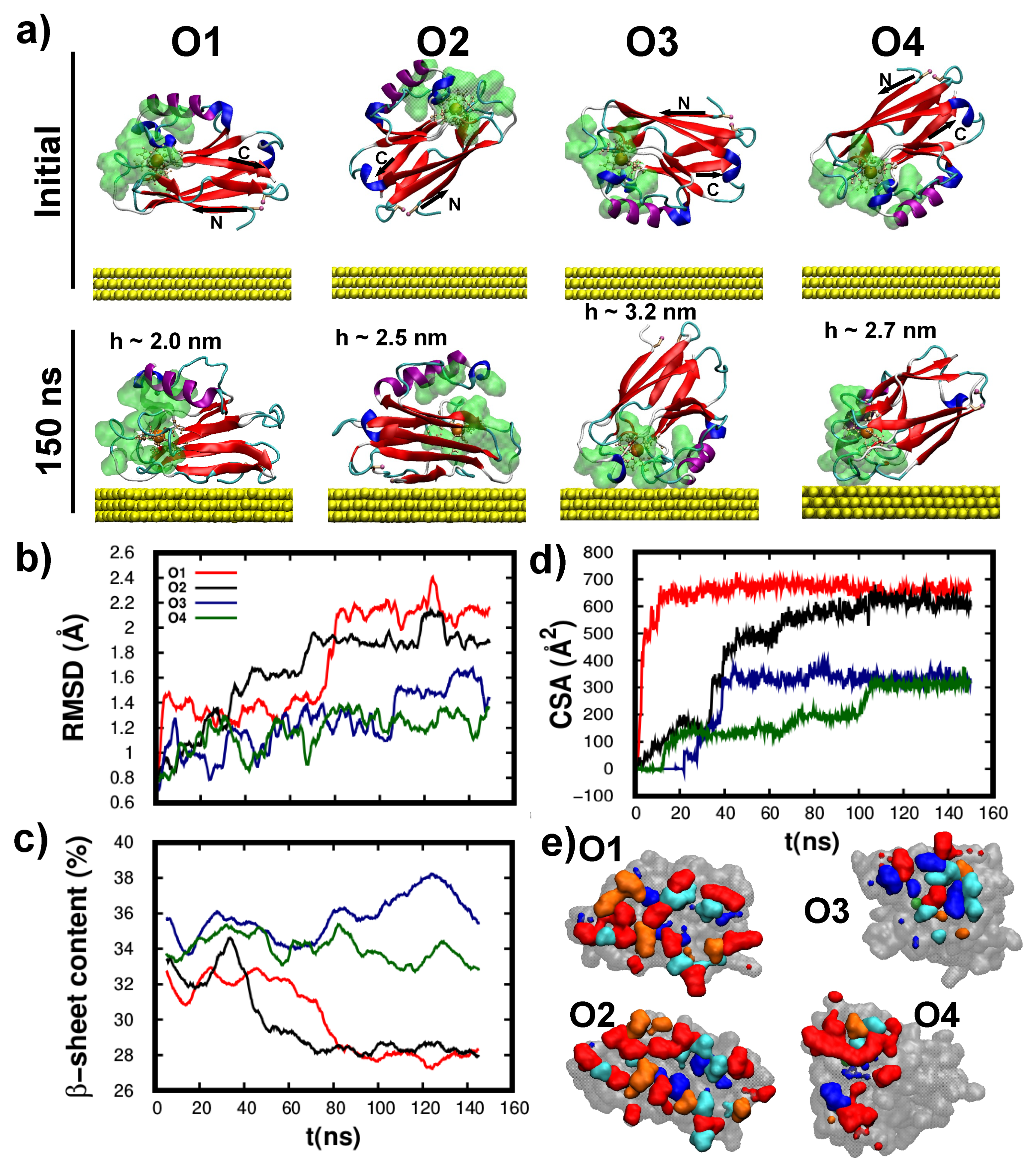
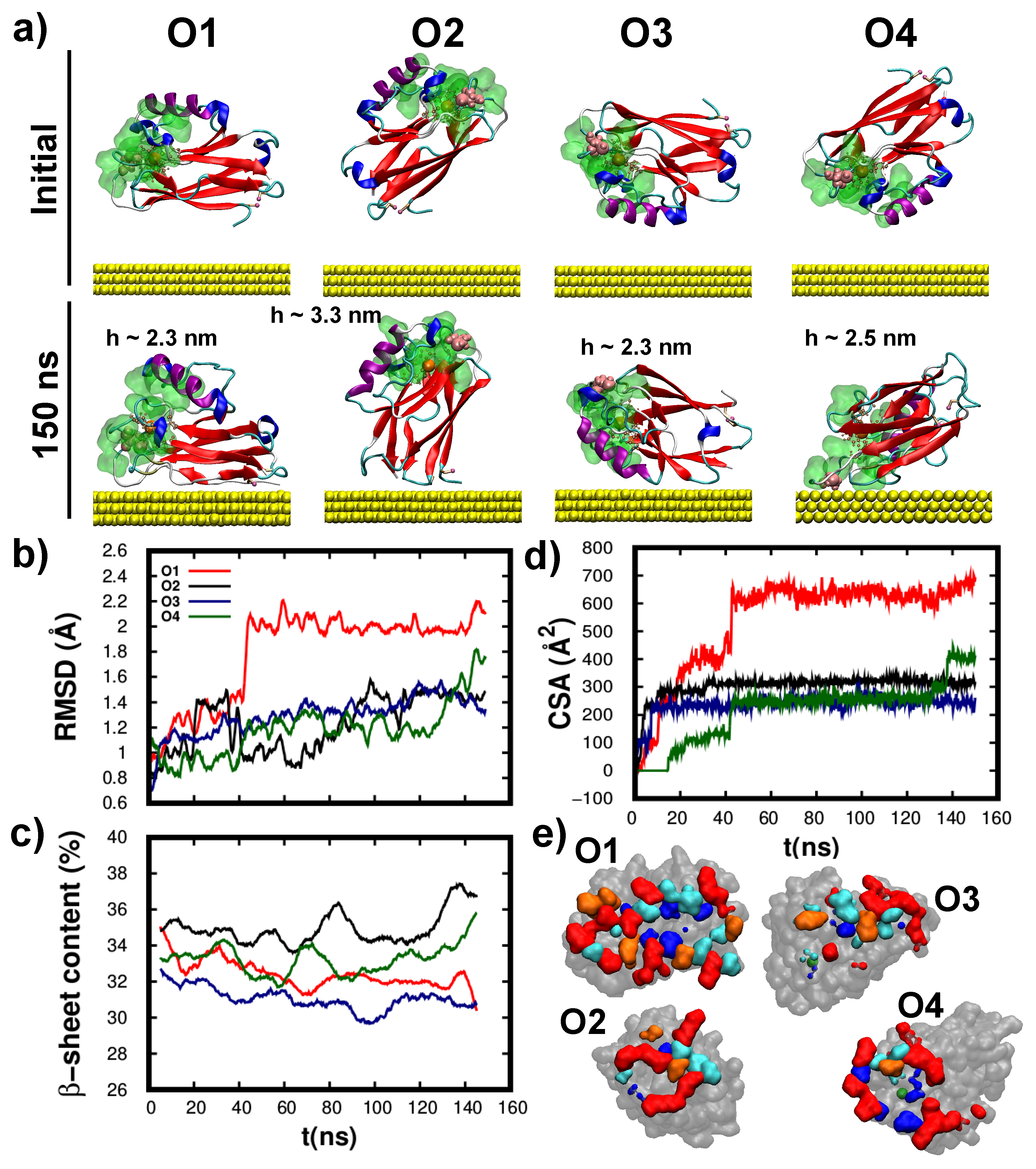
3.3. Structure/Conformation of Wild-Type and K41C Mutant upon Adsorption to Au(111)
3.4. Dynamics/Fluctuations of Wild-Type and K41C Mutant upon Adsorption to Au(111)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| CSA | Contact Surface Area |
| Rg | Radius of gyration |
| RMSD | Root Mean Square Displacement |
| RMSF | Root Mean Square Fluctuations |
References
- Amdursky, N.; Marchak, D.; Sepunaru, L.; Pecht, I.; Sheves, M.; Cahen, D. Electronic Transport via Proteins. Adv. Mater. 2014, 26, 7142–7161. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.P.; Aragonès, A.C.; Camarero, N.; Vilhena, J.G.; Ortega, M.; Zotti, L.A.; Pérez, R.; Cuevas, J.C.; Gorostiza, P.; Díez-Pérez, I. Bioengineering a Single-Protein Junction. J. Am. Chem. Soc. 2017, 139, 15337–15346. [Google Scholar] [CrossRef] [PubMed]
- Artés, J.M.; López-Martínez, M.; Díez-Pérez, I.; Sanz, F.; Gorostiza, P. Conductance Switching in Single Wired Redox Proteins. Small 2014, 10, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Ing, N.L.; El-Naggar, M.Y.; Hochbaum, A.I. Going the distance: Long-range conductivity in protein and peptide bioelectronic materials. J. Phys. Chem. B 2018, 122, 10403. [Google Scholar] [CrossRef] [PubMed]
- Fereiro, J.A.; Yu, X.; Pecht, I.; Sheves, M.; Cuevas, J.C.; Cahen, D. Tunneling explains efficient electron transport via protein junctions. Proc. Natl. Acad. Sci. USA 2018, 115, E4577. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lovrincic, R.; Sepunaru, L.; Li, W.; Vilan, A.; Pecht, I.; Sheves, M.; Cahen, D. Insights into Solid-State Electron Transport through Proteins from Inelastic Tunneling Spectroscopy: The Case of Azurin. ACS Nano 2015, 9, 9955. [Google Scholar] [CrossRef]
- Romero-Muñiz, C.; Ortega, M.; Vilhena, J.G.; Díez-Pérez, I.; Cuevas, J.C.; Pérez, R.; Zotti, L.A. Ab initio electronic structure calculations of entire blue copper azurins. Phys. Chem. Chem. Phys. 2018, 20, 30392–30402. [Google Scholar] [CrossRef] [Green Version]
- Valianti, S.; Cuevas, J.C.; Skourtis, S.S. Charge-Transport Mechanisms in Azurin-Based Monolayer Junctions. J. Phys. Chem. C. 2019, 123, 5907. [Google Scholar] [CrossRef]
- Bostick, C.D.; Mukhopadhyay, S.; Pecht, I.; Sheves, M.; Cahen, D.; Lederman, D. Protein bioelectronics: A review of what we do and do not know. Rep. Prog. Phys. 2018, 81, 26601. [Google Scholar] [CrossRef]
- Leary, E.; González, M.T.; van der Pol, C.; Bryce, M.R.; Filippone, S.; Martín, N.; Rubio-Bollinger, G.; Agraït, N. Unambiguous One-Molecule Conductance Measurements under Ambient Conditions. Nano Lett. 2011, 11, 2236–2241. [Google Scholar] [CrossRef]
- Latour, R.A. Molecular simulation of protein-surface interactions: Benefits, problems, solutions, and future directions. Biointerphases 2008, 3, FC2–FC12. [Google Scholar] [CrossRef] [PubMed]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of proteins-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, e4. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.C.; Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Di Felice, R.; Corni, S. Simulation of Peptide-Surface Recognition. J. Phys. Chem. Lett. 2011, 2, 1510–1519. [Google Scholar] [CrossRef]
- Rizzuti, B.; Sportelli, L.; Guzzi, R. Structural, dynamical and functional aspects of the inner motions in the blue copper protein azurin. Biophys. Chem. 2007, 125, 532–539. [Google Scholar] [CrossRef]
- Arcangeli, C.; Bizzarri, A.R.; Cannistraro, S. Long-term molecular dynamics simulation of copper azurin: Structure, dynamics and functionality. Biophys. Chem. 1999, 78, 247–257. [Google Scholar] [CrossRef]
- Bizzarri, A.R. Topological and dynamical properties of Azurin anchored to a gold substrate as investigated by molecular dynamics simulation. Biophys. Chem. 2006, 122, 206–214. [Google Scholar] [CrossRef]
- Venkat, A.S.; Corni, S.; Di Felice, R. Electronic Coupling Between Azurin and Gold at Different Protein/Substrate Orientations. Small 2007, 3, 1431–1437. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Baldacchini, C.; Cannistraro, S. Structure, Dynamics, and Electron Transfer of Azurin Bound to a Gold Electrode. Langmuir 2017, 33, 9190–9200. [Google Scholar] [CrossRef]
- Rizzuti, B.; Sportelli, L.; Guzzi, R. Evidence of reduced flexibility in disulfide bridge-depleted azurin: A molecular dynamics simulation study. Biophys. Chem. 2001, 94, 107–120. [Google Scholar] [CrossRef]
- Castellanos, M.; Pérez, R.; Carrasco, C.; Hernando-Pérez, M.; Gómez-Herrero, J.; de Pablo, P.J.; Mateu, M.G. Mechanical elasticity as a physical signature of conformational dynamics in a virus particle. Proc. Natl. Acad. Sci. USA 2012, 109, 12028–12033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, P.J.P.; Medrano, M.; Valbuena, A.; Rodríguez-Huete, A.; Castellanos, M.; Pérez, R.; Mateu, M.G. Amino Acid Side Chains Buried along Intersubunit Interfaces in a Viral Capsid Preserve Low Mechanical Stiffness Associated with Virus Infectivity. ACS Nano 2017, 11, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Hammann, C.; Messerschmidt, A.; Huber, R.; Nar, H.; Gilardi, G.; Canters, G.W. X-ray Crystal Structure of the Two Site-specific Mutants Ile7Ser and Phe110Ser of Azurin fromPseudomonas aeruginosa. J. Mol. Biol. 1996, 255, 362. [Google Scholar] [CrossRef] [PubMed]
- Kolczak, U.; Dennison, C.; Messerschmidt, A.; Canters, G. Azurin and Azurin Mutants. In Handbook of Metalloproteins; American Cancer Society: New York, NY, USA, 2006. [Google Scholar]
- Gabellieri, E.; Balestreri, E.; Galli, A.; Cioni, P. Cavity-Creating Mutations in Pseudomonas aeruginosa Azurin: Effects on Protein Dynamics and Stability. Biophys. J. 2008, 95, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Doss, C.G.P.; Rajith, B.; Garwasis, N.; Mathew, P.R.; Raju, A.S.; Apoorva, K.; William, D.; Sadhana, N.; Himani, T.; Dike, I. Screening of mutations affecting protein stability and dynamics of FGFR1—A simulation analysis. Appl. Transl. Genom. 2012, 1, 37. [Google Scholar] [CrossRef]
- Fiorani, P.; Bruselles, A.; Falconi, M.; Chillemi, G.; Desideri, A.; Benedetti, P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003, 278, 43268–43275. [Google Scholar] [CrossRef]
- Gekko, K. Compressibility gives new insight into protein dynamics and enzyme function. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2002, 1595, 382–386. [Google Scholar] [CrossRef]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Relationship between a point mutation S97C in CK1δ protein and its affect on ATP-binding affinity. J. Biomol. Struct. Dyn. 2014, 32, 394–405. [Google Scholar] [CrossRef]
- Fan, Y.; Cembran, A.; Ma, S.; Gao, J. Connecting Protein Conformational Dynamics with Catalytic Function as Illustrated in Dihydrofolate Reductase. Biochemistry 2013, 52, 2036–2049. [Google Scholar] [CrossRef]
- Behmard, E.; Abdolmaleki, P.; Asadabadi, E.B.; Jahandideh, S. Prevalent Mutations of Human Prion Protein: A Molecular Modeling and Molecular Dynamics Study. J. Biomol. Struct. Dyn. 2011, 29, 379–389. [Google Scholar] [CrossRef]
- Ortega, M.; Vilhena, J.G.; Rubio-Pereda, P.; Serena, P.A.; Pérez, R. Assessing the Accuracy of Different Solvation Models To Describe Protein Adsorption. J. Chem. Theory Comput. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235. [Google Scholar] [CrossRef] [PubMed]
- Nar, H.; Messerschmidt, A.; Huber, R.; van de Kamp, M.; Canters, G.W. Crystal structure analysis of oxidized Pseudomonas aeruginosa azurin at pH 5.5 and pH 9.0. J. Mol. Biol. 1991, 221, 765. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Van den Bosch, M.; Swart, M.; Snijders†, J.G.; Berendsen, H.J.C.; Mark, A.E.; Oostenbrink, C.; van Gunsteren, W.F.; Canters, G.W. Calculation of the Redox Potential of the Protein Azurin and Some Mutants. ChemBioChem 2005, 6, 738–746. [Google Scholar] [CrossRef]
- Paltrinieri, L.; Borsari, M.; Ranieri, A.; Battistuzzi, G.; Corni, S.; Bortolotti, C.A. The Active Site Loop Modulates the Reorganization Energy of Blue Copper Proteins by Controlling the Dynamic Interplay with Solvent. J. Phys. Chem. Lett. 2013, 4, 710–715. [Google Scholar] [CrossRef]
- Beedle, A.E.M.; Lezamiz, A.; Stirnemann, G.; Garcia-Manyes, S. The mechanochemistry of copper reports on the directionality of unfolding in model cupredoxin proteins. Nat. Commun. 2015, 6, 7894. [Google Scholar] [CrossRef] [Green Version]
- Zanetti-Polzi, L.; Bortolotti, C.A.; Daidone, I.; Aschi, M.; Amadei, A.; Corni, S. A few key residues determine the high redox potential shift in azurin mutants. Org. Biomol. Chem. 2015, 13, 11003–11013. [Google Scholar] [CrossRef]
- Zanetti-Polzi, L.; Corni, S.; Daidone, I.; Amadei, A. Extending the essential dynamics analysis to investigate molecular properties: Application to the redox potential of proteins. Phys. Chem. Chem. Phys. 2016, 18, 18450–18459. [Google Scholar] [CrossRef]
- Heinz, H.; Lin, T.J.; Kishore Mishra, R.; Emami, F.S. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir 2013, 29, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Ramezani-Dakhel, H. Simulations of inorganic-bioorganic interfaces to discover new materials: Insights, comparisons to experiment, challenges, and opportunities. Chem. Soc. Rev. 2016, 45, 412–448. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Vilhena, J.G.; Hinaut, A.; Meier, T.; Glatzel, T.; Baratoff, A.; Gnecco, E.; Pérez, R.; Meyer, E. Conformations and cryo-force spectroscopy of spray-deposited single-strand DNA on gold. Nat. Commun. 2019, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AMBER14. 2014. Available online: http://ambermd.org/ (accessed on 9 October 2019).
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Grand, S.L.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for {GPU} accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Di Felice, R.; Selloni, A.; Molinari, E. DFT Study of Cysteine Adsorption on Au(111). J. Phys. Chem. B 2003, 107, 1151–1156. [Google Scholar] [CrossRef]
- Alessandrini, A.; Corni, S.; Facci, P. Unravelling single metalloprotein electron transfer by scanning probe techniques. Phys. Chem. Chem. Phys. 2006, 8, 4383. [Google Scholar] [CrossRef] [PubMed]
- Artés, J.M.; Díez-Pérez, I.; Gorostiza, P. Transistor-like Behavior of Single Metalloprotein Junctions. Nano Lett. 2012, 12, 2679–2684. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Joosten, R.P.; te Beek, T.A.H.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.W.; Schneider, R.; Sander, C.; Vriend, G. A Series of PDB Related Databases for Everyday Needs. Nucleic Acids Res. 2010, 39, D411–D419. [Google Scholar] [CrossRef]
- Connolly, M.L. Analytical Molecular Surface Calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Rajapandian, V.; Hakkim, V.; Subramanian, V. Molecular Dynamics Studies on Native, Loop-Contracted, and Metal Ion-Substituted Azurins. J. Phys. Chem. B 2010, 114, 8474–8486. [Google Scholar] [CrossRef]
- Rubio-Pereda, P.; Vilhena, J.G.; Takeuchi, N.; Serena, P.A.; Pérez, R. Albumin (BSA) adsorption onto graphite stepped surfaces. J. Chem. Phys. 2017, 146, 214704. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, J.G.; Rubio-Pereda, P.; Vellosillo, P.; Serena, P.A.; Pérez, R. Albumin (BSA) Adsorption over Graphene in Aqueous Environment: Influence of Orientation, Adsorption Protocol, and Solvent Treatment. Langmuir 2016, 32, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, J.G.; Dumitru, A.C.; Herruzo, E.T.; Mendieta-Moreno, J.I.; Garcia, R.; Serena, P.A.; Perez, R. Adsorption Orientations and Immunological Recognition of Antibodies on Graphene. Nanoscale 2016, 8, 13463–13475. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, C.; Bizzarri, A.R.; Cannistraro, S. Concerted motions in copper plastocyanin and azurin: An essential dynamics study. Biophys. Chem. 2001, 90, 45–56. [Google Scholar] [CrossRef]
- Curtis, J.W. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 1999, 35, 307–312. [Google Scholar]
- Lim, W.A.; Sauer, R.T. The role of internal packing interactions in determining the structure and stability of a protein. J. Mol. Biol. 1991, 219, 359–376. [Google Scholar] [CrossRef]
- Garner, D.K.; Vaughan, M.D.; Hwang, H.J.; Savelieff, M.G.; Berry, S.M.; Honek, J.F.; Lu, Y. Reduction Potential Tuning of the Blue Copper Center in Pseudomonas aeruginosa Azurin by the Axial Methionine as Probed by Unnatural Amino Acids. J. Am. Chem. Soc. 2006, 128, 15608–15617. [Google Scholar] [CrossRef]
- Marshall, N.M.; Garner, D.K.; Wilson, T.D.; Gao, Y.G.; Robinson, H.; Nilges, M.J.; Lu, Y. Rationally tuning the reduction potential of a single cupredoxin beyond the natural range. Nature 2009, 462, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Petsko, G.A.; Ringe, D. Fluctuations in Protein Structure from X-ray Diffraction. Annu. Rev. Biophys. Bioeng. 1984, 13, 331–371. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Brunori, E.; Bonanni, B.; Cannistraro, S. Docking and molecular dynamics simulation of the Azurin–Cytochrome c551 electron transfer complex. J. Mol. Recognit. 2007, 20, 122–131. [Google Scholar] [CrossRef]
- Rizzuti, B.; Swart, M.; Sportelli, L.; Guzzi, R. Active site modeling in copper azurin molecular dynamics simulations. J. Mol. Model. 2004, 10, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Nar, H.; Messerschmidt, A.; Huber, R.; van de Kamp, M.; Canters, G.W. Crystal structure of Pseudomonas aeruginosa apo-azurin at 1.85 Å resolution. FEBS Lett. 1992, 306, 119–124. [Google Scholar] [CrossRef]
- Wei, T.; Carignano, M.A.; Szleifer, I. Lysozyme Adsorption on Polyethylene Surfaces: Why Are Long Simulations Needed? Langmuir 2011, 27, 12074–12081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanni, B.; Alliata, D.; Andolfi, L.; Bizzarri, A.; Cannistraro, S. Metalloproteins on Metal Surfaces as Hybrid Systems for Bionanodevices: An Extensive Characterization at the Single Molecule Level. In Surface Science Research Developments; Nova Science Pub Inc.: Hauppauge, NY, USA, 2005. [Google Scholar]
- Davis, J.J.; Hill, H.A.O. The scanning probe microscopy of metalloproteins and metalloenzymes. Chem. Commun. 2002, 5, 393–401. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RMSD (Å) | Rg (Å) | -Helix Content (%) | -Sheet Content (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | Mean | Mean | Mean | |||||
| wild-type | 1.086 | 0.158 | 14.049 | 0.051 | 10.156 | 1.893 | 36.149 | 1.601 |
| Apo | 1.099 | 0.137 | 14.061 | 0.062 | 11.151 | 2.128 | 35.299 | 2.002 |
| K41C | 1.021 | 0.121 | 14.051 | 0.021 | 10.912 | 1.952 | 35.916 | 1.722 |
| L120C | 0.980 | 0.107 | 14.036 | 0.065 | 11.049 | 1.986 | 34.446 | 1.491 |
| S89C | 1.100 | 0.110 | 14.062 | 0.060 | 9.319 | 1.532 | 35.674 | 1.463 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, M.; Vilhena, J.G.; Zotti, L.A.; Díez-Pérez, I.; Cuevas, J.C.; Pérez, R. Tuning Structure and Dynamics of Blue Copper Azurin Junctions via Single Amino-Acid Mutations. Biomolecules 2019, 9, 611. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100611
Ortega M, Vilhena JG, Zotti LA, Díez-Pérez I, Cuevas JC, Pérez R. Tuning Structure and Dynamics of Blue Copper Azurin Junctions via Single Amino-Acid Mutations. Biomolecules. 2019; 9(10):611. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100611
Chicago/Turabian StyleOrtega, Maria, J. G. Vilhena, Linda A. Zotti, Ismael Díez-Pérez, Juan Carlos Cuevas, and Rubén Pérez. 2019. "Tuning Structure and Dynamics of Blue Copper Azurin Junctions via Single Amino-Acid Mutations" Biomolecules 9, no. 10: 611. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100611