
Intestinal Barrier and Permeability in Health, Obesity and NAFLD

 , , and
, , and
Abstract
:1. Introduction
2. The Intestinal Barrier as an Integrated System of Multiple Elements
2.1. The Gut Microbiota
2.2. The Extracellular Barrier: The Gut Mucus
2.3. The Interplay between Gastrointestinal Motility and Secretions
2.4. The Epithelial Barrier and the Tight Junctions
2.5. The Immune-Competent Cells and Their Products (i.e., the Immune Barrier)
2.6. The Gut-Vascular Interface
2.7. The Hepatic Filter (i.e., the Liver Barrier)
3. Assessment of Intestinal Barrier In Vivo
4. Modifiers of the Intestinal Barrier
4.1. Fibers
4.1.1. Fibers as Source of Microbiota-Derived Short-Chain Fatty Acids (SCFA)
4.1.2. Additional Effects of Fibers
4.2. Polyphenols and Other Metabolites
4.3. Glutamine
4.4. Vitamin D and Zinc
4.5. Probiotics, Symbiotics and Prebiotics
4.6. Fats
4.7. Emulsifiers
4.8. Alcohol
5. The Burden of Obesity
6. The Burden of Non-Alcoholic Fatty Liver Disease (NAFLD)
7. The Intestinal Barrier: General Implications in Obesity and NAFLD
8. Intestinal Barrier Features in Obesity
Obesity and Gut Immunity
9. Intestinal Barrier Features in NAFLD
9.1. NAFLD as a Model of Systemic Inflammation
9.2. The Gut Microbiota
9.3. Bacterial Products
9.3.1. MAMPs/PAMPs
9.3.2. Alcohol
9.3.3. Bile Acids (BA)
9.3.4. SCFA including Propionate
9.3.5. Fasting-Induced Adipocyte Factor (Fiaf)
9.3.6. Trimethylamine (TMA)
9.3.7. Phenylacetate
9.3.8. Imidazole Propionate
9.3.9. Other Metabolites
10. NAFLD and Gut Permeability
11. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dommett, R.; Zilbauer, M.; George, J.T.; Bajaj-Elliott, M. Innate immune defence in the human gastrointestinal tract. Mol. Immunol. 2005, 42, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-liver axis at the frontier of host-microbial interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-Talk Between Bile Acids and Gastro-Intestinal and Thermogenic Hormones: Clues from Bariatric Surgery. Ann. Hepatol. 2017, 16, s68–s82. [Google Scholar] [CrossRef]
- Garruti, G.; Wang, D.Q.; Di Ciaula, A.; Portincasa, P. Cholecystectomy: A way forward and back to metabolic syndrome? Lab. Invest. 2018, 98, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 2018, 38, 5. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U.; Hornef, M.W.; Henriques-Normark, B.; Axelsson, L.G.; Midtvedt, T.; Putsep, K.; Andersson, M. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 2008, 57, 764–771. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef]
- Kallus, S.J.; Brandt, L.J. The intestinal microbiota and obesity. J. Clin. Gastroenterol. 2012, 46, 16–24. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- McGhee, J.R.; Fujihashi, K. Inside the mucosal immune system. PLoS Biol. 2012, 10, e1001397. [Google Scholar] [CrossRef] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. New Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- De Lacy Costello, B.; Amann, A.; Al-Kateb, H.; Flynn, C.; Filipiak, W.; Khalid, T.; Osborne, D.; Ratcliffe, N.M. A review of the volatiles from the healthy human body. J. Breath Res. 2014, 8, 014001. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Reynes, B.; Palou, M.; Rodriguez, A.M.; Palou, A. Regulation of Adaptive Thermogenesis and Browning by Prebiotics and Postbiotics. Front. Physiol. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Said, H.M.; Ortiz, A.; McCloud, E.; Dyer, D.; Moyer, M.P.; Rubin, S. Biotin uptake by human colonic epithelial NCM460 cells: A carrier-mediated process shared with pantothenic acid. Am. J. Physiol. 1998, 275, C1365–C1371. [Google Scholar] [CrossRef]
- Biesalski, H.K. Nutrition meets the microbiome: Micronutrients and the microbiota. Ann. N. Y. Acad. Sci. 2016, 1372, 53–64. [Google Scholar] [CrossRef]
- Belancic, A. Gut microbiome dysbiosis and endotoxemia—Additional pathophysiological explanation for increased COVID-19 severity in obesity. Obes. Med. 2020, 20, 100302. [Google Scholar] [CrossRef]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Backhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergstrom, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.; Schutte, A.; van der Post, S.; Svensson, F.; Rodriguez-Pineiro, A.M.; Nystrom, E.E.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Ismail, A.S.; Severson, K.M.; Vaishnava, S.; Behrendt, C.L.; Yu, X.; Benjamin, J.L.; Ruhn, K.A.; Hou, B.; DeFranco, A.L.; Yarovinsky, F. γδ intraepithelial lymphocytes are essential mediators of host–microbial homeostasis at the intestinal mucosal surface. Proc. Natl. Acad. Sci. USA 2011, 108, 8743–8748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011, 17, 584–593. [Google Scholar] [CrossRef]
- Gibbins, H.L.; Proctor, G.B.; Yakubov, G.E.; Wilson, S.; Carpenter, G.H. SIgA binding to mucosal surfaces is mediated by mucin-mucin interactions. PLoS ONE 2015, 10, e0119677. [Google Scholar] [CrossRef] [Green Version]
- Bergström, J.H.; Birchenough, G.M.H.; Katona, G.; Schroeder, B.O.; Schütte, A.; Ermund, A.; Johansson, M.E.V.; Hansson, G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 2016, 113, 13833–13838. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Vella, A. What to do about the leaky gut. Gut 2021. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Tsilingiri, K.; Barbosa, T.; Penna, G.; Caprioli, F.; Sonzogni, A.; Viale, G.; Rescigno, M. Probiotic and postbiotic activity in health and disease: Comparison on a novel polarised ex-vivo organ culture model. Gut 2012, 61, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Tsilingiri, K.; Rescigno, M. Postbiotics: What else. In Beneficial Microbes 4; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 101–107. [Google Scholar]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Mosca, F.; Gianni, M.L.; Rescigno, M. Can Postbiotics Represent a New Strategy for NEC. In Probiotics and Child Gastrointestinal Health; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Joncquel Chevalier-Curt, M.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Mastrodonato, M.; Mentino, D.; Portincasa, P.; Calamita, G.; Liquori, G.E.; Ferri, D. High-fat diet alters the oligosaccharide chains of colon mucins in mice. Histochem. Cell Biol. 2014, 142, 449–459. [Google Scholar] [CrossRef]
- Liquori, G.E.; Mastrodonato, M.; Mentino, D.; Scillitani, G.; Desantis, S.; Portincasa, P.; Ferri, D. In situ characterization of O-linked glycans of Muc2 in mouse colon. Acta Histochem. 2012, 114, 723–732. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131. [Google Scholar] [CrossRef]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Muller, M.; de Vos, W.M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.V. Fast renewal of the distal colonic mucus layers by the surface goblet cells as measured by in vivo labeling of mucin glycoproteins. PLoS ONE 2012, 7, e41009. [Google Scholar] [CrossRef]
- Johansson, M.E.; Sjovall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Ponziani, F.R.; Gerardi, V.; Gasbarrini, A. Diagnosis and treatment of small intestinal bacterial overgrowth. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 215–227. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Garruti, G.; Wang, H.H.; Bonfrate, L.; de Bari, O.; Wang, D.Q.; Portincasa, P. A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J. Lipids 2012, 2012, 304292. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hu, C.; Zhang, X.; Jia, W. Role of gut microbiota, bile acids and their cross-talk in the effects of bariatric surgery on obesity and type 2 diabetes. J. Diabetes Investig. 2018, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ory, D.S. Nuclear receptor signaling in the control of cholesterol homeostasis: Have the orphans found a home? Circ.Res. 2004, 95, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef]
- Nevo, S.; Kadouri, N.; Abramson, J. Tuft cells: From the mucosa to the thymus. Immunol. Lett. 2019, 210, 1–9. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Salzman, N.H. Paneth cell defensins and the regulation of the microbiome: Détente at mucosal surfaces. Gut Microbes 2010, 1, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–82. [Google Scholar] [CrossRef]
- Bennett, K.M.; Walker, S.L.; Lo, D.D. Epithelial microvilli establish an electrostatic barrier to microbial adhesion. Infect. Immun. 2014, 82, 2860–2871. [Google Scholar] [CrossRef] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Okawa, K.; Yano, T.; Tsukita, S.; Tsukita, S. Optimized proteomic analysis on gels of cell-cell adhering junctional membrane proteins. Biochemistry 2008, 47, 5378–5386. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef]
- Hollander, D.; Kaunitz, J.D. The "Leaky Gut": Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Schoultz, I.; Keita, Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Holmes, J.; Bridges, A.; Gookin, J.L.; Coccaro, M.R.; Proctor, W.; Colegio, O.R.; Anderson, J.M. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell Sci. 2008, 121, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Dzus, A.L.; Colgan, S.P. Autocrine regulation of epithelial permeability by hypoxia: Role for polarized release of tumor necrosis factor alpha. Gastroenterology 1998, 114, 657–668. [Google Scholar] [CrossRef]
- Madara, J.L.; Stafford, J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J. Clin. Investig. 1989, 83, 724–727. [Google Scholar] [CrossRef]
- Turner, J.R.; Rill, B.K.; Carlson, S.L.; Carnes, D.; Kerner, R.; Mrsny, R.J.; Madara, J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997, 273, C1378–C1385. [Google Scholar] [CrossRef]
- Hartmann, P.; Haimerl, M.; Mazagova, M.; Brenner, D.A.; Schnabl, B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 2012, 143, 1330–1340.e1331. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Rah, B.; Bastola, D.; Dhawan, P.; Singh, A.B. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci. Rep. 2017, 7, 5125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yue, R.; Chen, Y.; Huang, X.; Yang, M.; Shui, J.; Peng, Y. The Herbal Medicine Scutellaria-Coptis Alleviates Intestinal Mucosal Barrier Damage in Diabetic Rats by Inhibiting Inflammation and Modulating the Gut Microbiota. Evid. Based. Complement. Altern. Med. 2020, 2020, 4568629. [Google Scholar] [CrossRef]
- Nighot, M.; Ganapathy, A.S.; Saha, K.; Suchanec, E.; Castillo, E.; Gregory, A.; Shapiro, S.; Ma, T.; Nighot, P. Matrix Metalloproteinase MMP-12 promotes macrophage transmigration across intestinal epithelial tight junctions and increases severity of experimental colitis. J. Crohns Colitis 2021, 15, 1751–1765. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Cheroutre, H.; Lambolez, F.; Mucida, D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2011, 11, 445–456. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.D.; Jabri, B.; Bendelac, A. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2018, 18, 514–525. [Google Scholar] [CrossRef]
- Khan, S.; Luck, H.; Winer, S.; Winer, D.A. Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat. Commun. 2021, 12, 2598. [Google Scholar] [CrossRef]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.C.; Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 2006, 203, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Leeansyah, E.; Sandberg, J.K. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc. Natl. Acad. Sci. USA 2017, 114, E5434–E5443. [Google Scholar] [CrossRef] [Green Version]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Sandquist, I.; Kolls, J. Update on regulation and effector functions of Th17 cells. F1000Research 2018, 7, 205. [Google Scholar] [CrossRef]
- Hirota, K.; Turner, J.-E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of T H 17 cells in Peyer’s patches is responsible for the induction of T cell–dependent IgA responses. Nat. Immunol. 2013, 14, 372. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Rudra, D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef]
- Wojno, E.D.T.; Artis, D. Emerging concepts and future challenges in innate lymphoid cell biology. J. Exp. Med. 2016, 213, 2229–2248. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Eberl, G. Type 3 regulatory T cells at the interface of symbiosis. J. Microbiol. 2018, 56, 163–171. [Google Scholar] [CrossRef]
- Gautreaux, M.D.; Gelder, F.B.; Deitch, E.A.; Berg, R.D. Adoptive transfer of T lymphocytes to T-cell-depleted mice inhibits Escherichia coli translocation from the gastrointestinal tract. Infect. Immun. 1995, 63, 3827–3834. [Google Scholar] [CrossRef] [Green Version]
- Gautreaux, M.D.; Deitch, E.A.; Berg, R.D. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect. Immun. 1994, 62, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Spadoni, I.; Fornasa, G.; Rescigno, M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 2017, 17, 761–773. [Google Scholar] [CrossRef]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Alessandro, R.; Luchetti, M.M.; Milling, S.; Saieva, L.; Cypers, H.; Stampone, T.; Di Benedetto, P.; et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann. Rheum. Dis. 2017, 76, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 2014, 6, 237ra266. [Google Scholar] [CrossRef] [Green Version]
- Wood, N.J. Liver: The liver as a firewall--clearance of commensal bacteria that have escaped from the gut. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 391. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Mesmin, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Chassaing, B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 584–604. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Moriarty, T.J.; Wong, C.H.; Zhou, H.; Strieter, R.M.; van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Knook, D.L.; Barkway, C.; Sleyster, E.C. Lysosomal enzyme content of Kupffer and endothelial liver cells isolated from germfree and clean conventional rats. Infect. Immun. 1981, 33, 620–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, E.S.; Thomas, P.; Broitman, S.A. Clearance of gut-derived endotoxins by the liver. Release and modification of 3H, 14C-lipopolysaccharide by isolated rat Kupffer cells. Gastroenterology 1989, 96, 456–461. [Google Scholar] [CrossRef]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef]
- Schumann, R.R.; Kirschning, C.J.; Unbehaun, A.; Aberle, H.P.; Knope, H.P.; Lamping, N.; Ulevitch, R.J.; Herrmann, F. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol. Cell. Biol. 1996, 16, 3490–3503. [Google Scholar] [CrossRef] [Green Version]
- Pugin, J.; Schurer-Maly, C.C.; Leturcq, D.; Moriarty, A.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA 1993, 90, 2744–2748. [Google Scholar] [CrossRef] [Green Version]
- Landmann, R.; Knopf, H.P.; Link, S.; Sansano, S.; Schumann, R.; Zimmerli, W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect. Immun. 1996, 64, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Frey, E.A.; Miller, D.S.; Jahr, T.G.; Sundan, A.; Bazil, V.; Espevik, T.; Finlay, B.B.; Wright, S.D. Soluble CD14 participates in the response of cells to lipopolysaccharide. J. Exp. Med. 1992, 176, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Grover, M.; Camilleri, M.; Hines, J.; Burton, D.; Ryks, M.; Wadhwa, A.; Sundt, W.; Dyer, R.; Singh, R.J. 13C mannitol as a novel biomarker for measurement of intestinal permeability. Neurogastroenterol. Motil. 2016, 28, 1114–1119. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Nadeau, A.; Lamsam, J.; Nord, S.L.; Ryks, M.; Burton, D.; Sweetser, S.; Zinsmeister, A.R.; Singh, R. Understanding measurements of intestinal permeability in healthy humans with urine lactulose and mannitol excretion. Neurogastroenterol. Motil. 2010, 22, e15–e26. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.S.; Camilleri, M.; Eckert, D.J.; Busciglio, I.; Burton, D.D.; Ryks, M.; Wong, B.S.; Lamsam, J.; Singh, R.; Zinsmeister, A.R. Urine sugars for in vivo gut permeability: Validation and comparisons in irritable bowel syndrome-diarrhea and controls. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G919–G928. [Google Scholar] [CrossRef]
- Khoshbin, K.; Khanna, L.; Maselli, D.; Atieh, J.; Breen-Lyles, M.; Arndt, K.; Rhoten, D.; Dyer, R.B.; Singh, R.J.; Nayar, S. Development and Validation of Test for “Leaky Gut” Small Intestinal and Colonic Permeability Using Sugars in Healthy Adults. Gastroenterology 2021, 161, P463–P475. [Google Scholar] [CrossRef]
- Seethaler, B.; Basrai, M.; Neyrinck, A.M.; Nazare, J.-A.; Walter, J.; Delzenne, N.M.; Bischoff, S.C. Biomarkers for assessment of intestinal permeability in clinical practice. Am. J. Physiol.—Gastrointest. Liver Physiol. 2021, 321, G11–G17. [Google Scholar] [CrossRef]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 2020, 12, E564. [Google Scholar] [CrossRef] [Green Version]
- Suenaert, P.; Bulteel, V.; Lemmens, L.; Noman, M.; Geypens, B.; Van Assche, G.; Geboes, K.; Ceuppens, J.L.; Rutgeerts, P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 2000–2004. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Lombard, V.; Henrissat, B. Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE 2012, 7, e28742. [Google Scholar] [CrossRef] [Green Version]
- Institute of Medicine. Dietary, Functional, and Total Fiber. In Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids; National Academies Press: Washington, DC, USA, 2005. [Google Scholar]
- Soliman, G.A. Dietary Fiber, Atherosclerosis, and Cardiovascular Disease. Nutrients 2019, 11, 1155. [Google Scholar] [CrossRef] [Green Version]
- Titgemeyer, E.C.; Bourquin, L.D.; Fahey, G.C., Jr.; Garleb, K.A. Fermentability of various fiber sources by human fecal bacteria in vitro. Am. J. Clin. Nutr. 1991, 53, 1418–1424. [Google Scholar] [CrossRef]
- Swann, O.G.; Kilpatrick, M.; Breslin, M.; Oddy, W.H. Dietary fiber and its associations with depression and inflammation. Nutr. Rev. 2019, 78, 394–411. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Hytting-Andreasen, R.; Balk-Møller, E.; Hartmann, B.; Pedersen, J.; Windeløv, J.A.; Holst, J.J.; Kissow, H. Endogenous glucagon-like peptide- 1 and 2 are essential for regeneration after acute intestinal injury in mice. PLoS ONE 2018, 13, e0198046. [Google Scholar] [CrossRef]
- Maruta, K.; Takajo, T.; Akiba, Y.; Said, H.; Irie, E.; Kato, I.; Kuwahara, A.; Kaunitz, J.D. GLP-2 Acutely Prevents Endotoxin-Related Increased Intestinal Paracellular Permeability in Rats. Dig. Dis. Sci. 2020, 65, 2605–2618. [Google Scholar] [CrossRef]
- Hunt, J.E.; Hartmann, B.; Schoonjans, K.; Holst, J.J.; Kissow, H. Dietary Fiber Is Essential to Maintain Intestinal Size, L-Cell Secretion, and Intestinal Integrity in Mice. Front. Endocrinol. 2021, 12, 640602. [Google Scholar] [CrossRef]
- Genda, T.; Sasaki, Y.; Kondo, T.; Hino, S.; Nishimura, N.; Tsukahara, T.; Sonoyama, K.; Morita, T. Fructo-oligosaccharide-Induced Transient Increases in Cecal Immunoglobulin A Concentrations in Rats Are Associated with Mucosal Inflammation in Response to Increased Gut Permeability. J. Nutr. 2017, 147, 1900–1908. [Google Scholar] [CrossRef]
- Chen, T.; Ma, Y.; Xu, L.; Sun, C.; Xu, H.; Zhu, J. Soluble Dietary Fiber Reduces Feeding Intolerance in Severe Acute Pancreatitis: A Randomized Study. JPEN J. Parenter Enter. Nutr. 2021, 45, 125–135. [Google Scholar] [CrossRef]
- Wilms, E.; Gerritsen, J.; Smidt, H.; Besseling-Van Der Vaart, I.; Rijkers, G.T.; Garcia Fuentes, A.R.; Masclee, A.A.M.; Troost, F.J. Effects of Supplementation of the Synbiotic Ecologic® 825/FOS P6 on Intestinal Barrier Function in Healthy Humans: A Randomized Controlled Trial. PLoS ONE 2016, 11, e0167775. [Google Scholar] [CrossRef] [Green Version]
- Drabińska, N.; Krupa-Kozak, U.; Jarocka-Cyrta, E. Intestinal Permeability in Children with Celiac Disease after the Administration of Oligofructose-Enriched Inulin into a Gluten-Free Diet—Results of a Randomized, Placebo-Controlled, Pilot Trial. Nutrients 2020, 12, 1736. [Google Scholar] [CrossRef] [PubMed]
- Ganda Mall, J.-P.; Fart, F.; Sabet, J.A.; Lindqvist, C.M.; Nestestog, R.; Hegge, F.T.; Keita, Å.V.; Brummer, R.J.; Schoultz, I. Effects of Dietary Fibres on Acute Indomethacin-Induced Intestinal Hyperpermeability in the Elderly: A Randomised Placebo Controlled Parallel Clinical Trial. Nutrients 2020, 12, 1954. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1125–1136.e1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Rios-Covian, D.; Gueimonde, M.; Duncan, S.H.; Flint, H.J.; de los Reyes-Gavilan, C.G. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol. Lett. 2015, 362, fnv176. [Google Scholar] [CrossRef] [Green Version]
- Mahowald, M.A.; Rey, F.E.; Seedorf, H.; Turnbaugh, P.J.; Fulton, R.S.; Wollam, A.; Shah, N.; Wang, C.; Magrini, V.; Wilson, R.K.; et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. USA 2009, 106, 5859–5864. [Google Scholar] [CrossRef] [Green Version]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.K.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [Green Version]
- Zambell, K.L.; Fitch, M.D.; Fleming, S.E. Acetate and Butyrate Are the Major Substrates for De Novo Lipogenesis in Rat Colonic Epithelial Cells. J. Nutr. 2003, 133, 3509–3515. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Usuda, H.; Okamoto, T.; Wada, K. Leaky Gut: Effect of Dietary Fiber and Fats on Microbiome and Intestinal Barrier. Int. J. Mol. Sci. 2021, 22, 7613. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 190–205. [Google Scholar] [CrossRef]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [Green Version]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Huang, X.; Oshima, T.; Tomita, T.; Fukui, H.; Miwa, H. Butyrate Alleviates Cytokine-Induced Barrier Dysfunction by Modifying Claudin-2 Levels. Biology 2021, 10, 205. [Google Scholar] [CrossRef]
- Gaudier, E.; Jarry, A.; Blottiere, H.M.; de Coppet, P.; Buisine, M.P.; Aubert, J.P.; Laboisse, C.; Cherbut, C.; Hoebler, C. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1168–G1174. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Vernero, M.; De Blasio, F.; Ribaldone, D.G.; Bugianesi, E.; Pellicano, R.; Saracco, G.M.; Astegiano, M.; Caviglia, G.P. The Usefulness of Microencapsulated Sodium Butyrate Add-On Therapy in Maintaining Remission in Patients with Ulcerative Colitis: A Prospective Observational Study. J. Clin. Med. 2020, 9, 3941. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.C.; Wang, Y.; Wang, Z.B.; Liu, W.Y.; Sun, S.; Li, L.; Su, D.F.; Zhang, L.C. Propionate Ameliorates Dextran Sodium Sulfate-Induced Colitis by Improving Intestinal Barrier Function and Reducing Inflammation and Oxidative Stress. Front. Pharm. 2016, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Marchix, J.; Goddard, G.; Helmrath, M.A. Host-Gut Microbiota Crosstalk in Intestinal Adaptation. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [Green Version]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal. Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br. J. Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Suligoj, T.; Vigsnaes, L.K.; Abbeele, P.V.D.; Apostolou, A.; Karalis, K.; Savva, G.M.; McConnell, B.; Juge, N. Effects of Human Milk Oligosaccharides on the Adult Gut Microbiota and Barrier Function. Nutrients 2020, 12, 2808. [Google Scholar] [CrossRef]
- Swanson, G.R.; Siskin, J.; Gorenz, A.; Shaikh, M.; Raeisi, S.; Fogg, L.; Forsyth, C.; Keshavarzian, A. Disrupted diurnal oscillation of gut-derived Short chain fatty acids in shift workers drinking alcohol: Possible mechanism for loss of resiliency of intestinal barrier in disrupted circadian host. Transl. Res. J. Lab. Clin. Med. 2020, 221, 97–109. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Brussow, H.; Parkinson, S.J. You are what you eat. Nat. Biotechnol. 2014, 32, 243–245. [Google Scholar] [CrossRef]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.T.; Calatayud, M.; Rotsaert, C.; Seifert, N.; Richard, N.; Van den Abbeele, P.; Marzorati, M.; Steinert, R.E. Antioxidant Vitamins and Prebiotic FOS and XOS Differentially Shift Microbiota Composition and Function and Improve Intestinal Epithelial Barrier In Vitro. Nutrients 2021, 13, 1125. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Lahti, L.; Salojarvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.H.; Date, P.; Farquharson, F.; Johnstone, A.M.; Lobley, G.E.; et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.W.; Duncan, S.H.; McWilliam Leitch, E.C.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Zapata, R.C.; Pezeshki, A.; Reidelberger, R.D.; Chelikani, P.K. Inulin fiber dose-dependently modulates energy balance, glucose tolerance, gut microbiota, hormones and diet preference in high-fat-fed male rats. J. Nutr. Biochem. 2018, 59, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Wang, J.; Sailer, M.; Theis, S.; Verbeke, K.; Raes, J. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 2017, 66, 1968–1974. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Zhang, T.; Li, P.; Wu, X.; Lu, G.; Marcella, C.; Ji, X.; Ji, G.; Zhang, F. Alterations of Akkermansia muciniphila in the inflammatory bowel disease patients with washed microbiota transplantation. Appl. Microbiol. Biotechnol. 2020, 104, 10203–10215. [Google Scholar] [CrossRef] [PubMed]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Bian, X.; Wu, W.; Yang, L.; Lv, L.; Wang, Q.; Li, Y.; Ye, J.; Fang, D.; Wu, J.; Jiang, X.; et al. Administration of Akkermansia muciniphila Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Front. Microbiol. 2019, 10, 2259. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Xue, X.; Zhang, L.; Yang, X.; Zhao, L.; Zhang, C. Strain-Specific Anti-inflammatory Properties of Two Akkermansia muciniphila Strains on Chronic Colitis in Mice. Front. Cell. Infect. Microbiol. 2019, 9, 239. [Google Scholar] [CrossRef]
- Wang, L.; Tang, L.; Feng, Y.; Zhao, S.; Han, M.; Zhang, C.; Yuan, G.; Zhu, J.; Cao, S.; Wu, Q.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8(+) T cells in mice. Gut 2020, 69, 1988–1997. [Google Scholar] [CrossRef] [Green Version]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietila, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef]
- Guglielmetti, S.; Bernardi, S.; Del Bo, C.; Cherubini, A.; Porrini, M.; Gargari, G.; Hidalgo-Liberona, N.; Gonzalez-Dominguez, R.; Peron, G.; Zamora-Ros, R.; et al. Effect of a polyphenol-rich dietary pattern on intestinal permeability and gut and blood microbiomics in older subjects: Study protocol of the MaPLE randomised controlled trial. BMC Geriatr. 2020, 20, 77. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, J.; Li, D.; Ke, W.; Chen, F.; Hu, X. Targeting the gut microbiota with resveratrol: A demonstration of novel evidence for the management of hepatic steatosis. J. Nutr. Biochem. 2020, 81, 108363. [Google Scholar] [CrossRef]
- Fan, J.; Zhao, X.H.; Li, T.J. Heat treatment of galangin and kaempferol inhibits their benefits to improve barrier function in rat intestinal epithelial cells. J. Nutr. Biochem. 2021, 87, 108517. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Morales, P.; Gotteland, M. Polyphenols Protect the Epithelial Barrier Function of Caco-2 Cells Exposed to Indomethacin through the Modulation of Occludin and Zonula Occludens-1 Expression. J. Agric. Food Chem. 2013, 61, 5291–5297. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, H. Quercetin Enhances Intestinal Barrier Function through the Assembly of Zonnula Occludens-2, Occludin, and Claudin-1 and the Expression of Claudin-4 in Caco-2 Cells. J. Nutr. 2009, 139, 965–974. [Google Scholar] [CrossRef]
- Cremonini, E.; Daveri, E.; Mastaloudis, A.; Adamo, A.M.; Mills, D.; Kalanetra, K.; Hester, S.N.; Wood, S.M.; Fraga, C.G.; Oteiza, P.I. Anthocyanins protect the gastrointestinal tract from high fat diet-induced alterations in redox signaling, barrier integrity and dysbiosis. Redox Biol. 2019, 26, 101269. [Google Scholar] [CrossRef]
- Lyall, K.A.; Hurst, S.M.; Cooney, J.; Jensen, D.; Lo, K.; Hurst, R.D.; Stevenson, L.M. Short-term blackcurrant extract consumption modulates exercise-induced oxidative stress and lipopolysaccharide-stimulated inflammatory responses. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R70–R81. [Google Scholar] [CrossRef] [Green Version]
- Smeriglio, A.; Barreca, D.; Bellocco, E.; Trombetta, D. Proanthocyanidins and hydrolysable tannins: Occurrence, dietary intake and pharmacological effects. Br. J. Pharm. 2017, 174, 1244–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Long, X.; Yang, J.; Du, L.; Zhang, X.; Li, J.; Hou, C. Pomegranate peel polyphenols reduce chronic low-grade inflammatory responses by modulating gut microbiota and decreasing colonic tissue damage in rats fed a high-fat diet. Food Funct. 2019, 10, 8273–8285. [Google Scholar] [CrossRef]
- Hering, N.A.; Luettig, J.; Jebautzke, B.; Schulzke, J.D.; Rosenthal, R. The Punicalagin Metabolites Ellagic Acid and Urolithin A Exert Different Strengthening and Anti-Inflammatory Effects on Tight Junction-Mediated Intestinal Barrier Function In Vitro. Front. Pharm. 2021, 12, 610164. [Google Scholar] [CrossRef]
- Zhou, Q.; Verne, M.L.; Fields, J.Z.; Lefante, J.J.; Basra, S.; Salameh, H.; Verne, G.N. Randomised placebo-controlled trial of dietary glutamine supplements for postinfectious irritable bowel syndrome. Gut 2019, 68, 996–1002. [Google Scholar] [CrossRef]
- Benjamin, J.; Makharia, G.; Ahuja, V.; Anand Rajan, K.D.; Kalaivani, M.; Gupta, S.D.; Joshi, Y.K. Glutamine and whey protein improve intestinal permeability and morphology in patients with Crohn’s disease: A randomized controlled trial. Dig. Dis. Sci. 2012, 57, 1000–1012. [Google Scholar] [CrossRef]
- Anderson, P.M.; Lalla, R.V. Glutamine for Amelioration of Radiation and Chemotherapy Associated Mucositis during Cancer Therapy. Nutrients 2020, 12, 1675. [Google Scholar] [CrossRef] [PubMed]
- Linsalata, M.; Riezzo, G.; Orlando, A.; D’Attoma, B.; Prospero, L.; Tutino, V.; Notarnicola, M.; Russo, F. The Relationship between Low Serum Vitamin D Levels and Altered Intestinal Barrier Function in Patients with IBS Diarrhoea Undergoing a Long-Term Low-FODMAP Diet: Novel Observations from a Clinical Trial. Nutrients 2021, 13, 1011. [Google Scholar] [CrossRef] [PubMed]
- Raftery, T.; Martineau, A.R.; Greiller, C.L.; Ghosh, S.; McNamara, D.; Bennett, K.; Meddings, J.; O’Sullivan, M. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: Results from a randomised double-blind placebo-controlled study. United Eur. Gastroenterol. J. 2015, 3, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; FitzGerald, A.J.; Marchbank, T.; Ntatsaki, E.; Murray, D.; Ghosh, S.; Playford, R.J. Zinc carnosine, a health food supplement that stabilises small bowel integrity and stimulates gut repair processes. Gut 2007, 56, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M. Human Intestinal Barrier: Effects of Stressors, Diet, Prebiotics, and Probiotics. Clin. Transl. Gastroenterol. 2021, 12, e00308. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Usami, M.; Komurasaki, T.; Hanada, A.; Kinoshita, K.; Ohata, A. Effect of γ-linolenic acid or docosahexaenoic acid on tight junction permeability in intestinal monolayer cells and their mechanism by protein kinase C activation and/or eicosanoid formation. Nutrition 2003, 19, 150–156. [Google Scholar] [CrossRef]
- Usami, M.; Muraki, K.; Iwamoto, M.; Ohata, A.; Matsushita, E.; Miki, A. Effect of eicosapentaenoic acid (EPA) on tight junction permeability in intestinal monolayer cells. Clin. Nutr. 2001, 20, 351–359. [Google Scholar] [CrossRef]
- Willemsen, L.E.; Koetsier, M.A.; Balvers, M.; Beermann, C.; Stahl, B.; van Tol, E.A. Polyunsaturated fatty acids support epithelial barrier integrity and reduce IL-4 mediated permeability in vitro. Eur. J. Nutr. 2008, 47, 183–191. [Google Scholar] [CrossRef]
- Lindmark, T.; Nikkila, T.; Artursson, P. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 1995, 275, 958–964. [Google Scholar]
- Anderberg, E.K.; Lindmark, T.; Artursson, P. Sodium caprate elicits dilatations in human intestinal tight junctions and enhances drug absorption by the paracellular route. Pharm. Res. 1993, 10, 857–864. [Google Scholar] [CrossRef]
- De La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Tian, B.; Zhao, J.; Zhang, M.; Chen, Z.; Ma, Q.; Liu, H.; Nie, C.; Zhang, Z.; An, W.; Li, J. Lycium ruthenicum Anthocyanins Attenuate High-Fat Diet-Induced Colonic Barrier Dysfunction and Inflammation in Mice by Modulating the Gut Microbiota. Mol. Nutr. Food Res. 2021, 65, e2000745. [Google Scholar] [CrossRef]
- Mujawdiya, P.K.; Sharma, P.; Sharad, S.; Kapur, S. Reversal of Increase in Intestinal Permeability by Mangifera indica Seed Kernel Extract in High-Fat Diet-Induced Obese Mice. Pharmaceuticals 2020, 13, 190. [Google Scholar] [CrossRef]
- Nascimento, J.C.; Matheus, V.A.; Oliveira, R.B.; Tada, S.F.S.; Collares-Buzato, C.B. High-Fat Diet Induces Disruption of the Tight Junction-Mediated Paracellular Barrier in the Proximal Small Intestine Before the Onset of Type 2 Diabetes and Endotoxemia. Dig. Dis. Sci. 2020, 66, 3359–3374. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Yang, H.; Zhou, Y.; Tang, L. Autophagy induction by rapamycin ameliorates experimental colitis and improves intestinal epithelial barrier function in IL-10 knockout mice. Int. Immunopharmacol. 2020, 81, 105977. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Denizot, J.; Thevenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [Green Version]
- Muhomah, T.A.; Nishino, N.; Katsumata, E.; Haoming, W.; Tsuruta, T. High-fat diet reduces the level of secretory immunoglobulin A coating of commensal gut microbiota. Biosci Microbiota Food Health 2019, 38, 55–64. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Luben, R.; Shrestha, S.S.; Welch, A.; Khaw, K.T.; Hart, A.R. Dietary n-3 polyunsaturated fatty acids and the aetiology of ulcerative colitis: A UK prospective cohort study. Eur. J. Gastroenterol. Hepatol. 2010, 22, 602–606. [Google Scholar] [CrossRef]
- Schreiner, P.; Martinho-Grueber, M.; Studerus, D.; Vavricka, S.R.; Tilg, H.; Biedermann, L.; on behalf of Swiss IBDnet, an official working group of the Swiss Society of Gastroenterology. Nutrition in Inflammatory Bowel Disease. Digestion 2020, 101 (Suppl. 1), 120–135. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Chapkin, R.S.; Davidson, L.A.; Ly, L.; Weeks, B.R.; Lupton, J.R.; McMurray, D.N. Immunomodulatory effects of (n-3) fatty acids: Putative link to inflammation and colon cancer. J. Nutr. 2007, 137, 200S–204S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferrieres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef] [Green Version]
- Bowser, S.M.; McMillan, R.P.; Boutagy, N.E.; Tarpey, M.D.; Smithson, A.T.; Osterberg, K.L.; Neilson, A.P.; Davy, B.M.; Davy, K.P.; Hulver, M.W. Serum endotoxin, gut permeability and skeletal muscle metabolic adaptations following a short term high fat diet in humans. Metab. Clin. Exp. 2020, 103, 154041. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhong, Z.; Wang, B.; Xia, X.; Yao, W.; Huang, L.; Wang, Y.; Ding, W. Early-life high-fat diet-induced obesity programs hippocampal development and cognitive functions via regulation of gut commensal Akkermansia muciniphila. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2019, 44, 2054–2064. [Google Scholar] [CrossRef]
- Lassenius, M.I.; Pietilainen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjanen, J.; Forsblom, C.; Porsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Jia, L.; Vianna, C.R.; Fukuda, M.; Berglund, E.D.; Liu, C.; Tao, C.; Sun, K.; Liu, T.; Harper, M.J.; Lee, C.E.; et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 3878. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Wang, P.W.; Yang, I.H.; Huang, H.M.; Chang, C.S.; Wu, C.L.; Chuang, J.H. High-fat diet induces toll-like receptor 4-dependent macrophage/microglial cell activation and retinal impairment. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3041–3050. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Subramanian, S.; Montes, V.N.; Goodspeed, L.; Wang, S.; Han, C.; Teresa, A.S., 3rd; Kim, J.; O’Brien, K.D.; Chait, A. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced NAFLD mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, P1100–P1101. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Jeong, S.Y.; Choi, A.J.; Kim, S.J. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunol. Lett. 2015, 165, 10–19. [Google Scholar] [CrossRef]
- Shen, T.; Chen, X.; Li, Y.; Tang, X.; Jiang, X.; Yu, C.; Zheng, Y.; Guo, H.; Ling, W. Interleukin-17A exacerbates high-fat diet-induced hepatic steatosis by inhibiting fatty acid beta-oxidation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1510–1518. [Google Scholar] [CrossRef]
- Hassan, A.M.; Mancano, G.; Kashofer, K.; Frohlich, E.E.; Matak, A.; Mayerhofer, R.; Reichmann, F.; Olivares, M.; Neyrinck, A.M.; Delzenne, N.M.; et al. High-fat diet induces depression-like behaviour in mice associated with changes in microbiome, neuropeptide Y, and brain metabolome. Nutr. Neurosci. 2019, 22, 877–893. [Google Scholar] [CrossRef] [Green Version]
- Munch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, S.; Avila-Pacheco, J.; Soto, M.; Kostic, A.; Dreyfuss, J.M.; Pan, H.; Ussar, S.; Altindis, E.; Li, N.; Bry, L.; et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018, 22, 3072–3086. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P.J. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe 2019, 26, 265–272. [Google Scholar] [CrossRef]
- Wei, L.; Yue, F.; Xing, L.; Wu, S.; Shi, Y.; Li, J.; Xiang, X.; Lam, S.M.; Shui, G.; Russell, R.; et al. Constant Light Exposure Alters Gut Microbiota and Promotes the Progression of Steatohepatitis in High Fat Diet Rats. Front. Microbiol. 2020, 11, 1975. [Google Scholar] [CrossRef]
- Ashrafian, F.; Shahriary, A.; Behrouzi, A.; Moradi, H.R.; Keshavarz Azizi Raftar, S.; Lari, A.; Hadifar, S.; Yaghoubfar, R.; Ahmadi Badi, S.; Khatami, S.; et al. Akkermansia muciniphila-Derived Extracellular Vesicles as a Mucosal Delivery Vector for Amelioration of Obesity in Mice. Front. Microbiol. 2019, 10, 2155. [Google Scholar] [CrossRef]
- Chelakkot, C.; Choi, Y.; Kim, D.K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.S.; Jee, Y.K.; Gho, Y.S.; et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef] [PubMed]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Tanabe, S.; Suzuki, T. High-fat Diet-induced Intestinal Hyperpermeability is Associated with Increased Bile Acids in the Large Intestine of Mice. J. Food Sci. 2016, 81, H216–H222. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Pathak, P.; Liu, H.; Donepudi, A.; Ferrell, J.; Boehme, S. Intestinal Farnesoid X Receptor and Takeda G Protein Couple Receptor 5 Signaling in Metabolic Regulation. Dig. Dis. 2017, 35, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchiano, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Huang, M.; Kong, B.; Zhang, M.; Rizzolo, D.; Armstrong, L.E.; Schumacher, J.D.; Chow, M.D.; Lee, Y.H.; Joseph, L.B.; Stofan, M.; et al. Enhanced alcoholic liver disease in mice with intestine-specific farnesoid X receptor deficiency. Lab. Invest. 2020, 100, 1158–1168. [Google Scholar] [CrossRef]
- Glade, M.J.; Meguid, M.M. A glance at... dietary emulsifiers, the human intestinal mucus and microbiome, and dietary fiber. Nutrition 2016, 32, 609–614. [Google Scholar] [CrossRef]
- Lock, J.Y.; Carlson, T.L.; Wang, C.M.; Chen, A.; Carrier, R.L. Acute Exposure to Commonly Ingested Emulsifiers Alters Intestinal Mucus Structure and Transport Properties. Sci. Rep. 2018, 8, 10008. [Google Scholar] [CrossRef]
- Svolos, V.; Hansen, R.; Nichols, B.; Quince, C.; Ijaz, U.Z.; Papadopoulou, R.T.; Edwards, C.A.; Watson, D.; Alghamdi, A.; Brejnrod, A. Treatment of active Crohn’s disease with an ordinary food-based diet that replicates exclusive enteral nutrition. Gastroenterology 2019, 156, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.; Wine, E.; Assa, A.; Sigall Boneh, R.; Shaoul, R.; Kori, M.; Cohen, S.; Peleg, S.; Shamaly, H.; On, A.; et al. Crohn’s Disease Exclusion Diet Plus Partial Enteral Nutrition Induces Sustained Remission in a Randomized Controlled Trial. Gastroenterology 2019, 157, 440–450.e448. [Google Scholar] [CrossRef] [Green Version]
- Sigall-Boneh, R.; Pfeffer-Gik, T.; Segal, I.; Zangen, T.; Boaz, M.; Levine, A. Partial enteral nutrition with a Crohn’s disease exclusion diet is effective for induction of remission in children and young adults with Crohn’s disease. Inflamm. Bowel. Dis. 2014, 20, 1353–1360. [Google Scholar] [CrossRef]
- Sandall, A.M.; Cox, S.R.; Lindsay, J.O.; Gewirtz, A.T.; Chassaing, B.; Rossi, M.; Whelan, K. Emulsifiers Impact Colonic Length in Mice and Emulsifier Restriction is Feasible in People with Crohn’s Disease. Nutrients 2020, 12, 2827. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Shumard, T.; Xie, H.; Dodda, A.; Varady, K.A.; Feferman, L.; Halline, A.G.; Goldstein, J.L.; Hanauer, S.B.; Tobacman, J.K. A randomized trial of the effects of the no-carrageenan diet on ulcerative colitis disease activity. Nutr. Healthy Aging 2017, 4, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Starkel, P.; Leclercq, S.; de Timary, P.; Schnabl, B. Intestinal dysbiosis and permeability: The yin and yang in alcohol dependence and alcoholic liver disease. Clin. Sci. 2018, 132, 199–212. [Google Scholar] [CrossRef]
- Palasciano, G.; Portincasa, P.; Di Ciaula, A.; Palmieri, V. Prolonged consumption of moderate doses of alcohol and in vitro gastro-duodenal and ileal contractility in the rat. Eur. J. Clin. Investig. 1995, 25, 171–175. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Grattagliano, I.; Portincasa, P. Chronic alcoholics retain dyspeptic symptoms, pan-enteric dysmotility, and autonomic neuropathy before and after abstinence. J. Dig. Dis. 2016, 17, 735–746. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, J.; Chang, B.; Wang, B.; Zhang, D.; Wang, B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol. Med. Rep. 2014, 9, 2352–2356. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Gottfried, E.B.; Korsten, M.A.; Lieber, C.S. Alcohol-induced gastric and duodenal lesions in man. Am. J. Gastroenterol. 1978, 70, 587–592. [Google Scholar]
- Brozinsky, S.; Fani, K.; Grosberg, S.J.; Wapnick, S. Alcohol ingestion-induced changes in the human rectal mucosa: Light and electron microscopic studies. Dis. Colon. Rectum. 1978, 21, 329–335. [Google Scholar] [CrossRef]
- Elamin, E.; Masclee, A.; Troost, F.; Pieters, H.-J.; Keszthelyi, D.; Aleksa, K.; Dekker, J.; Jonkers, D. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: A combined in vivo and in vitro approach. PLoS ONE 2014, 9, e107421. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef]
- Lang, S.; Duan, Y.; Liu, J.; Torralba, M.G.; Kuelbs, C.; Ventura-Cots, M.; Abraldes, J.G.; Bosques-Padilla, F.; Verna, E.C.; Brown, R.S., Jr.; et al. Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients With Alcoholic Hepatitis. Hepatology 2020, 71, 522–538. [Google Scholar] [CrossRef]
- Parlesak, A.; Schafer, C.; Schutz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky gut in alcoholic cirrhosis: A possible mechanism for alcohol-induced liver damage. Am. J. Gastroenterol. 1999, 94, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Peters, T.J.; Wise, R.J. The leaky gut of alcoholism: Possible route of entry for toxic compounds. Lancet 1984, 1, 179–182. [Google Scholar] [CrossRef]
- González-Muniesa, P.; Mártinez-González, M.A.; Hu, F.B.; Després, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- Collaboration, N.C.D.R.F. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- The Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: A pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet 2013, 383, P970–P983. [Google Scholar] [CrossRef] [Green Version]
- Faienza, M.F.; Chiarito, M.; Molina-Molina, E.; Shanmugam, H.; Lammert, F.; Krawczyk, M.; D’Amato, G.; Portincasa, P. Childhood obesity, cardiovascular and liver health: A growing epidemic with age. World J. Pediatrics 2020, 16, 438–445. [Google Scholar] [CrossRef]
- Faienza, M.F.; Wang, D.Q.H.; Frühbeck, G.; Garruti, G.; Portincasa, P. The dangerous link between childhood and adulthood predictors of obesity and metabolic syndrome. Intern. Emerg. Med. 2016, 11, 175–182. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef] [Green Version]
- De Meyts, P.; Delzenne, N. Editorial: The Brain—Gut—Microbiome Network in Metabolic Regulation and Dysregulation. Front. Endocrinol. 2021, 12, 760558. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Overweight and Obesity: Adult Obesity Facts. Available online: https://www.cdc.gov/obesity/data/adult.html (accessed on 28 August 2021).
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018. NCHS Data Brief. 2020, 360, 1–8. [Google Scholar]
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. New Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Baldini, F.; Fabbri, R.; Eberhagen, C.; Voci, A.; Portincasa, P.; Zischka, H.; Vergani, L. Adipocyte hypertrophy parallels alterations of mitochondrial status in a cell model for adipose tissue dysfunction in obesity. Life Sci. 2021, 265, 118812. [Google Scholar] [CrossRef]
- Grattagliano, I.; Di Ciaula, A.; Baj, J.; Molina-Molina, E.; Shanmugam, H.; Garruti, G.; Wang, D.Q.; Portincasa, P. Protocols for Mitochondria as the Target of Pharmacological Therapy in the Context of Nonalcoholic Fatty Liver Disease (NAFLD). Methods Mol. Biol. 2021, 2310, 201–246. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Grasselli, E.; Baldini, F.; Vecchione, G.; Oliveira, P.J.; Sardao, V.A.; Voci, A.; Portincasa, P.; Vergani, L. Excess fructose and fatty acids trigger a model of nonalcoholic fatty liver disease progression in vitro: Protective effect of the flavonoid silybin. Int. J. Mol. Med. 2019, 44, 705–712. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Maurice, J.; Manousou, P. Non-alcoholic fatty liver disease. Clin. Med. 2018, 18, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Dang, K.; Ladhani, S.; Singal, A.K.; Wong, R.J. Prevalence of Alcoholic Fatty Liver Disease Among Adults in the United States, 2001–2016. JAMA 2019, 321, 1723–1725. [Google Scholar] [CrossRef] [Green Version]
- Li, J.F.; Qu, F.; Zheng, S.J.; Wu, H.L.; Liu, M.; Liu, S.; Ren, Y.; Ren, F.; Chen, Y.; Duan, Z.P.; et al. Elevated plasma sphingomyelin (d18:1/22:0) is closely related to hepatic steatosis in patients with chronic hepatitis C virus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1725–1732. [Google Scholar] [CrossRef]
- Yasui, K.; Harano, Y.; Mitsuyoshi, H.; Tsuji, K.; Endo, M.; Nakajima, T.; Minami, M.; Itoh, Y.; Zen, Y.; Nakanuma, Y.; et al. Steatosis and hepatic expression of genes regulating lipid metabolism in Japanese patients infected with hepatitis C virus. J. Gastroenterol. 2010, 45, 95–104. [Google Scholar] [CrossRef]
- Jian Wu, Y.; Shu Chen, L.; Gui Qiang, W. Effects of fatty liver and related factors on the efficacy of combination antiviral therapy in patients with chronic hepatitis C. Liver Int. 2006, 26, 166–172. [Google Scholar] [CrossRef]
- Hwang, S.J.; Luo, J.C.; Chu, C.W.; Lai, C.R.; Lu, C.L.; Tsay, S.H.; Wu, J.C.; Chang, F.Y.; Lee, S.D. Hepatic steatosis in chronic hepatitis C virus infection: Prevalence and clinical correlation. J. Gastroenterol. Hepatol. 2001, 16, 190–195. [Google Scholar] [CrossRef]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Stattermayer, A.F.; Traussnigg, S.; Dienes, H.P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef]
- Jordan, T.; Popovic, P.; Rotovnik Kozjek, N. Liver steatosis in adult patients on home parenteral nutrition. Eur. J. Clin. Nutr. 2020, 74, 255–260. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Kuwajima, V.; Nadelson, J.; Atiq, O.; Sanyal, A.J. Drug-induced fatty liver disease: An overview of pathogenesis and management. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef]
- Liu, J.; Ghaziani, T.T.; Wolf, J.L. Acute Fatty Liver Disease of Pregnancy: Updates in Pathogenesis, Diagnosis, and Management. Am. J. Gastroenterol. 2017, 112, 838–846. [Google Scholar] [CrossRef]
- Chapman, J.; Arnold, J.K. Reye Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Soullane, S.; Lee, G.E.; Auger, N. Perinatal Risk Factors for Pediatric Nonalcoholic Fatty Liver Disease: Impact of Inborn Errors of Metabolism. Clin. Gastroenterol. Hepatol. 2021, in press. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.E9. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. Mayo Clin. 1980, 55, 434–438. [Google Scholar]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e121. [Google Scholar] [CrossRef] [Green Version]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Torbenson, M.S.; Yeh, M.M. Steatohepatitic hepatocellular carcinoma. Hepatoma Res. 2021, 7, 38. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988-1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.E1. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-Alcoholic Fatty Liver Disease in Non-Obese Individuals: Prevalence, Pathogenesis and Treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, F.; Wang, W.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Zhu, L.; Cai, J.; Li, H. Epidemiological Features of NAFLD From 1999 to 2018 in China. Hepatology 2020, 71, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Yoshitaka, H.; Hamaguchi, M.; Kojima, T.; Fukuda, T.; Ohbora, A.; Fukui, M. Nonoverweight nonalcoholic fatty liver disease and incident cardiovascular disease: A post hoc analysis of a cohort study. Medicine 2017, 96, e6712. [Google Scholar] [CrossRef]
- Palmentieri, B.; de Sio, I.; La Mura, V.; Masarone, M.; Vecchione, R.; Bruno, S.; Torella, R.; Persico, M. The role of bright liver echo pattern on ultrasound B-mode examination in the diagnosis of liver steatosis. Dig. Liver Dis. 2006, 38, 485–489. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease-An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Caussy, C.; Soni, M.; Cui, J.; Bettencourt, R.; Schork, N.; Chen, C.H.; Ikhwan, M.A.; Bassirian, S.; Cepin, S.; Gonzalez, M.P.; et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Investig. 2017, 127, 2697–2704. [Google Scholar] [CrossRef] [Green Version]
- Stender, S.; Loomba, R. PNPLA3 Genotype and Risk of Liver and All-Cause Mortality. Hepatology 2020, 71, 777–779. [Google Scholar] [CrossRef]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Kaser, S.; Tilg, H. Non-alcoholic steatohepatitis: A microbiota-driven disease. Trends Endocrinol. Metab. 2013, 24, 537–545. [Google Scholar] [CrossRef]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Díaz-Orozco, L.; Córdova-Gallardo, J. Redefinition of fatty liver disease from NAFLD to MAFLD raised disease awareness: Mexican experience. J. Hepatol. 2021, 75, 221–222. [Google Scholar] [CrossRef]
- Nan, Y.; An, J.; Bao, J.; Chen, H.; Chen, Y.; Ding, H.; Dou, X.; Duan, Z.; Fan, J.; Gao, Y.; et al. The Chinese Society of Hepatology position statement on the redefinition of fatty liver disease. J. Hepatol. 2021, 75, P454–P461. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.; Fan, J.G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919. [Google Scholar] [CrossRef]
- Shiha, G.; Korenjak, M.; Eskridge, W.; Casanovas, T.; Velez-Moller, P.; Hogstrom, S.; Richardson, B.; Munoz, C.; Sigurethardottir, S.; Coulibaly, A.; et al. Redefining fatty liver disease: An international patient perspective. Lancet. Gastroenterol. Hepatol. 2021, 6, 73–79. [Google Scholar] [CrossRef]
- Shiha, G.; Alswat, K.; Al Khatry, M.; Sharara, A.I.; Ormeci, N.; Waked, I.; Benazzouz, M.; Al-Ali, F.; Hamed, A.E.; Hamoudi, W.; et al. Nomenclature and definition of metabolic-associated fatty liver disease: A consensus from the Middle East and north Africa. Lancet. Gastroenterol. Hepatol. 2021, 6, 57–64. [Google Scholar] [CrossRef]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Carbone, F.; Shanmugham, H.; Molina-Molina, E.; Bonfrate, L.; Ministrini, S.; Montecucco, F.; Portincasa, P. Adiponectin involved in portal flow hepatic extraction of 13C-metacethin in obesity and non-alcoholic fatty liver. Eur. J. Intern. Med. 2021, 89, P56–P64. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Lammert, F.; Thompson, R.J. Genetics of liver disease: From pathophysiology to clinical practice. J. Hepatol. 2015, 62, S6–S14. [Google Scholar] [CrossRef] [Green Version]
- Albillos, A.; Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2019, 72, P558–P577. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Yoo, E.R.; Li, A.A.; Cholankeril, G.; Tighe, S.P.; Kim, W.; Harrison, S.A.; Ahmed, A. Elevated urinary bisphenol A levels are associated with non-alcoholic fatty liver disease among adults in the United States. Liver Int. 2019, 39, 1335–1342. [Google Scholar] [CrossRef]
- Franco, M.E.; Fernandez-Luna, M.T.; Ramirez, A.J.; Lavado, R. Metabolomic-based assessment reveals dysregulation of lipid profiles in human liver cells exposed to environmental obesogens. Toxicol. Appl. Pharmacol. 2020, 398, 115009. [Google Scholar] [CrossRef]
- Wahlang, B.; Appana, S.; Falkner, K.C.; McClain, C.J.; Brock, G.; Cave, M.C. Insecticide and metal exposures are associated with a surrogate biomarker for non-alcoholic fatty liver disease in the National Health and Nutrition Examination Survey 2003-2004. Environ. Sci. Pollut. Res. Int. 2020, 27, 6476–6487. [Google Scholar] [CrossRef]
- Milosevic, N.; Milanovic, M.; Sudji, J.; Bosic Zivanovic, D.; Stojanoski, S.; Vukovic, B.; Milic, N.; Medic Stojanoska, M. Could phthalates exposure contribute to the development of metabolic syndrome and liver disease in humans? Environ. Sci. Pollut. Res. Int. 2020, 27, 772–784. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Zhu, P.; Wu, Y.; Jin, Y.; Yu, S.; Wei, H.; Qian, M.; Cao, W.; Xu, S.; et al. Prenatal exposure to diesel exhaust PM2.5 programmed non-alcoholic fatty liver disease differently in adult male offspring of mice fed normal chow and a high-fat diet. Environ. Pollut. 2019, 255, 113366. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xu, Y.; Xu, C.; Shu, Y.; Ma, S.; Lu, C.; Mo, X. Associations between mercury exposure and the risk of nonalcoholic fatty liver disease (NAFLD) in US adolescents. Environ. Sci. Pollut. Res. Int. 2019, 26, 31384–31391. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Yuan, C.; Si, B.; Wang, M.; Da, S.; Bai, L.; Wu, W. Combined effects of ambient particulate matter exposure and a high-fat diet on oxidative stress and steatohepatitis in mice. PLoS ONE 2019, 14, e0214680. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.X.; Ge, C.X.; Qin, Y.T.; Gu, T.T.; Lou, D.S.; Li, Q.; Hu, L.F.; Feng, J.; Huang, P.; Tan, J. Prolonged PM2.5 exposure elevates risk of oxidative stress-driven nonalcoholic fatty liver disease by triggering increase of dyslipidemia. Free Radic. Biol. Med. 2019, 130, 542–556. [Google Scholar] [CrossRef]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef] [Green Version]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of faecal Bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef] [Green Version]
- Pozo-Rubio, T.; Mujico, J.R.; Marcos, A.; Puertollano, E.; Nadal, I.; Sanz, Y.; Nova, E. Immunostimulatory effect of faecal Bifidobacterium species of breast-fed and formula-fed infants in a peripheral blood mononuclear cell/Caco-2 co-culture system. Br. J. Nutr. 2011, 106, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Benno, Y.; Sawada, K.; Mitsuoka, T. The intestinal microflora of infants: Composition of fecal flora in breast-fed and bottle-fed infants. Microbiol. Immunol. 1984, 28, 975–986. [Google Scholar] [CrossRef]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Cholesterol lowering and inhibition of sterol absorption by Lactobacillus reuteri NCIMB 30242: A randomized controlled trial. Eur. J. Clin. Nutr. 2012, 66, 1234–1241. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.R.; Probert, H.M.; Loo, J.V.; Rastall, R.A.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Updating the concept of prebiotics. Nutr. Res. Rev. 2004, 17, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.T.; Steed, H.; Macfarlane, S. Bacterial metabolism and health-related effects of galacto-oligosaccharides and other prebiotics. J. Appl. Microbiol. 2008, 104, 305–344. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, J.; Lange, B.; Frick, J.S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef]
- Kim, M.S.; Hwang, S.S.; Park, E.J.; Bae, J.W. Strict vegetarian diet improves the risk factors associated with metabolic diseases by modulating gut microbiota and reducing intestinal inflammation. Environ. Microbiol. Rep. 2013, 5, 765–775. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Chen, D.; Yang, Z.; Chen, X.; Huang, Y.; Yin, B.; Guo, F.; Zhao, H.; Huang, J.; Wu, Y.; Gu, R. Effect of Lactobacillus rhamnosus hsryfm 1301 on the Gut Microbiota and Lipid Metabolism in Rats Fed a High-Fat Diet. J. Microbiol. Biotechnol. 2015, 25, 687–695. [Google Scholar] [CrossRef]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef]
- Machado, M.V.; Cortez-Pinto, H. Diet, Microbiota, Obesity, and NAFLD: A Dangerous Quartet. Int. J. Mol. Sci. 2016, 17, 481. [Google Scholar] [CrossRef] [Green Version]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Moreira, G.V.; Azevedo, F.F.; Ribeiro, L.M.; Santos, A.; Guadagnini, D.; Gama, P.; Liberti, E.A.; Saad, M.; Carvalho, C. Liraglutide modulates gut microbiota and reduces NAFLD in obese mice. J. Nutr. Biochem. 2018, 62, 143–154. [Google Scholar] [CrossRef]
- Brandt, A.; Hernandez-Arriaga, A.; Kehm, R.; Sanchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Wang, H.; Zhang, P.; Gao, C.; Tao, J.; Ge, Z.; Zhu, D.; Bi, Y. Modulation of gut microbiota contributes to curcumin-mediated attenuation of hepatic steatosis in rats. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Chi, L.; Mahbub, R.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Multi-Omics Reveals that Lead Exposure Disturbs Gut Microbiome Development, Key Metabolites, and Metabolic Pathways. Chem. Res. Toxicol. 2017, 30, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Bian, X.; Mahbub, R.; Lu, K. Sex-Specific Effects of Organophosphate Diazinon on the Gut Microbiome and Its Metabolic Functions. Environ. Health Perspect. 2017, 125, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Joly, C.; Gay-Queheillard, J.; Leke, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 2013, 20, 2726–2734. [Google Scholar] [CrossRef]
- Joly Condette, C.; Bach, V.; Mayeur, C.; Gay-Queheillard, J.; Khorsi-Cauet, H. Chlorpyrifos Exposure During Perinatal Period Affects Intestinal Microbiota Associated With Delay of Maturation of Digestive Tract in Rats. J. Pediatric Gastroenterol. Nutr. 2015, 61, 30–40. [Google Scholar] [CrossRef]
- Wahlang, B.; Jin, J.; Beier, J.I.; Hardesty, J.E.; Daly, E.F.; Schnegelberger, R.D.; Falkner, K.C.; Prough, R.A.; Kirpich, I.A.; Cave, M.C. Mechanisms of Environmental Contributions to Fatty Liver Disease. Curr. Environ. Health Rep. 2019, 6, 80–94. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e27. [Google Scholar] [CrossRef] [Green Version]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef] [Green Version]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [Green Version]
- Ray, K. NAFLD. Leaky guts: Intestinal permeability and NASH. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 123. [Google Scholar] [CrossRef]
- Miele, L.; Marrone, G.; Lauritano, C.; Cefalo, C.; Gasbarrini, A.; Day, C.; Grieco, A. Gut-liver axis and microbiota in NAFLD: Insight pathophysiology for novel therapeutic target. Curr. Pharm. Des. 2013, 19, 5314–5324. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.; Wylie, A.T.; Tucker, K.L.; Hamp, T.J.; Gharaibeh, R.Z.; Fodor, A.A.; Cullen, J.M. Dietary fructose induces endotoxemia and hepatic injury in calorically controlled primates. Am. J. Clin. Nutr. 2013, 98, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e712. [Google Scholar] [CrossRef] [Green Version]
- Bluemel, S.; Wang, L.; Martino, C.; Lee, S.; Wang, Y.; Williams, B.; Horvath, A.; Stadlbauer, V.; Zengler, K.; Schnabl, B. The Role of Intestinal C-type Regenerating Islet Derived-3 Lectins for Nonalcoholic Steatohepatitis. Hepatol. Commun. 2018, 2, 393–406. [Google Scholar] [CrossRef]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int. J. Hepatol. 2014, 2014, 560620. [Google Scholar] [CrossRef]
- Bifulco, M. Mediterranean diet: The missing link between gut microbiota and inflammatory diseases. Eur. J. Clin. Nutr. 2015, 69, 1078. [Google Scholar] [CrossRef] [Green Version]
- Biolato, M.; Manca, F.; Marrone, G.; Cefalo, C.; Racco, S.; Miggiano, G.A.; Valenza, V.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability after Mediterranean diet and low-fat diet in non-alcoholic fatty liver disease. World J. Gastroenterol. 2019, 25, 509–520. [Google Scholar] [CrossRef]
- Enomoto, N.; Yamashina, S.; Kono, H.; Schemmer, P.; Rivera, C.A.; Enomoto, A.; Nishiura, T.; Nishimura, T.; Brenner, D.A.; Thurman, R.G. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology 1999, 29, 1680–1689. [Google Scholar] [CrossRef]
- Pappo, I.; Bercovier, H.; Berry, E.M.; Haviv, Y.; Gallily, R.; Freund, H.R. Polymyxin B reduces total parenteral nutrition-associated hepatic steatosis by its antibacterial activity and by blocking deleterious effects of lipopolysaccharide. JPEN J. Parenter Enter. Nutr. 1992, 16, 529–532. [Google Scholar] [CrossRef]
- Pappo, I.; Becovier, H.; Berry, E.M.; Freund, H.R. Polymyxin B reduces cecal flora, TNF production and hepatic steatosis during total parenteral nutrition in the rat. J. Surg. Res. 1991, 51, 106–112. [Google Scholar] [CrossRef]
- Drenick, E.J.; Fisler, J.; Johnson, D. Hepatic steatosis after intestinal bypass--prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology 1982, 82, 535–548. [Google Scholar] [CrossRef]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuny, F.; Richet, H.; Casalta, J.P.; Angelakis, E.; Habib, G.; Raoult, D. Vancomycin treatment of infective endocarditis is linked with recently acquired obesity. PLoS ONE 2010, 5, e9074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saari, A.; Virta, L.J.; Sankilampi, U.; Dunkel, L.; Saxen, H. Antibiotic exposure in infancy and risk of being overweight in the first 24 months of life. Pediatrics 2015, 135, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014, 158, 705–721. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Cox, L.M.; Blaser, M.J. Pathways in microbe-induced obesity. Cell Metab. 2013, 17, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Liou, A.P.; Paziuk, M.; Luevano, J.-M.; Machineni, S.; Turnbaugh, P.J.; Kaplan, L.M. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 2013, 5, ra141–ra178. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.; Bell, R.; Klag, K.A.; Lee, S.-H.; Soto, R.; Ghazaryan, A.; Buhrke, K.; Ekiz, H.A.; Ost, K.S.; Boudina, S. T cell–mediated regulation of the microbiota protects against obesity. Science 2019, 365, eaat9351. [Google Scholar] [CrossRef]
- Luck, H.; Khan, S.; Kim, J.H.; Copeland, J.K.; Revelo, X.S.; Tsai, S.; Chakraborty, M.; Cheng, K.; Chan, Y.T.; Nøhr, M.K. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat. Commun. 2019, 10, 3650. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Fernandes, J.; Su, W.; Rahat-Rozenbloom, S.; Wolever, T.M.; Comelli, E.M. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr. Diabetes 2014, 4, e121. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef]
- Yamashita, H.; Fujisawa, K.; Ito, E.; Idei, S.; Kawaguchi, N.; Kimoto, M.; Hiemori, M.; Tsuji, H. Improvement of obesity and glucose tolerance by acetate in Type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Biosci. Biotechnol. Biochem. 2007, 71, 1236–1243. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Inoue, D.; Hirano, K.; Tsujimoto, G. The SCFA Receptor GPR43 and Energy Metabolism. Front. Endocrinol. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.S.; Morrison, D.J.; Frost, G. Control of appetite and energy intake by SCFA: What are the potential underlying mechanisms? Proc. Nutr. Soc. 2015, 74, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Kaji, I.; Karaki, S.; Kuwahara, A. Short-chain fatty acid receptor and its contribution to glucagon-like peptide-1 release. Digestion 2014, 89, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Conterno, L.; Fava, F.; Viola, R.; Tuohy, K.M. Obesity and the gut microbiota: Does up-regulating colonic fermentation protect against obesity and metabolic disease? Genes Nutr. 2011, 6, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher f, M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E740–E747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannides, I.; Pothoulakis, C. Obesity, innate immunity and gut inflammation. Curr. Opin. Gastroenterol. 2007, 23, 661–666. [Google Scholar] [CrossRef]
- Kim, S.J.; Choi, Y.; Choi, Y.H.; Park, T. Obesity activates toll-like receptor-mediated proinflammatory signaling cascades in the adipose tissue of mice. J. Nutr. Biochem. 2012, 23, 113–122. [Google Scholar] [CrossRef]
- Ye, D.; Li, F.Y.; Lam, K.S.; Li, H.; Jia, W.; Wang, Y.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012, 61, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Rodes, L.; Khan, A.; Paul, A.; Coussa-Charley, M.; Marinescu, D.; Tomaro-Duchesneau, C.; Shao, W.; Kahouli, I.; Prakash, S. Effect of probiotics Lactobacillus and Bifidobacterium on gut-derived lipopolysaccharides and inflammatory cytokines: An in vitro study using a human colonic microbiota model. J. Microbiol. Biotechnol. 2013, 23, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, M.; Iborra, S.; Conde-Garrosa, R.; Mastrangelo, A.; Danne, C.; Mann, E.R.; Reid, D.M.; Gaboriau-Routhiau, V.; Chaparro, M.; Lorenzo, M.P.; et al. Microbiota Sensing by Mincle-Syk Axis in Dendritic Cells Regulates Interleukin-17 and -22 Production and Promotes Intestinal Barrier Integrity. Immunity 2019, 50, 446–461.e449. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef]
- Yusta, B.; Baggio, L.L.; Koehler, J.; Holland, D.; Cao, X.; Pinnell, L.J.; Johnson-Henry, K.C.; Yeung, W.; Surette, M.G.; Bang, K.A. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes 2015, 64, 2537–2549. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; Winer, S.; Winer, D.A. Gut T cells feast on GLP-1 to modulate cardiometabolic disease. Cell Metab. 2019, 29, 787–789. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Seier Poulsen, S.; Han, S. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564. [Google Scholar] [CrossRef]
- Tai, N.; Wong, F.S.; Wen, L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev. Endocr. Metab. Disord. 2015, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front. Endocrinol. 2012, 3, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dali-Youcef, N.; Mecili, M.; Ricci, R.; Andres, E. Metabolic inflammation: Connecting obesity and insulin resistance. Ann. Med. 2013, 45, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Amyot, J.; Semache, M.; Ferdaoussi, M.; Fontes, G.; Poitout, V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-Like Receptor-4 and NF-kappaB signalling. PLoS ONE 2012, 7, e36200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut Dysfunction and Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, C.H.; Schiffer, T.A.; Li, X.; Weitzberg, E.; Carlstrom, M.; Lundberg, J.O. Germ-free mice are not protected against diet-induced obesity and metabolic dysfunction. Acta Physiol. 2021, 231, e13581. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062. [Google Scholar] [CrossRef]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Pihlajamaki, J.; Kuulasmaa, T.; Kaminska, D.; Simonen, M.; Karja, V.; Gronlund, S.; Kakela, P.; Paakkonen, M.; Kainulainen, S.; Punnonen, K.; et al. Serum interleukin 1 receptor antagonist as an independent marker of non-alcoholic steatohepatitis in humans. J. Hepatol. 2012, 56, 663–670. [Google Scholar] [CrossRef]
- Zimmermann, E.; Anty, R.; Tordjman, J.; Verrijken, A.; Gual, P.; Tran, A.; Iannelli, A.; Gugenheim, J.; Bedossa, P.; Francque, S.; et al. C-reactive protein levels in relation to various features of non-alcoholic fatty liver disease among obese patients. J. Hepatol. 2011, 55, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.H.; Huang, C.C.; Chan, W.L.; Chen, J.W.; Leu, H.B. The severity of non-alcoholic fatty liver disease correlates with high sensitivity C-reactive protein value and is independently associated with increased cardiovascular risk in healthy population. Clin. Biochem. 2010, 43, 1399–1404. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Damas, J.K.; Konopski, Z.; Loberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjoro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Machado, M.V.; Diehl, A.M. Fibrosis in nonalcoholic Fatty liver disease: Mechanisms and clinical implications. Semin. Liver Dis. 2015, 35, 132–145. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Moschen, A.R.; Molnar, C.; Enrich, B.; Geiger, S.; Ebenbichler, C.F.; Tilg, H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 2011, 17, 840–845. [Google Scholar] [CrossRef]
- Ballak, D.B.; van Diepen, J.A.; Moschen, A.R.; Jansen, H.J.; Hijmans, A.; Groenhof, G.J.; Leenders, F.; Bufler, P.; Boekschoten, M.V.; Muller, M.; et al. IL-37 protects against obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 4711. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Shanmugam, H.; Di Ciaula, A.; Grattagliano, I.; Di Palo, D.M.; Palmieri, V.O.; Portincasa, P. ((13)C)-Methacetin breath test provides evidence of subclinical liver dysfunction linked to fat storage but not lifestyle. JHEP Rep. 2021, 3, 100203. [Google Scholar] [CrossRef]
- Serino, M.; Luche, E.; Chabo, C.; Amar, J.; Burcelin, R. Intestinal microflora and metabolic diseases. Diabetes Metab. 2009, 35, 262–272. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Janssen, A.W.F.; Houben, T.; Katiraei, S.; Dijk, W.; Boutens, L.; van der Bolt, N.; Wang, Z.; Brown, J.M.; Hazen, S.L.; Mandard, S.; et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: A potential role for bile acids. J. Lipid Res. 2017, 58, 1399–1416. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lezana, T.; Raurell, I.; Bravo, M.; Torres-Arauz, M.; Salcedo, M.T.; Santiago, A.; Schoenenberger, A.; Manichanh, C.; Genesca, J.; Martell, M.; et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018, 67, 1485–1498. [Google Scholar] [CrossRef]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [Green Version]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.K.; Gwak, G.Y.; Choi, M.S.; Koh, K.C.; Paik, S.W.; Yoo, B.C.; Lee, J.H. A case of nonalcoholic steatohepatitis and small intestinal bacterial overgrowth with peripheral edema caused by intestinal bypass surgery and relieved by repair. Gut Liver 2012, 6, 520–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanab, A.A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: Association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig. Dis. Sci. 2011, 56, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1466. [Google Scholar] [CrossRef] [Green Version]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019, 68, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Lelouvier, B.; Servant, F.; Paisse, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef]
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 5688. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Nwe, P.K.; Yang, Y.; Rosen, C.E.; Bielecka, A.A.; Kuchroo, M.; Cline, G.W.; Kruse, A.C.; Ring, A.M.; Crawford, J.M.; et al. A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 2019, 177, 1217–1231.e1218. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Cohen, L.J.; Esterhazy, D.; Kim, S.H.; Lemetre, C.; Aguilar, R.R.; Gordon, E.A.; Pickard, A.J.; Cross, J.R.; Emiliano, A.B.; Han, S.M.; et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 2017, 549, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.S.; Lee, F.Y.; Lee, S.D.; Tsai, Y.T.; Lin, H.C.; Lu, R.H.; Hsu, W.C.; Huang, C.C.; Wang, S.S.; Lo, K.J. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol. 1995, 22, 165–172. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Lee, F.Y.; Barden, G.E.; Cartun, R.; West, A.B. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology 1995, 108, 1835–1841. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Bellot, P.; Garcia-Pagan, J.C.; Frances, R.; Abraldes, J.G.; Navasa, M.; Perez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Ramadori, G.; Moriconi, F.; Malik, I.; Dudas, J. Physiology and pathophysiology of liver inflammation, damage and repair. J. Physiol. Pharm. 2008, 59 (Suppl. 1), 107–117. [Google Scholar]
- Kudo, H.; Takahara, T.; Yata, Y.; Kawai, K.; Zhang, W.; Sugiyama, T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J. Hepatol. 2009, 51, 168–175. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68-69, 452–462. [Google Scholar] [CrossRef]
- Benyon, R.C.; Arthur, M.J. Extracellular matrix degradation and the role of hepatic stellate cells. Semin. Liver Dis. 2001, 21, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Knittel, T.; Mehde, M.; Kobold, D.; Saile, B.; Dinter, C.; Ramadori, G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-alpha and TGF-beta1. J. Hepatol. 1999, 30, 48–60. [Google Scholar] [CrossRef]
- Miele, L.; Forgione, A.; La Torre, G.; Vero, V.; Cefalo, C.; Racco, S.; Vellone, V.G.; Vecchio, F.M.; Gasbarrini, G.; Rapaccini, G.L.; et al. Serum levels of hyaluronic acid and tissue metalloproteinase inhibitor-1 combined with age predict the presence of nonalcoholic steatohepatitis in a pilot cohort of subjects with nonalcoholic fatty liver disease. Transl. Res. J. Lab. Clin. Med. 2009, 154, 194–201. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef]
- Gil-Cardoso, K.; Gines, I.; Pinent, M.; Ardevol, A.; Terra, X.; Blay, M. A cafeteria diet triggers intestinal inflammation and oxidative stress in obese rats. Br. J. Nutr. 2017, 117, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Van Ampting, M.T.; Schonewille, A.J.; Vink, C.; Brummer, R.J.; van der Meer, R.; Bovee-Oudenhoven, I.M. Intestinal barrier function in response to abundant or depleted mucosal glutathione in Salmonella-infected rats. BMC Physiol. 2009, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [Green Version]
- Utzeri, E.; Usai, P. Role of non-steroidal anti-inflammatory drugs on intestinal permeability and nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 3954–3963. [Google Scholar] [CrossRef]
- Ramachandran, A.; Prabhu, R.; Thomas, S.; Reddy, J.B.; Pulimood, A.; Balasubramanian, K.A. Intestinal mucosal alterations in experimental cirrhosis in the rat: Role of oxygen free radicals. Hepatology 2002, 35, 622–629. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Nieto, N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology 2006, 44, 1487–1501. [Google Scholar] [CrossRef]
- Krause, P.; Morris, V.; Greenbaum, J.A.; Park, Y.; Bjoerheden, U.; Mikulski, Z.; Muffley, T.; Shui, J.W.; Kim, G.; Cheroutre, H.; et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat. Commun. 2015, 6, 7055. [Google Scholar] [CrossRef]
- Gomez-Hurtado, I.; Moratalla, A.; Moya-Perez, A.; Peiro, G.; Zapater, P.; Gonzalez-Navajas, J.M.; Gimenez, P.; Such, J.; Sanz, Y.; Frances, R. Role of interleukin 10 in norfloxacin prevention of luminal free endotoxin translocation in mice with cirrhosis. J. Hepatol. 2014, 61, 799–808. [Google Scholar] [CrossRef]
- Thompson, K.; Maltby, J.; Fallowfield, J.; McAulay, M.; Millward-Sadler, H.; Sheron, N. Interleukin-10 expression and function in experimental murine liver inflammation and fibrosis. Hepatology 1998, 28, 1597–1606. [Google Scholar] [CrossRef]
- De Souza-Cruz, S.; Victoria, M.B.; Tarrago, A.M.; da Costa, A.G.; Pimentel, J.P.; Pires, E.F.; Araujo Lde, P.; Coelho-dos-Reis, J.G.; Gomes Mde, S.; Amaral, L.R.; et al. Liver and blood cytokine microenvironment in HCV patients is associated to liver fibrosis score: A proinflammatory cytokine ensemble orchestrated by TNF and tuned by IL-10. BMC Microbiol. 2016, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Minemura, M.; Shimizu, Y. Gut microbiota and liver diseases. World J. Gastroenterol. 2015, 21, 1691–1702. [Google Scholar] [CrossRef]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Pappo, I.; Bercovier, H.; Berry, E.; Gallilly, R.; Feigin, E.; Freund, H.R. Antitumor necrosis factor antibodies reduce hepatic steatosis during total parenteral nutrition and bowel rest in the rat. JPEN J. Parenter. Enter. Nutr. 1995, 19, 80–82. [Google Scholar] [CrossRef]
- Kirsch, R.; Clarkson, V.; Verdonk, R.C.; Marais, A.D.; Shephard, E.G.; Ryffel, B.; de la, M.H.P. Rodent nutritional model of steatohepatitis: Effects of endotoxin (lipopolysaccharide) and tumor necrosis factor alpha deficiency. J. Gastroenterol. Hepatol. 2006, 21, 174–182. [Google Scholar] [CrossRef]
- Jin, X.; Yu, C.H.; Lv, G.C.; Li, Y.M. Increased intestinal permeability in pathogenesis and progress of nonalcoholic steatohepatitis in rats. World J. Gastroenterol. 2007, 13, 1732–1736. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Corazza, G.R.; Gasbarrini, G.; Montalto, M.; Di Stefano, M.; Basilisco, G.; Parodi, A.; Usai-Satta, P.; Vernia, P.; Anania, C.; et al. Methodology and indications of H2-breath testing in gastrointestinal diseases: The Rome Consensus Conference. Aliment. Pharmacol. Ther. 2009, 29 (Suppl. 1), 1–49. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Lauritano, E.C.; Gabrielli, M.; Scarpellini, E.; Lupascu, A.; Ojetti, V.; Gasbarrini, G. Small intestinal bacterial overgrowth: Diagnosis and treatment. Dig. Di.s 2007, 25, 237–240. [Google Scholar] [CrossRef]
- De Wit, N.J.; Afman, L.A.; Mensink, M.; Muller, M. Phenotyping the effect of diet on non-alcoholic fatty liver disease. J. Hepatol. 2012, 57, 1370–1373. [Google Scholar] [CrossRef]
- O’Sullivan, A.; He, X.; McNiven, E.M.; Haggarty, N.W.; Lonnerdal, B.; Slupsky, C.M. Early diet impacts infant rhesus gut microbiome, immunity, and metabolism. J. Proteome Res. 2013, 12, 2833–2845. [Google Scholar] [CrossRef]
- Wang, H.H.; Lee, D.K.; Liu, M.; Portincasa, P.; Wang, D.Q.H. Novel Insights into the Pathogenesis and Management of the Metabolic Syndrome. Pediatric Gastroenterol. Hepatol. Nutr. 2020, 23, 189–230. [Google Scholar] [CrossRef]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Kim, H.N.; Lee, E.J.; Ryu, S.; Chang, Y.; Shin, H.; Kim, H.L.; Kim, T.H.; Yoo, K.; Kim, H.Y. Fecal and blood microbiota profiles and presence of nonalcoholic fatty liver disease in obese versus lean subjects. PLoS ONE 2019, 14, e0213692. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef]
- Caussy, C.; Hsu, C.; Lo, M.T.; Liu, A.; Bettencourt, R.; Ajmera, V.H.; Bassirian, S.; Hooker, J.; Sy, E.; Richards, L.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018, 68, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef]
- Grattagliano, I.; Diogo, C.V.; Mastrodonato, M.; de Bari, O.; Persichella, M.; Wang, D.Q.; Liquori, A.; Ferri, D.; Carratu, M.R.; Oliveira, P.J.; et al. A silybin-phospholipids complex counteracts rat fatty liver degeneration and mitochondrial oxidative changes. World J. Gastroenterol. 2013, 19, 3007–3017. [Google Scholar] [CrossRef]
- Mastrodonato, M.; Calamita, G.; Rossi, R.; Mentino, D.; Bonfrate, L.; Portincasa, P.; Ferri, D.; Liquori, G.E. Altered distribution of caveolin-1 in early liver steatosis. Eur. J. Clin. Investig. 2011, 41, 642–651. [Google Scholar] [CrossRef]
- Pacelli, C.; Coluccia, A.; Grattagliano, I.; Cocco, T.; Petrosillo, G.; Paradies, G.; De Nitto, E.; Massaro, A.; Persichella, M.; Borracci, P.; et al. Dietary choline deprivation impairs rat brain mitochondrial function and behavioral phenotype. J. Nutr. 2010, 140, 1072–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Et Biophys. Acta 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtmann, T.M.; Inzaugarat, M.E.; Knorr, J.; Geisler, L.; Schulz, M.; Bieghs, V.; Frissen, M.; Feldstein, A.E.; Tacke, F.; Trautwein, C.; et al. Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner. Cells 2021, 10, 2618. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.D.; Li, R.; Demaster, E.G.; Elson, M.; Furne, J.; Levitt, D.G. Use of measurements of ethanol absorption from stomach and intestine to assess human ethanol metabolism. Am. J. Physiol.—Gastrointest. Liver Physiol. 1997, 273, G951–G957. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [Green Version]
- Ansari, R.A.; Husain, K.; Rizvi, S.A. Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism. Biomolecules 2016, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Hamarneh, S.R.; Kim, B.M.; Kaliannan, K.; Morrison, S.A.; Tantillo, T.J.; Tao, Q.; Mohamed, M.M.R.; Ramirez, J.M.; Karas, A.; Liu, W.; et al. Intestinal Alkaline Phosphatase Attenuates Alcohol-Induced Hepatosteatosis in Mice. Dig. Dis. Sci. 2017, 62, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Lee, H.R.; Lee, Y.J. Alcoholic liver disease: Focus on prodromal gut health. J. Dig. Dis. 2016, 17, 493–500. [Google Scholar] [CrossRef]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Starkel, P.; Windey, K.; Tremaroli, V.; Backhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Ginès, P.; Nevens, F.; Fernández, J.; To, U.; García-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Primers 2016, 2, 16041. [Google Scholar] [CrossRef] [Green Version]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef] [Green Version]
- Basuroy, S.; Sheth, P.; Mansbach, C.M.; Rao, R.K. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: Protection by EGF and L-glutamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G367–G375. [Google Scholar] [CrossRef]
- Samak, G.; Aggarwal, S.; Rao, R.K. ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G50–G59. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, P.; Chen, P.; Wang, H.J.; Wang, L.; McCole, D.F.; Brandl, K.; Starkel, P.; Belzer, C.; Hellerbrand, C.; Tsukamoto, H.; et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 2013, 58, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Starkel, P.; van Pijkeren, J.P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214.e216. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Jeong, D.; Kang, I.B.; Kim, H.; Song, K.Y.; Seo, K.H. Dual function of Lactobacillus kefiri DH5 in preventing high-fat-diet-induced obesity: Direct reduction of cholesterol and upregulation of PPAR-alpha in adipose tissue. Mol. Nutr. Food Res. 2017, 61, 1700252. [Google Scholar] [CrossRef]
- Xie, G.; Zhong, W.; Zheng, X.; Li, Q.; Qiu, Y.; Li, H.; Chen, H.; Zhou, Z.; Jia, W. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J. Proteome Res. 2013, 12, 3297–3306. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Baraona, E.; Julkunen, R.; Tannenbaum, L.; Lieber, C.S. Role of intestinal bacterial overgrowth in ethanol production and metabolism in rats. Gastroenterology 1986, 90, 103–110. [Google Scholar] [CrossRef]
- Mezey, E.; Imbembo, A.L.; Potter, J.J.; Rent, K.C.; Lombardo, R.; Holt, P.R. Endogenous ethanol production and hepatic disease following jejunoileal bypass for morbid obesity. Am. J. Clin. Nutr. 1975, 28, 1277–1283. [Google Scholar] [CrossRef]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Mottaran, E.; Stewart, S.F.; Rolla, R.; Vay, D.; Cipriani, V.; Moretti, M.; Vidali, M.; Sartori, M.; Rigamonti, C.; Day, C.P.; et al. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 38–45. [Google Scholar] [CrossRef]
- Couch, R.D.; Dailey, A.; Zaidi, F.; Navarro, K.; Forsyth, C.B.; Mutlu, E.; Engen, P.A.; Keshavarzian, A. Alcohol induced alterations to the human fecal VOC metabolome. PLoS ONE 2015, 10, e0119362. [Google Scholar] [CrossRef] [Green Version]
- Kaji, H.; Asanuma, Y.; Yahara, O.; Shibue, H.; Hisamura, M.; Saito, N.; Kawakami, Y.; Murao, M. Intragastrointestinal alcohol fermentation syndrome: Report of two cases and review of the literature. J. Forensic. Sci. Soc. 1984, 24, 461–471. [Google Scholar] [CrossRef]
- Salaspuro, M. Bacteriocolonic pathway for ethanol oxidation: Characteristics and implications. Ann. Med. 1996, 28, 195–200. [Google Scholar] [CrossRef]
- Dawes, E.A.; Foster, S.M. The formation of ethanol in Escherichia coli. Biochim. Et Biophys. Acta 1956, 22, 253–265. [Google Scholar] [CrossRef]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Durr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genom. 2014, 15, 1066. [Google Scholar] [CrossRef] [Green Version]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxidative Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.H.; Huo, L.J.; Li, T.T. Effect of interleukin-22 on proliferation and activation of hepatic stellate cells induced by acetaldehyde and related mechanism. Zhonghua Gan Zang Bing Za Zhi 2017, 25, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Wang, S.; Xu, R.; Lv, X. Purinergic P2X7 receptor mediates acetaldehyde-induced hepatic stellate cells activation via PKC-dependent GSK3beta pathway. Int. Immunopharmacol. 2017, 43, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lázaro, M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol. 2016, 62, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, M.; Schwartz, D.M.; Tandon, M.; Son, A.; Zeng, M.; Swaim, W.; Eckhaus, M.; Hoffman, V.; Cui, Y.; Xiao, B.; et al. Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. Cell Metab. 2017, 25, 635–646. [Google Scholar] [CrossRef] [Green Version]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Albaugh, V.L.; Banan, B.; Antoun, J.; Xiong, Y.; Guo, Y.; Ping, J.; Alikhan, M.; Clements, B.A.; Abumrad, N.N.; Flynn, C.R. Role of Bile Acids and GLP-1 in Mediating the Metabolic Improvements of Bariatric Surgery. Gastroenterology 2019, 156, 1041–1051.e1044. [Google Scholar] [CrossRef] [Green Version]
- Thoni, V.; Pfister, A.; Melmer, A.; Enrich, B.; Salzmann, K.; Kaser, S.; Lamina, C.; Ebenbichler, C.F.; Hackl, H.; Tilg, H.; et al. Dynamics of Bile Acid Profiles, GLP-1, and FGF19 After Laparoscopic Gastric Banding. J. Clin. Endocrinol. Metab. 2017, 102, 2974–2984. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S.t. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Sawicki, C.M.; Livingston, K.A.; Obin, M.; Roberts, S.B.; Chung, M.; McKeown, N.M. Dietary Fiber and the Human Gut Microbiota: Application of Evidence Mapping Methodology. Nutrients 2017, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, J.; Li, J.V.; Zhou, N.Y.; Tang, H.; Wang, Y. Gut microbiota composition modifies fecal metabolic profiles in mice. J. Proteome Res. 2013, 12, 2987–2999. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Bellahcene, M.; O’Dowd, J.F.; Wargent, E.T.; Zaibi, M.S.; Hislop, D.C.; Ngala, R.A.; Smith, D.M.; Cawthorne, M.A.; Stocker, C.J.; Arch, J.R. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br. J. Nutr. 2013, 109, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Weidemann, M.J.; Hems, R.; Williams, D.L.; Spray, G.H.; Krebs, H.A. Gluconeogenesis from propionate in kidney and liver of the vitamin B12-deficient rat. Biochem. J. 1970, 117, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Ilan, Y.; Maron, R.; Tukpah, A.M.; Maioli, T.U.; Murugaiyan, G.; Yang, K.; Wu, H.Y.; Weiner, H.L. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9765–9770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Zeisel, S.H. Choline’s role in maintaining liver function: New evidence for epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Grattagliano, I.; Caraceni, P.; Portincasa, P.; Domenicali, M.; Palmieri, V.O.; Trevisani, F.; Bernardi, M.; Palasciano, G. Adaptation of subcellular glutathione detoxification system to stress conditions in choline-deficient diet induced rat fatty liver. Cell Biol. Toxicol. 2003, 19, 355–366. [Google Scholar] [CrossRef]
- Xie, G.; Yan, A.; Lin, P.; Wang, Y.; Guo, L. Trimethylamine N-oxide-a marker for atherosclerotic vascular disease. Rev. Cardiovasc. Med. 2021, 22, 787–797. [Google Scholar] [CrossRef]
- Cretoiu, D.; Ionescu, R.F.; Enache, R.M.; Cretoiu, S.M.; Voinea, S.C. Gut Microbiome, Functional Food, Atherosclerosis, and Vascular Calcifications-Is There a Missing Link? Microorganisms 2021, 9, 1913. [Google Scholar] [CrossRef]
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II-induced hypertension. Redox. Biol. 2021, 46, 102115. [Google Scholar] [CrossRef]
- Shen, X.; Li, L.; Sun, Z.; Zang, G.; Zhang, L.; Shao, C.; Wang, Z. Gut Microbiota and Atherosclerosis-Focusing on the Plaque Stability. Front. Cardiovasc. Med. 2021, 8, 668532. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H. A Gut Feeling about Thrombosis. New Engl. J. Med. 2016, 374, 2494–2496. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J. Gut microbiota, the genome, and diet in atherogenesis. New Engl. J. Med. 2013, 368, 1647–1649. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Li, P.; Zhong, C.; Li, S.; Sun, T.; Huang, H.; Chen, X.; Zhu, Y.; Hu, X.; Peng, X.; Zhang, X.; et al. Plasma concentration of trimethylamine-N-oxide and risk of gestational diabetes mellitus. Am. J. Clin. Nutr. 2018, 108, 603–610. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Li, X.S.; Fan, Y.; Li, D.S.; Wu, Y.; Hazen, S.L. Increased Trimethylamine N-Oxide Portends High Mortality Risk Independent of Glycemic Control in Patients with Type 2 Diabetes Mellitus. Clin. Chem. 2017, 63, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yao, P.; Cheng, J.; et al. Association between microbiota-dependent metabolite trimethylamine-N-oxide and type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e917. [Google Scholar] [CrossRef] [Green Version]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 2008, 135, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Sumagin, R.; Rankin, C.R.; Leoni, G.; Mina, M.J.; Reiter, D.M.; Stehle, T.; Dermody, T.S.; Schaefer, S.A.; Hall, R.A.; et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol. Biol. Cell 2013, 24, 2849–2860. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal. Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Nazim, M.; Stamp, G.; Hodgson, H.J. Non-alcoholic steatohepatitis associated with small intestinal diverticulosis and bacterial overgrowth. Hepatogastroenterology 1989, 36, 349–351. [Google Scholar]
- Lichtman, S.N.; Sartor, R.B.; Keku, J.; Schwab, J.H. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology 1990, 98, 414–423. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R.B. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- DeMeo, M.T.; Mutlu, E.A.; Keshavarzian, A.; Tobin, M.C. Intestinal permeation and gastrointestinal disease. J. Clin. Gastroenterol. 2002, 34, 385–396. [Google Scholar] [CrossRef]
- Arslan, G.; Atasever, T.; Cindoruk, M.; Yildirim, I.S. (51)CrEDTA colonic permeability and therapy response in patients with ulcerative colitis. Nucl. Med. Commun. 2001, 22, 997–1001. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Zocco, M.A.; Cerrito, L.; Gasbarrini, A.; Pompili, M. Bacterial translocation in patients with liver cirrhosis: Physiology, clinical consequences, and practical implications. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 641–656. [Google Scholar] [CrossRef]
- Nier, A.; Engstler, A.J.; Maier, I.B.; Bergheim, I. Markers of intestinal permeability are already altered in early stages of non-alcoholic fatty liver disease: Studies in children. PLoS ONE 2017, 12, e0183282. [Google Scholar] [CrossRef] [Green Version]
- Cariello, R.; Federico, A.; Sapone, A.; Tuccillo, C.; Scialdone, V.R.; Tiso, A.; Miranda, A.; Portincasa, P.; Carbonara, V.; Palasciano, G.; et al. Intestinal permeability in patients with chronic liver diseases: Its relationship with the aetiology and the entity of liver damage. Dig. Liver Dis. 2010, 42, 200–204. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Tsamandas, A.C.; Tsiaoussis, G.I.; Karatza, E.; Triantos, C.; Vagianos, C.E.; Spiliopoulou, I.; Kaltezioti, V.; Charonis, A.; Nikolopoulou, V.N.; et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur. J. Clin. Investig. 2012, 42, 439–446. [Google Scholar] [CrossRef]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]




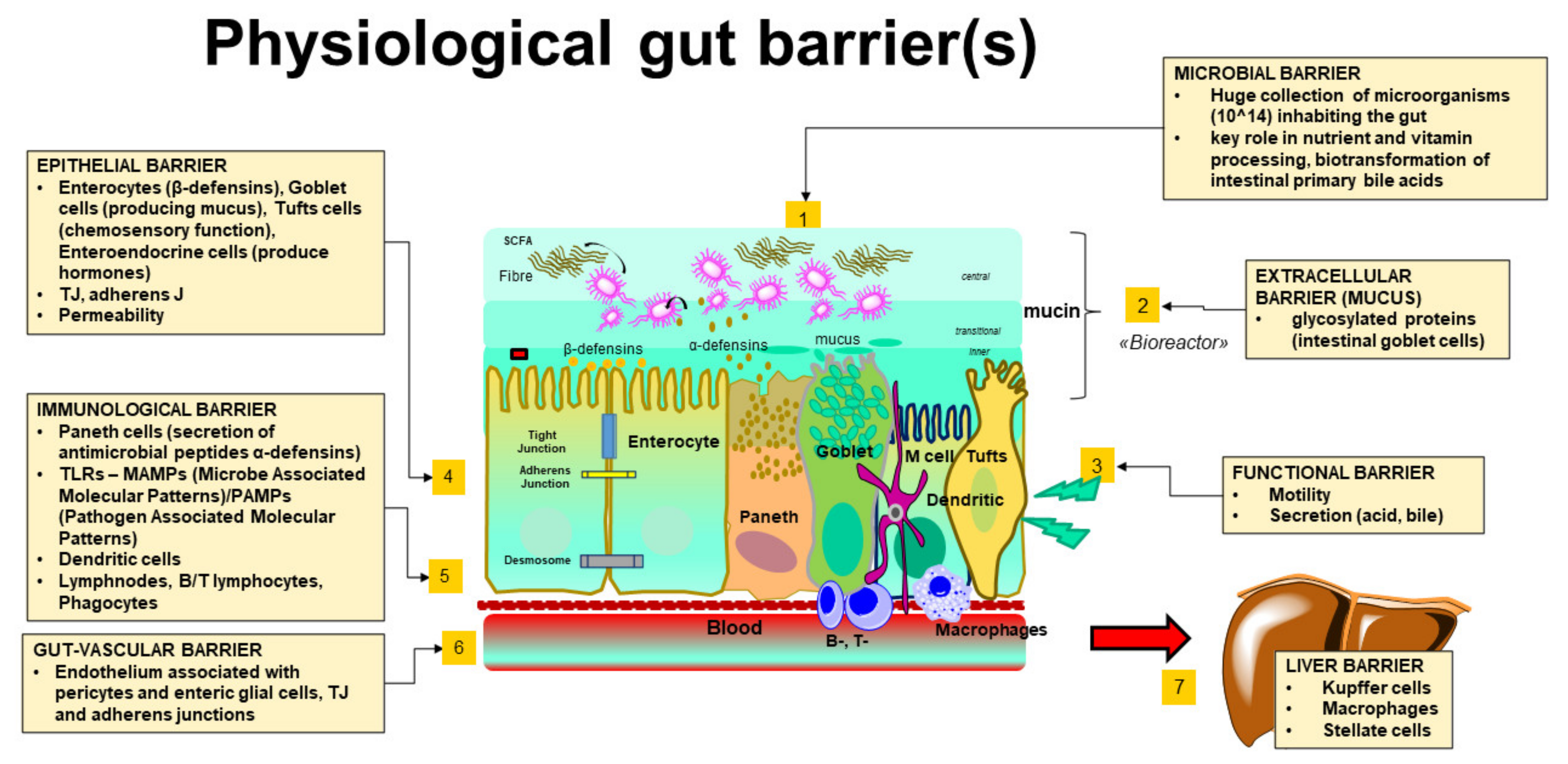{kind=link}
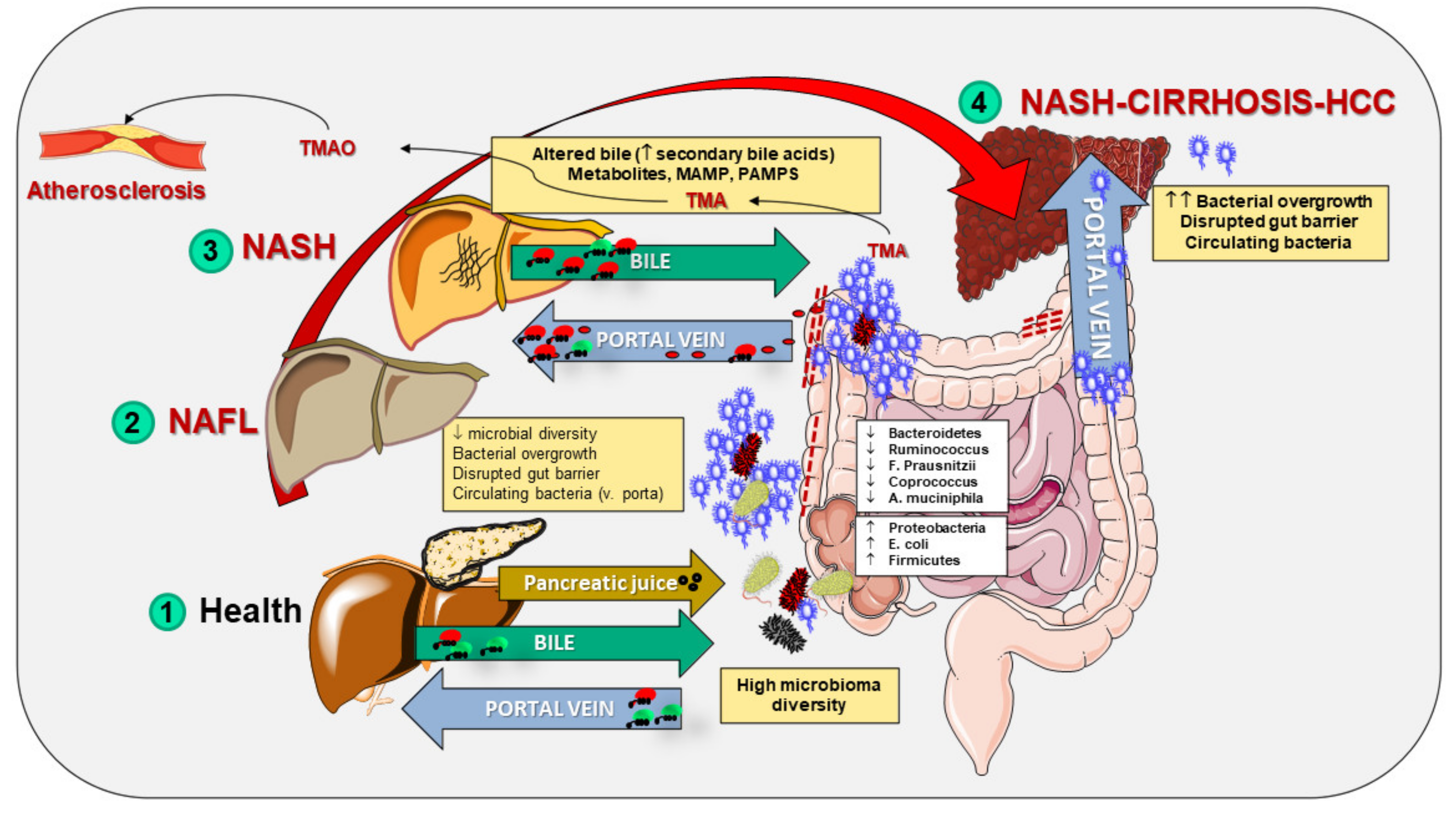{kind=link}
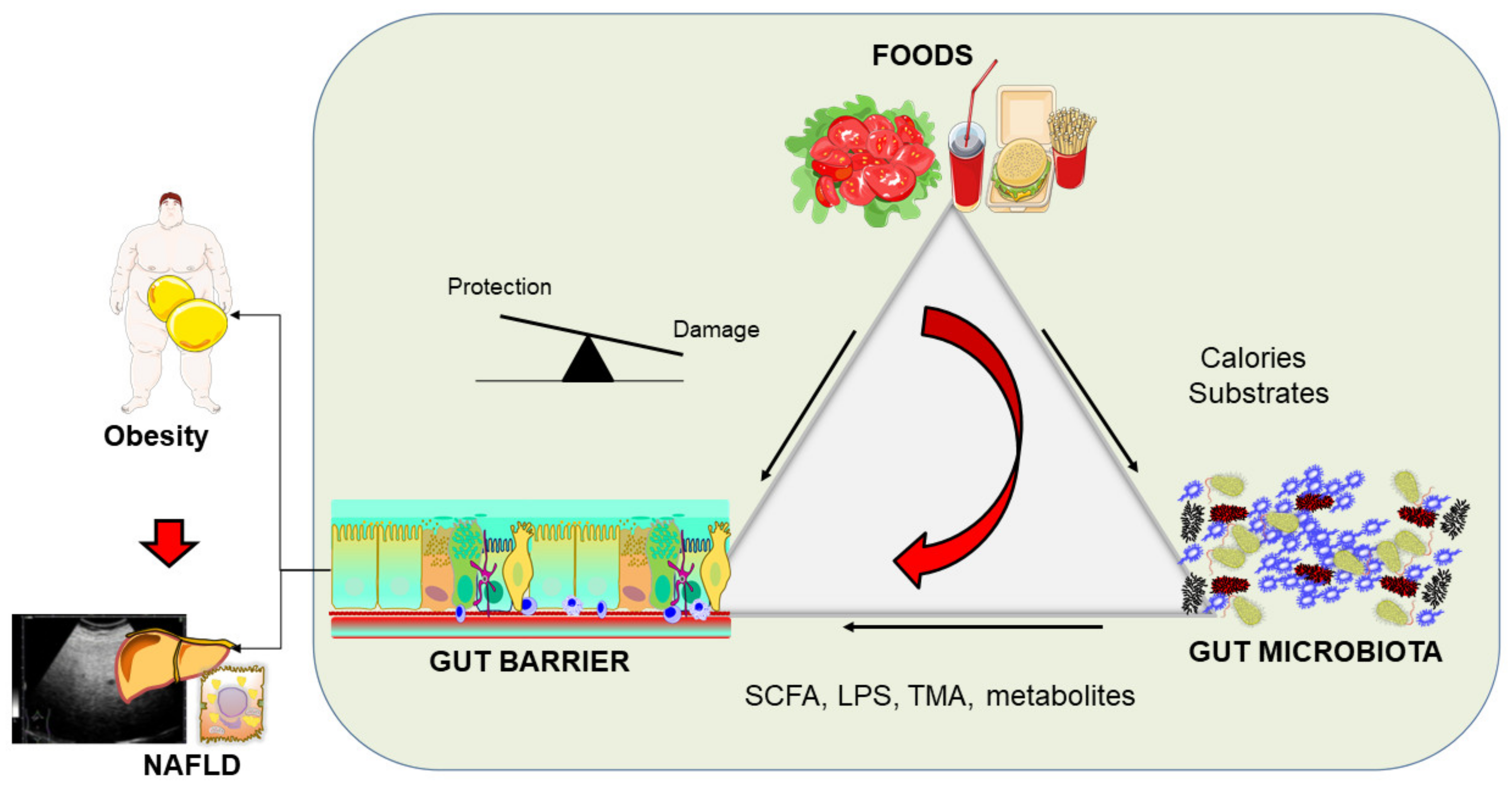{kind=link}
| Food Group | Soluble Fibers | Insoluble Fibers |
|---|---|---|
| Cereals and grains | Nonstarch polysaccharides | Nonstarch polysaccharides |
| Hemicellulose Arabinoxylan β-glucan | Hemicellulose Cellulose Lignin | |
| Resistant oligosaccharides | Resistant starch | |
| Inulin | ||
| Fruits and vegetables | Nonstarch polysaccharides | Nonstarch polysaccharides |
| Hemicellulose | Hemicellulose | |
| Pectin | Cellulose | |
| Pectin | ||
| Resistant oligosaccharides | Lignin | |
| Inulin | Resistant starch | |
| Legumes and pulses | Nonstarch polysaccharides | Nonstarch polysaccharides |
| Hemicellulose | Hemicellulose | |
| Pectin | Pectin | |
| Gum | ||
| Lignin | ||
| Resistant starch |
| Metabolic, nonalcoholic fatty liver disease (NAFLD, 23–69%) [307] |
| Alcoholic fatty liver disease (ALD) (about 5%) [308] |
| Viral hepatitis B and C (especially genotype 3) (30–80%) [309,310,311,312] |
| Lipodystrophy (80%) [313] |
| Wilson’s disease (about 50%) [314] |
| Starvation (prevalence undetermined) |
| Parenteral nutrition (about 28%) [315] |
| Abetalipoproteinemia (prevalence undetermined) |
| Hepatotoxic drugs (about 2%) [316] (amiodarone, anti-retroviral agents for HIV, glucocorticoids, methotrexate, tamoxifen, valproate) |
| Pregnancy (incidence 1:7000–15,000 pregnancies) [317] |
| Reye syndrome (100%) [318] |
| Inborn errors of metabolism (about 40% of children hospitalized for nonalcoholic fatty liver disease) [319] (lecithin-cholesterol acyltransferase deficiency, cholesterol ester storage disease, Wolman disease) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portincasa, P.; Bonfrate, L.; Khalil, M.; Angelis, M.D.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.-H.; Di Ciaula, A. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines 2022, 10, 83. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10010083
Portincasa P, Bonfrate L, Khalil M, Angelis MD, Calabrese FM, D’Amato M, Wang DQ-H, Di Ciaula A. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines. 2022; 10(1):83. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10010083
Chicago/Turabian StylePortincasa, Piero, Leonilde Bonfrate, Mohamad Khalil, Maria De Angelis, Francesco Maria Calabrese, Mauro D’Amato, David Q.-H. Wang, and Agostino Di Ciaula. 2022. "Intestinal Barrier and Permeability in Health, Obesity and NAFLD" Biomedicines 10, no. 1: 83. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10010083