
Effect of N-3 Polyunsaturated Fatty Acids on Lipid Composition in Familial Hypercholesterolemia: A Randomized Crossover Trial

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
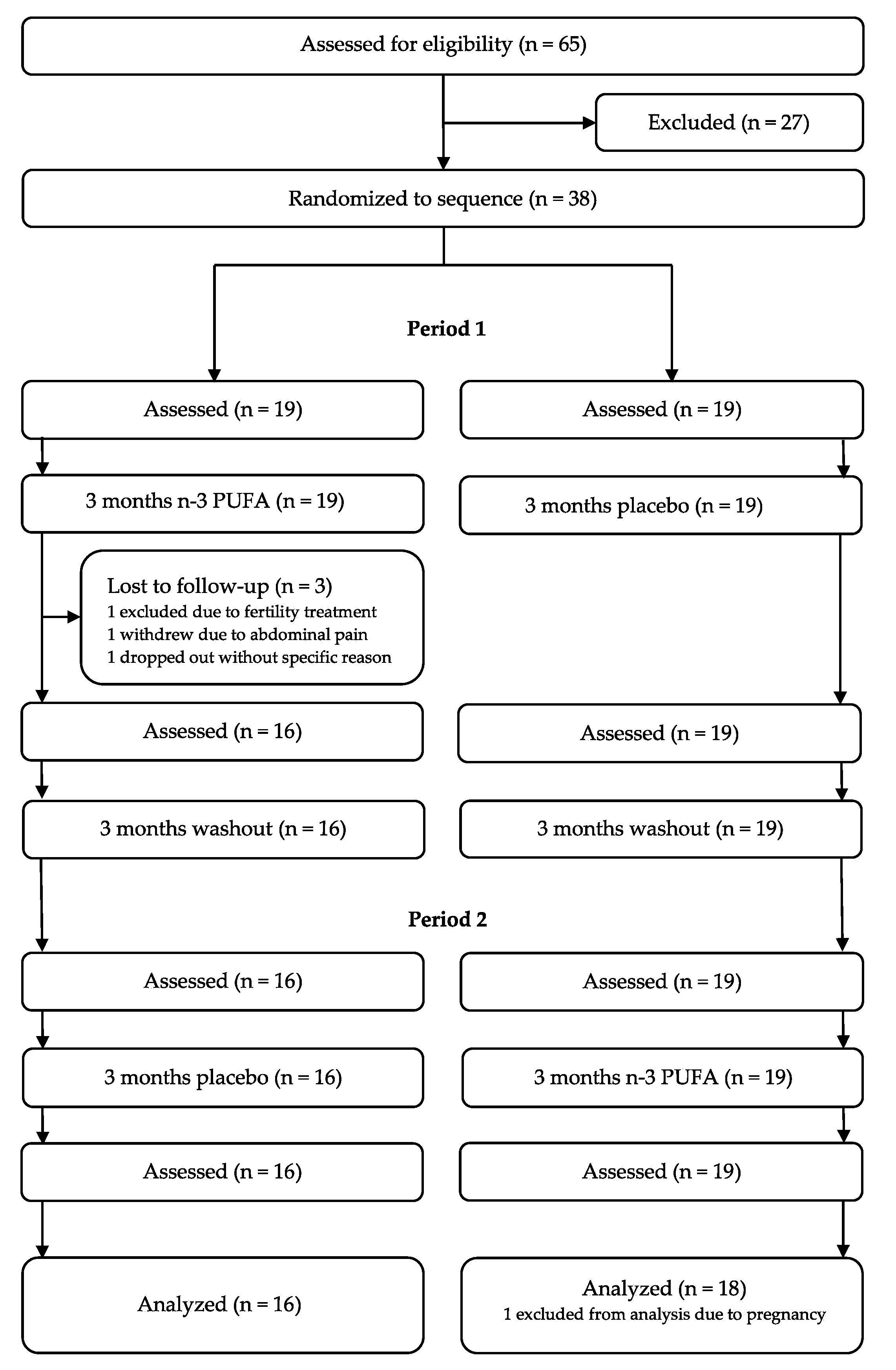
2.1. Trial Design and Interventions
2.2. Blood Samples
2.3. Lipoprotein Measurements
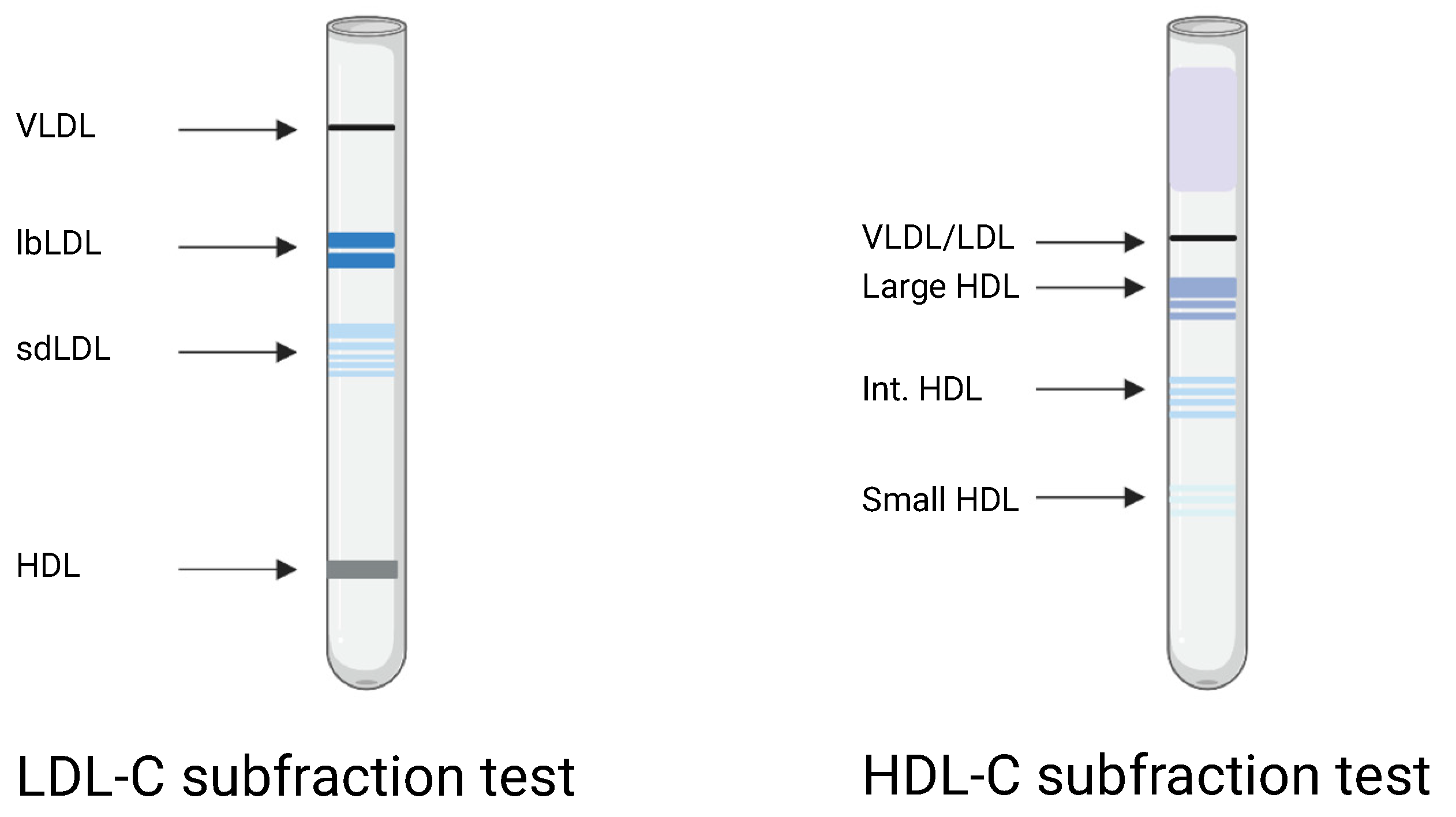
2.4. Lipoprotein Subfractions
2.5. HDL-C Function
2.6. Statistical Analysis
3. Results
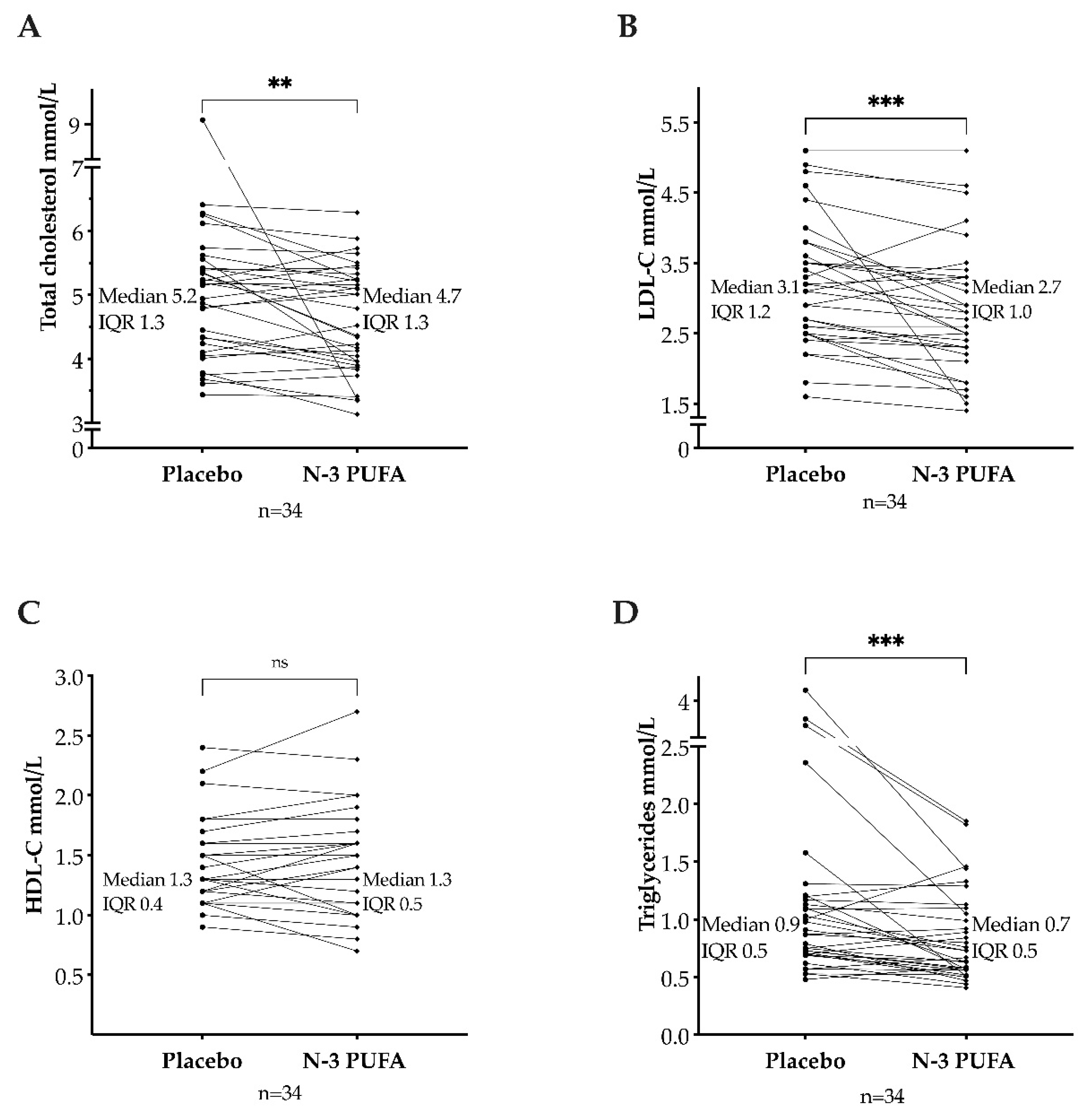
3.1. Total Cholesterol, Triglycerides, and Lipoproteins
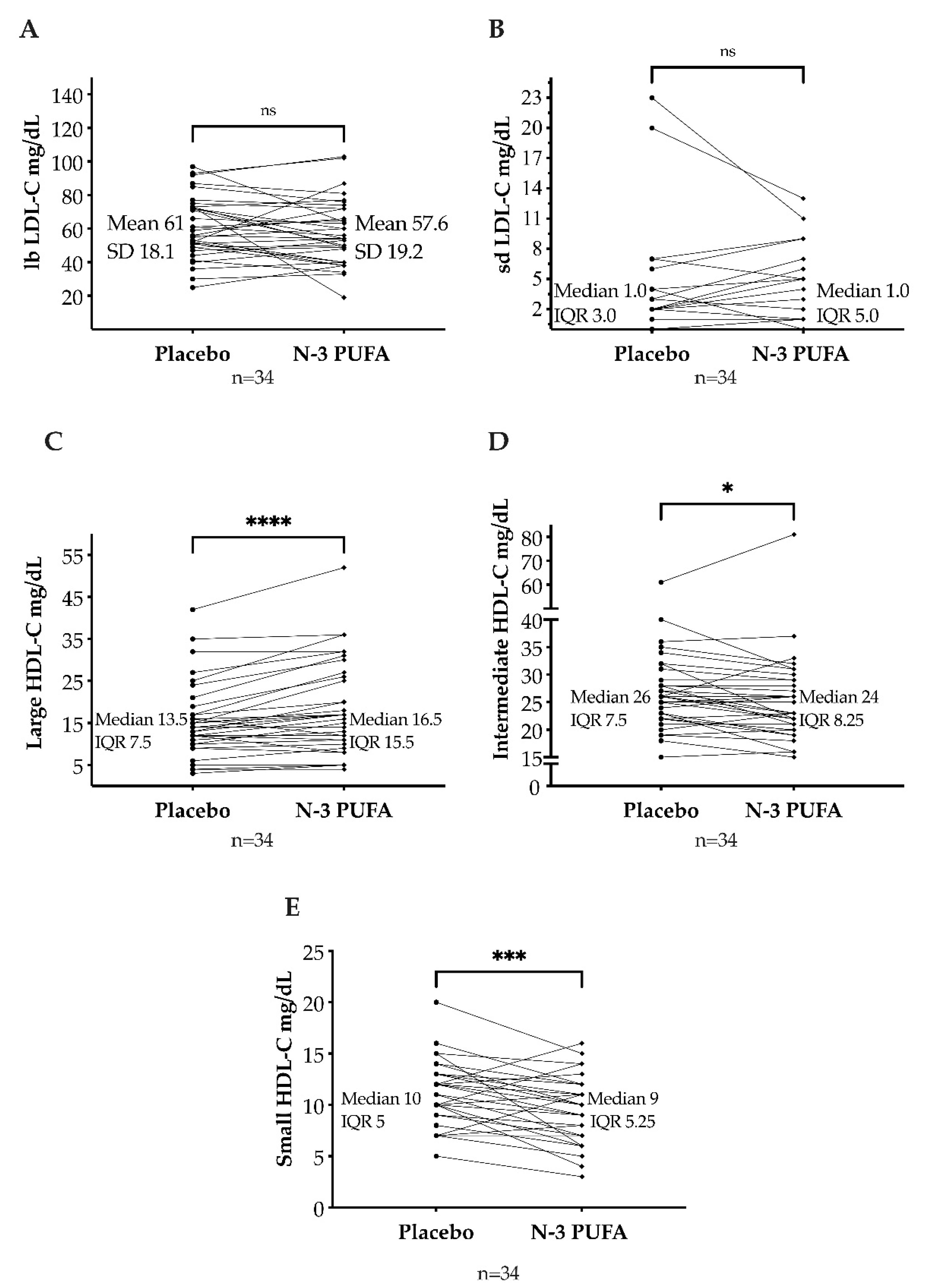
3.2. LDL and HDL Cholesterol Subfractions
3.3. Apolipoproteins and HDL-C Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Duell, P.B.; Gidding, S.S.; Andersen, R.L.; Knickelbine, T.; Anderson, L.; Gianos, E.; Shrader, P.; Kindt, I.; O’Brien, E.C.; McCann, D.; et al. Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: The CASCADE FH registry. Atherosclerosis 2019, 289, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 1999, 354, 447–455.
- Kromhout, D.; Giltay, E.J.; Geleijnse, J.M. n-3 Fatty Acids and Cardiovascular Events after Myocardial Infarction. N. Engl. J. Med. 2010, 363, 2015–2026. [Google Scholar] [CrossRef]
- Rauch, B.; Schiele, R.; Schneider, S.; Diller, F.; Victor, N.; Gohlke, H.; Gottwik, M.; Steinbeck, G.; Del Castillo, U.; Sack, R.; et al. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation 2010, 122, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.P.; Koenig, W.; McGuire, D.K.; et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020, 324, 2268–2280. [Google Scholar] [CrossRef]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.; Summerbell, C.D.; Worthington, H.V.; Song, F.; et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2020, 3, CD003177. [Google Scholar] [CrossRef]
- Nordestgaard, B.G. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ. Res. 2016, 118, 547–563. [Google Scholar] [CrossRef]
- Balestrieri, G.P.; Maffi, V.; Sleiman, I.; Spandrio, S.; Di Stefano, O.; Salvi, A.; Scalvini, T. Fish oil supplementation in patients with heterozygous familial hypercholesterolemia. Recenti Progress. Med. 1996, 87, 102–105. [Google Scholar]
- Engler, M.M.; Engler, M.B.; Malloy, M.; Chiu, E.; Besio, D.; Paul, S.; Stuehlinger, M.; Morrow, J.; Ridker, P.; Rifai, N.; et al. Docosahexaenoic acid restores endothelial function in children with hyperlipidemia: Results from the EARLY study. Int. J. Clin. Pharmacol. Ther. 2004, 42, 672–679. [Google Scholar] [CrossRef]
- Chan, D.C.; Pang, J.; Barrett, P.H.; Sullivan, D.R.; Burnett, J.R.; van Bockxmeer, F.M.; Watts, G.F. omega-3 Fatty Acid Ethyl Esters Diminish Postprandial Lipemia in Familial Hypercholesterolemia. J. Clin. Endocrinol. Metab. 2016, 101, 3732–3739. [Google Scholar] [CrossRef] [Green Version]
- Hande, L.N.; Thunhaug, H.; Enebakk, T.; Ludviksen, J.; Pettersen, K.; Hovland, A.; Lappegård, K.T. Addition of marine omega-3 fatty acids to statins in familial hypercholesterolemia does not affect in vivo or in vitro endothelial function. J. Clin. Lipidol. 2019, 13, 762–770. [Google Scholar] [CrossRef]
- Kjellmo, C.A.; Karlsson, H.; Nestvold, T.K.; Ljunggren, S.; Cederbrant, K.; Marcusson-Stahl, M.; Mathisen, M.; Lappegard, K.T.; Hovland, A. Bariatric surgery improves lipoprotein profile in morbidly obese patients by reducing LDL cholesterol, apoB, and SAA/PON1 ratio, increasing HDL cholesterol, but has no effect on cholesterol efflux capacity. J. Clin. Lipidol. 2018, 12, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.Y.; Jacobson, T.A. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: A systematic review and meta-analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef]
- Dunbar, R.L.; Nicholls, S.J.; Maki, K.C.; Roth, E.M.; Orloff, D.G.; Curcio, D.; Johnson, J.; Kling, D.; Davidson, M.H. Effects of omega-3 carboxylic acids on lipoprotein particles and other cardiovascular risk markers in high-risk statin-treated patients with residual hypertriglyceridemia: A randomized, controlled, double-blind trial. Lipids Health Dis. 2015, 14, 98. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.; Stone, N.J.; Ballantyne, C.; Bittner, V.; Criqui, M.H.; Ginsberg, H.N.; Goldberg, A.C.; Howard, W.J.; Jacobson, M.S.; Kris-Etherton, P.M.; et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2011, 123, 2292–2333. [Google Scholar] [CrossRef] [Green Version]
- Ip, S.; Lichtenstein, A.H.; Chung, M.; Lau, J.; Balk, E.M. Systematic review: Association of low-density lipoprotein subfractions with cardiovascular outcomes. Ann. Intern. Med. 2009, 150, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.D.; Sanders, T.A.; Davies, I.G.; Morgan, L.M.; Millward, D.J.; Lewis, F.; Slaughter, S.; Cooper, J.A.; Miller, G.J.; Griffin, B.A. Effects of altering the ratio of dietary n−6 to n−3 fatty acids on insulin sensitivity, lipoprotein size, and postprandial lipemia in men and postmenopausal women aged 45–70 y: The OPTILIP Study. Am. J. Clin. Nutr. 2006, 84, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Hartwich, J.; Malec, M.M.; Partyka, L.; Perez-Martinez, P.; Marin, C.; Lopez-Miranda, J.; Tierney, A.C.; Mc Monagle, J.; Roche, H.M.; Defoort, C.; et al. The effect of the plasma n-3/n-6 polyunsaturated fatty acid ratio on the dietary LDL phenotype transformation—Insights from the LIPGENE study. Clin. Nutr. 2009, 28, 510–515. [Google Scholar] [CrossRef]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid. Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef] [Green Version]
- Hafiane, A.; Genest, J. High density lipoproteins: Measurement techniques and potential biomarkers of cardiovascular risk. BBA Clin. 2015, 3, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Cuchel, M.; Rader, D.J. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [Green Version]
- Versmissen, J.; Vongpromek, R.; Yahya, R.; van der Net, J.B.; van Vark-van der Zee, L.; Blommesteijn-Touw, J.; Wattimena, D.; Rietveld, T.; Pullinger, C.R.; Christoffersen, C.; et al. Familial hypercholesterolaemia: Cholesterol efflux and coronary disease. Eur. J. Clin. Investig. 2016, 46, 643–650. [Google Scholar] [CrossRef]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association Between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients With Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Favari, E.; Calabresi, L.; Adorni, M.P.; Jessup, W.; Simonelli, S.; Franceschini, G.; Bernini, F. Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 2009, 48, 11067–11074. [Google Scholar] [CrossRef]
- Du, X.M.; Kim, M.J.; Hou, L.; Le Goff, W.; Chapman, M.J.; Van Eck, M.; Curtiss, L.K.; Burnett, J.R.; Cartland, S.P.; Quinn, C.M.; et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ. Res. 2015, 116, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- James, R.W. A long and winding road: Defining the biological role and clinical importance of paraoxonases. Clin. Chem. Lab. Med. 2006, 44, 1052–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, H.; Kontush, A.; James, R.W. Functionality of HDL: Antioxidation and Detoxifying Effects. In High Density Lipoproteins: From Biological Understanding to Clinical Exploitation; von Eckardstein, A., Kardassis, D., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 207–228. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, T.; Nicholls, S.J.; Topol, E.J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D.M.; Ellis, S.G.; et al. Relationship of Paraoxonase 1 (PON1) Gene Polymorphisms and Functional Activity With Systemic Oxidative Stress and Cardiovascular Risk. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Hartiala, J.; Fan, Y.; Wu, Y.; Stewart, A.F.; Erdmann, J.; Kathiresan, S.; Consortium, C.A.; Roberts, R.; McPherson, R.; et al. Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Idrees, M.; Siddiq, A.R.; Ajmal, M.; Akram, M.; Khalid, R.R.; Hussain, A.; Qamar, R.; Bokhari, H. Decreased serum PON1 arylesterase activity in familial hypercholesterolemia patients with a mutated LDLR gene. Genet. Mol. Biol. 2018, 41, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Song, W.; Prendergast, C.J.; Smith, L.; Ifrim, A.; Meador, B.; Ding, L.; Zhang, Y.; Yancey, P.; Linton, M.F. Levels of High Density Lipoprotein-Associated Proteins, Myeloperoxidase and Serum Amyloid A1, and Cholesterol Efflux Capacity in Heterozygous Familial Hypercholesterolemic Patients. Available online: https://www.ahajournals.org/doi/10.1161/atvb.40.suppl_1.235 (accessed on 4 May 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline | ||||||
|---|---|---|---|---|---|---|
| Treatment Sequence | Treatment Sequence | |||||
| Variable | N-3 PUFAs First, Then Placebo n = 16 | Placebo First, Then N-3 PUFAs n = 18 | Total n = 34 | N-3 PUFAs First, Then Placebo n = 16 | Placebo First, Then N-3 PUFAs n = 18 | Total n = 34 |
| Before Treatment 1 (Month 0) | Before Treatment 2 (Month 6) | |||||
| TC mmol/L | 4.8 ± 1.0 | 4.8 (4.4, 5.4) | 4.7 (4.4, 5.4) | 5.0 ± 1.2 | 5.0 ± 1.1 | 5.0 ± 1.2 |
| LDL-C mmol/L | 3.0 (2.7, 3.3) | 3.1 (2.9, 3.5) | 3.0 (2.8, 3.3) | 3.2 ± 0.9 | 3.1 (2.6, 3.6) | 3.2 (2.5, 3.7) |
| HDL-C mmol/L | 1.3 (1.1, 1.5) | 1.2 (1.0, 1.5) | 1.2 (1.1, 1.5) | 1.3 (1.1, 1.6) | 1.2 (1.0, 1.6) | 1.2 (1.1, 1.6) |
| TG mmol/L | 0.7 (0.6, 1.3) | 1.0 (0.8, 1.6) | 0.8 (0.7, 1.4) | 0.8 (0.6, 1.4) | 0.9 (0.8, 1.3) | 0.9 (0.7, 1.4) |
| LbLDL-C mmol/L | 1.45 (1.2, 1.7) | 1.45 (1.2, 1.6) | 1.45 (1.2, 1.7) | 1.55 ± 0.6 | 1.58 (1.0, 1.8) | 1.58 (1.1, 1.9) |
| LbLDL-C mg/dL | 56 (47, 67) | 56 (46, 63) | 56 (47, 65) | 60 ± 22.3 | 61 (39, 68) | 61 (41, 74) |
| SdLDL-C mmol/L | 0.04 (0, 0.1) | 0.05 (0.002, 0.1) | 0.05 (0, 0.1) | 0.03 (0, 0.1) | 0 (0, 0.1) | 0.01 (0, 0.1) |
| SdLDL-C mg/dL | 1.5 (0, 3.0) | 2.0 (0.8, 2.3) | 2.0 (0, 3.0) | 1.0 (0, 3.8) | 0 (0, 4) | 0.5 (0, 4.0) |
| Large HDL-C mmol/L | 0.38 (0.2, 0.5) | 0.34 (0.2, 0.5) | 0.34 (0.2, 0.5) | 0.44 ± 0.2 | 0.28 (0.2, 0.5) | 0.3 (0.2, 0.6) |
| Large HDL-C mg/dL | 14.5 (9.3, 20.5) | 13 (6.5, 18.8) | 13 (8.0, 19.5) | 17.1 ± 8.8 | 11 (6.5, 17.8) | 12 (9, 22) |
| Int. HDL-C mmol/L | 0.68 ± 0.1 | 0.65 ± 0.2 | 0.66 ± 0.2 | 0.63 (0.6, 0.8) | 0.65 ± 0.2 | 0.62 (0.5, 0.8) |
| Int. HDL-C mg/dL | 26.3 ± 5.1 | 25.1 ± 7.9 | 25.7 ± 6.6 | 24.5 (22, 30) | 25.1 ± 7.2 | 24.0 (21, 30) |
| Small HDL-C mmol/L | 0.25 ± 0.1 | 0.3 ± 0.1 | 0.28 ± 0.1 | 0.25 ± 0.1 | 0.28 ± 0.1 | 0.27 ± 0.1 |
| Small HDL-C mg/dL | 9.7 ± 2.9 | 11.6 ± 3.2 | 10.7 ± 3.2 | 9.8 ± 4.2 | 10.9 ± 2.5 | 10.4 ± 3.4 |
| ApoA1 (µg/L) | 1.52 (1.3, 1.9) | 1.49 (1.3, 1.8) | 1.50 (1.3, 1.8) | 1.48 (1.3, 1.8) | 1.52 (1.2, 1.8 | 1.5 (1.3, 1.8) |
| ApoB (µg/L) | 0.94 (0.8, 1.1) | 1.0 (0.9, 1.1) | 0.99 (0.8, 1.1) | 0.95 (0.7, 1.2) | 1.01 (0.9, 1.2) | 1.0 (0.8, 1.2) |
| ApoB/ApoA1 | 0.61 (0.5, 0.7) | 0.65 (0.5, 0.9) | 0.62 (0.5, 0.8) | 0.62 (0.5, 0.8) | 0.64 (0.5, 0.9) | 0.6 (0.5, 0.9) |
| Total | Group Starting with N-3 PUFAs | Group Starting with Placebo | |
|---|---|---|---|
| Number of patients, n (n female) | 34 (17) | 16 (7) | 18 (10) |
| LDL-C < 1.8 mmol/L | 1 (0) | 0 | 1 (0) |
| LDL-C 1.8 to <2.6 | 5 (2) | 3(1) | 2(1) |
| LDL-C 2.6 to <3.0 | 7 (5) | 5 (3) | 2 (2) |
| LDL-C 3.0 to <4.9 | 19 (9) | 7 (3) | 12 (6) |
| LDL-C ≥ 4.9 | 2 (1) | 1 (0) | 1(1) |
| Triglycerides < 1.7 mmol/L | 29 (14) | 15 (6) | 14 (8) |
| Triglycerides ≥ 1.7 mmol/L | 5 (3) | 1(1) | 4 (2) |
| ApoB/ApoA1 > 0.9 | 4 (2) | 1 (0) | 3 (2) |
| Established ASCVD | 9 (4) | 4 (1) | 5 (3) |
| Baseline | ||||||
|---|---|---|---|---|---|---|
| Treatment Sequence | Treatment Sequence | |||||
| Variable | N-3 PUFAs First, Then Placebo n = 16 | Placebo First, Then N-3 PUFAs n = 18 | Total n = 34 | N-3 PUFAs First, Then Placebo n = 16 | Placebo First, Then N-3 PUFAs n = 18 | Total n = 34 |
| Before Treatment 1 (Month 0) | Before Treatment 2 (Month 6) | |||||
| SAA1 (µg/mL) | 0.99 (0.5, 2.4) | 2.2 (0.9, 4.1) | 1.4 (0.7, 3.1) | 1.1 (0.9, 1.8) | 2.4 (1.4, 3.7) | 1.7 (0.9, 2.5) |
| PON1 (U/mL) | 111 ± 33.2 | 109.9 ± 28.8 | 110.4 ± 30.5 | 115.7 ± 29.2 | 108.7 ± 23.3 | 112.0 ± 26.1 |
| SAA1/PON1 | 0.01 (0.005, 0.02) | 0.02 (0.01, 0.04) | 0.01 (0.01, 0.03) | 0.01 (0.01, 0.02) | 0.02 (0.01, 0.04) | 0.02 (0.01, 0.03) |
| CEC (%) | 37.5 ± 4.2 | 38.6 ± 5.0 | 39.1 ± 4.6 | 38.1 ± 4.0 | 39.5 ± 3.9 | 38.9 ± 4.0 |
| N-3 PUFAs n = 34 | Placebo n = 34 | Treatment Difference (N-3 PUFAs−Placebo) with 95% Confidence Interval | p | |
|---|---|---|---|---|
| ApoA1 (µg/L) | 1.51 (1.3, 1.7) | 1.55 (1.3, 1.9) | −0.07 [−0.12 to 0.05] | 0.29 |
| ApoB (µg/L) | 0.91 (0.8, 1.2) | 1.03 (0.8, 1.2) | −0.06 [−0.13 to 0.01] | 0.09 |
| ApoB/ApoA1 | 0.57 (0.5, 0.7) | 0.58 (0.5, 0.8) | −0.009 [−0.04 to 0.04] | 0.91 |
| SAA1 (µg/mL) | 1.36 (1.0, 2.6) | 1.68 (1.0, 2.7) | −0.11 [−0.59 to 0.11] | 0.15 |
| PON1 (U/mL) | 107 ± 25.5 | 113 ± 31.0 | −5.9 [−12.9 to 1.1] | 0.10 |
| SAA1/PON1 | 0.01 (0.01, 0.02) | 0.02 (0.01, 0.2) | −0.002 [−0.006 to 0.003] | 0.38 |
| CEC (%) | 38.5 ± 3.2 | 38.9 ± 3.5 | −0.41 [−1.8 to 1.0] | 0.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hande, L.N.; Kjellmo, C.; Pettersen, K.; Ljunggren, S.; Karlsson, H.; Cederbrant, K.; Marcusson-Ståhl, M.; Hovland, A.; Lappegård, K.T. Effect of N-3 Polyunsaturated Fatty Acids on Lipid Composition in Familial Hypercholesterolemia: A Randomized Crossover Trial. Biomedicines 2022, 10, 1809. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081809
Hande LN, Kjellmo C, Pettersen K, Ljunggren S, Karlsson H, Cederbrant K, Marcusson-Ståhl M, Hovland A, Lappegård KT. Effect of N-3 Polyunsaturated Fatty Acids on Lipid Composition in Familial Hypercholesterolemia: A Randomized Crossover Trial. Biomedicines. 2022; 10(8):1809. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081809
Chicago/Turabian StyleHande, Liv Nesse, Christian Kjellmo, Kristin Pettersen, Stefan Ljunggren, Helen Karlsson, Karin Cederbrant, Maritha Marcusson-Ståhl, Anders Hovland, and Knut Tore Lappegård. 2022. "Effect of N-3 Polyunsaturated Fatty Acids on Lipid Composition in Familial Hypercholesterolemia: A Randomized Crossover Trial" Biomedicines 10, no. 8: 1809. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081809