
Immunotherapy against the Cystine/Glutamate Antiporter xCT Improves the Efficacy of APR-246 in Preclinical Breast Cancer Models
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cell Viability Assay
2.3. Immunoblot
2.4. Cancer Stem Cell Self-Renewal Assay
2.5. FACS Analysis
2.6. In Vivo Treatments
2.7. ELISA Assay
2.8. Antibody Dependent Cell Cytotoxicity (ADCC)
2.9. Statistical Analysis
3. Results
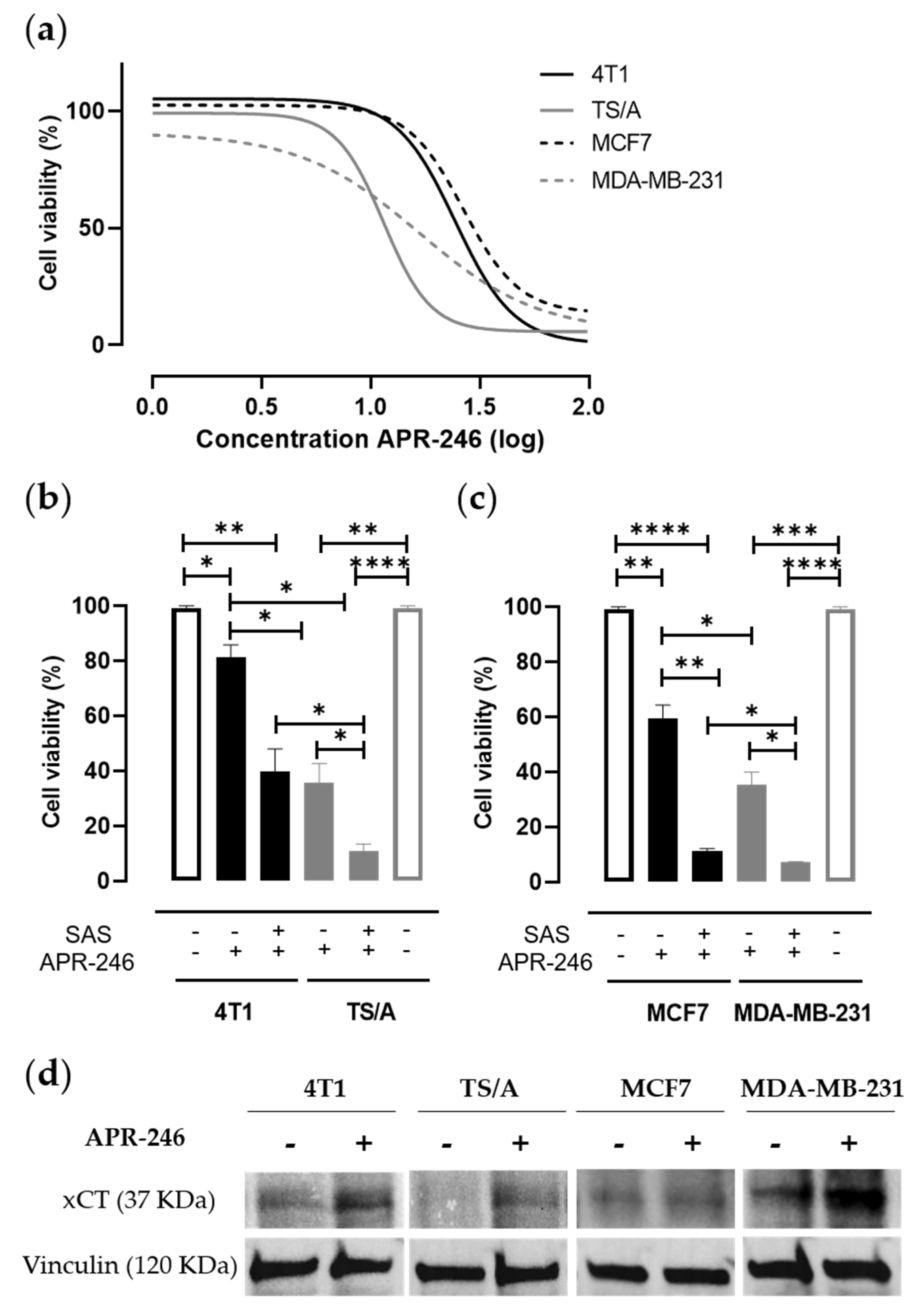
3.1. Pharmacological Inhibition of xCT Combined with APR-246 Treatment Effectively Reduces Tumor Cell Viability by Altering Intracellular Redox Balance
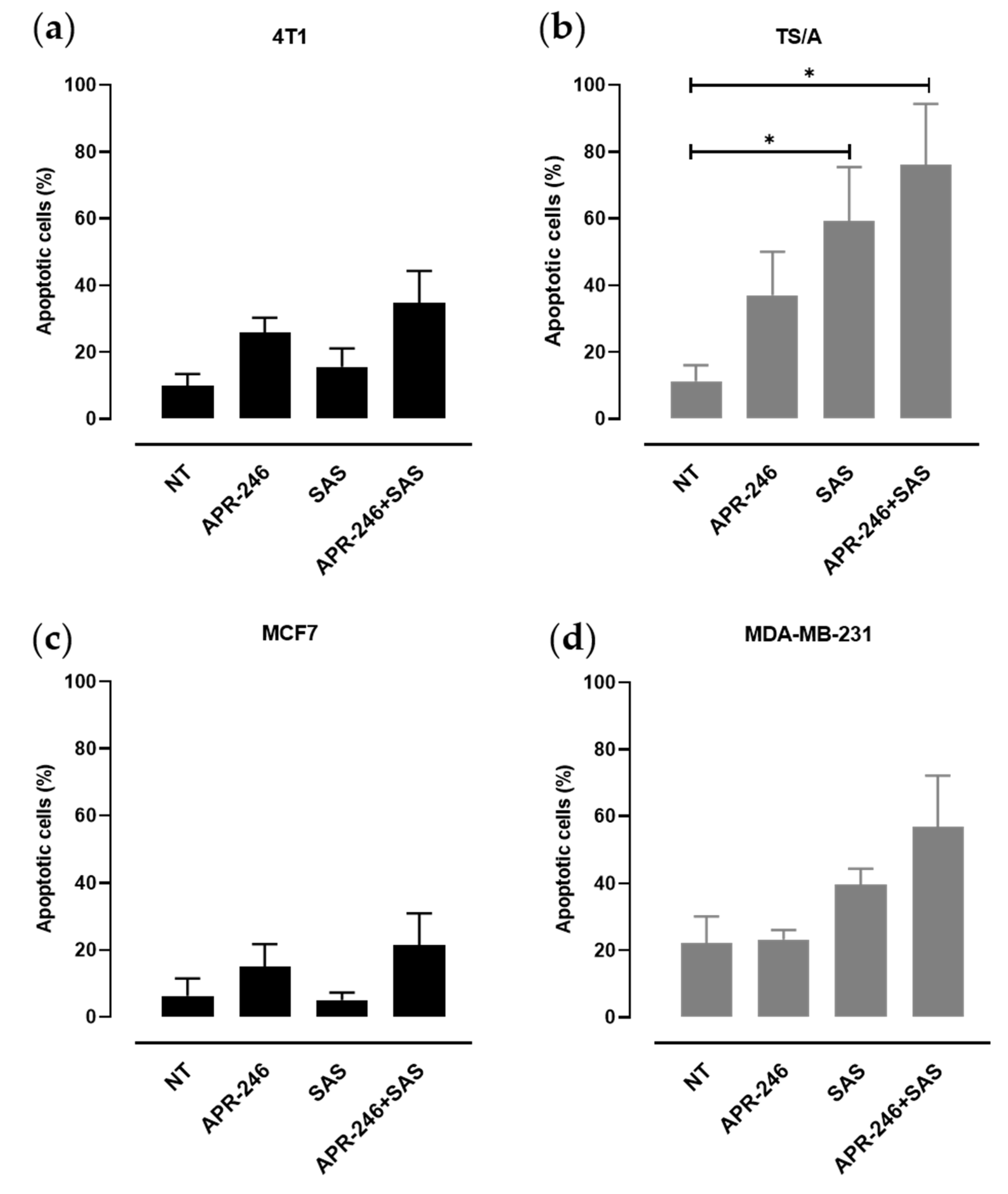
3.2. xCT Inhibition Potentiates APR-246 Effectiveness by Inducing Apoptosis of Tumor Cells
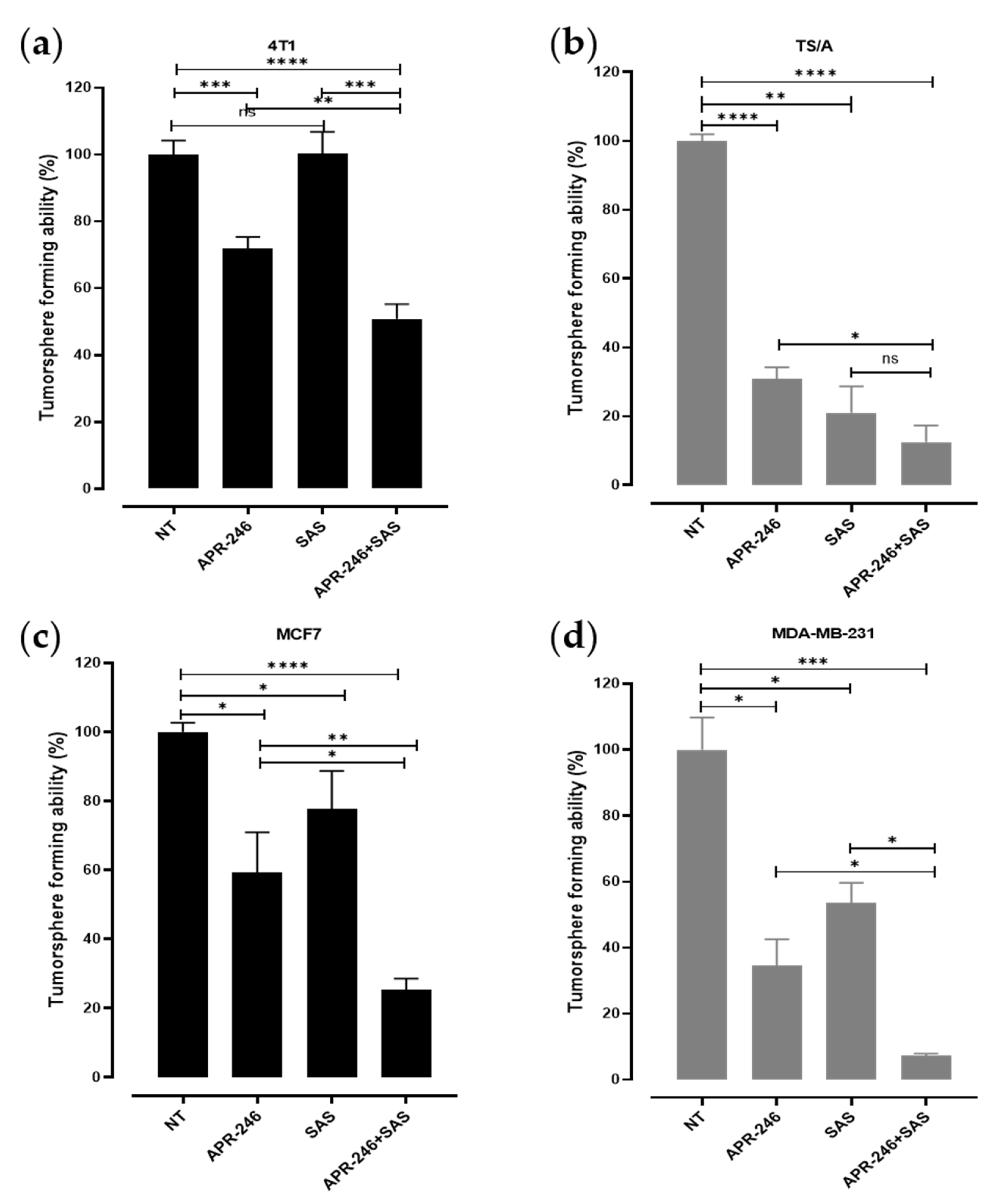
3.3. The Combinatorial Treatment with APR-246 and SAS Synergistically Inhibits the Generation of Tumorspheres
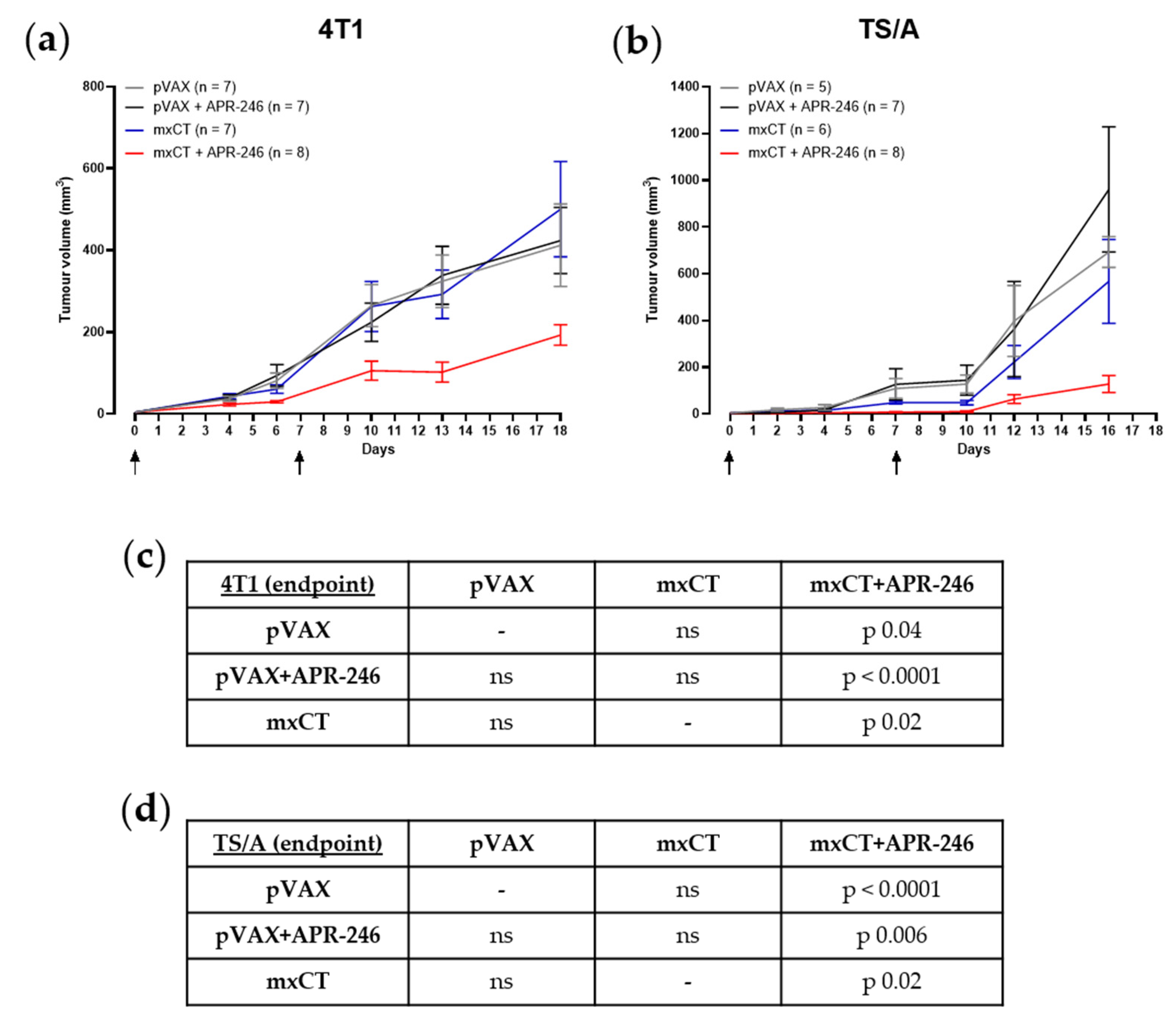
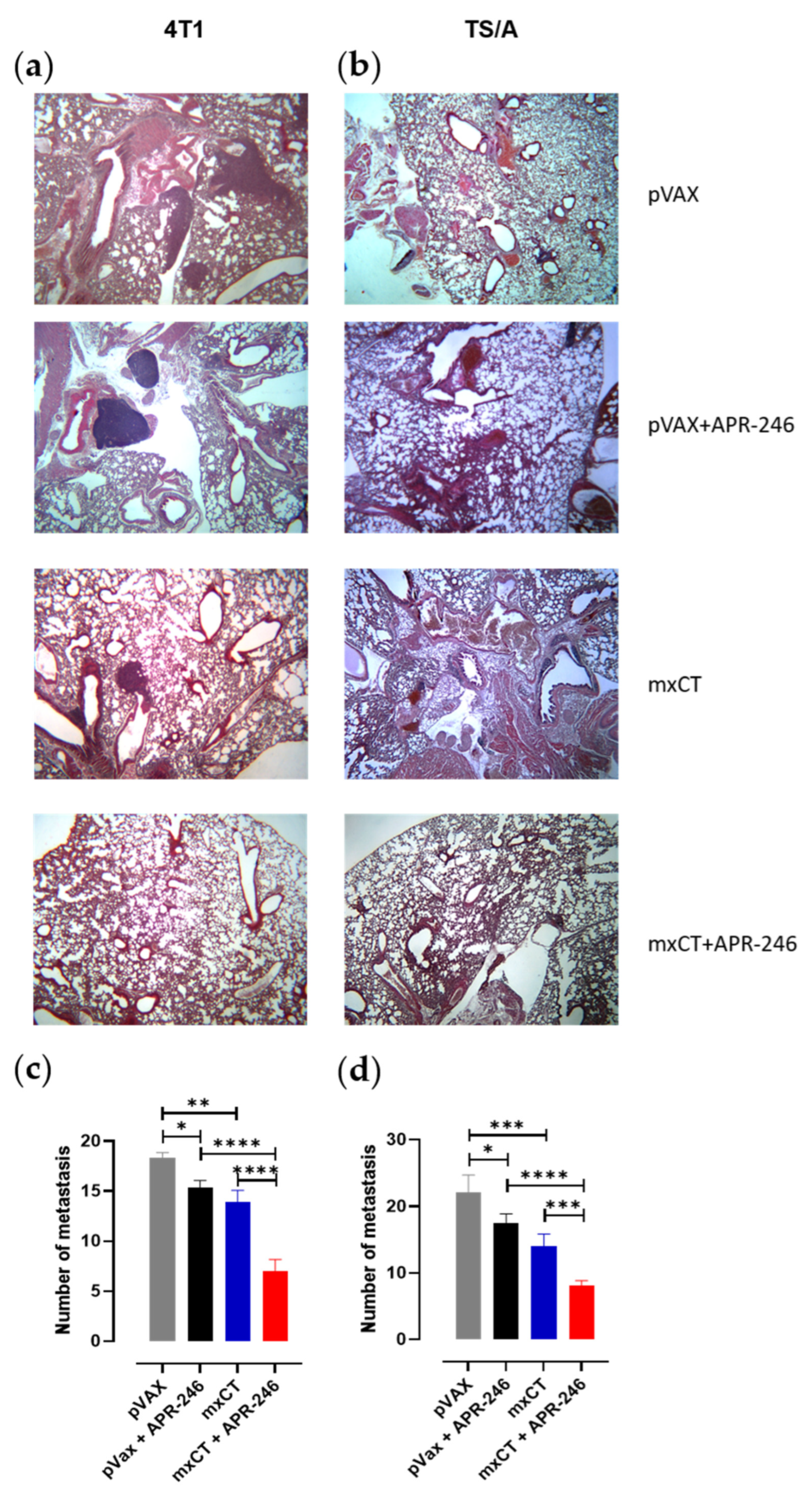
3.4. xCT Immune Targeting Coupled with APR-246 Treatment Hampers Tumor Growth and Lung Metastasis Formation in Preclinical Models of Breast Cancer
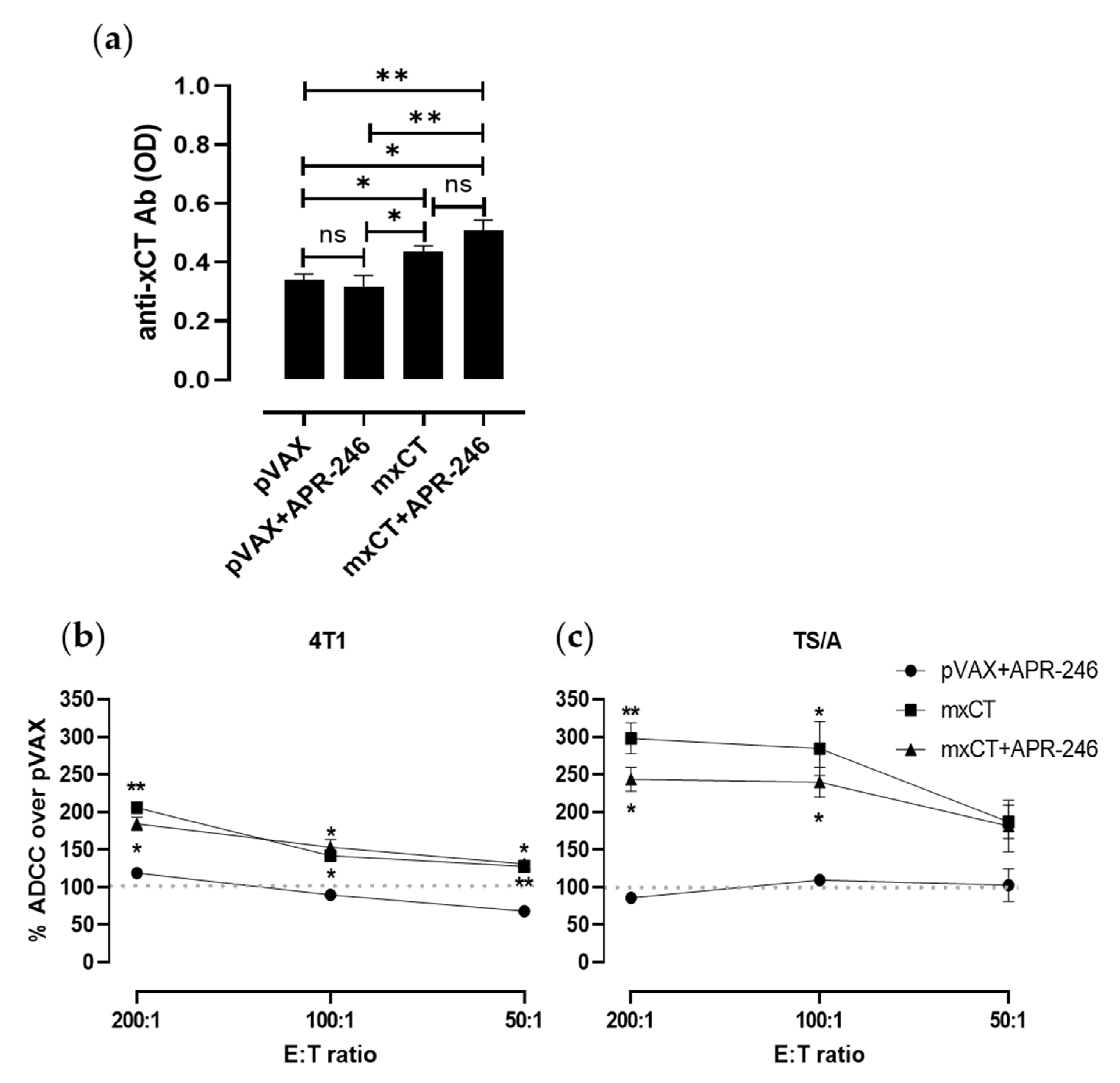
3.5. DNA Vaccination against xCT Induces an Antibody Response Driving ADCC
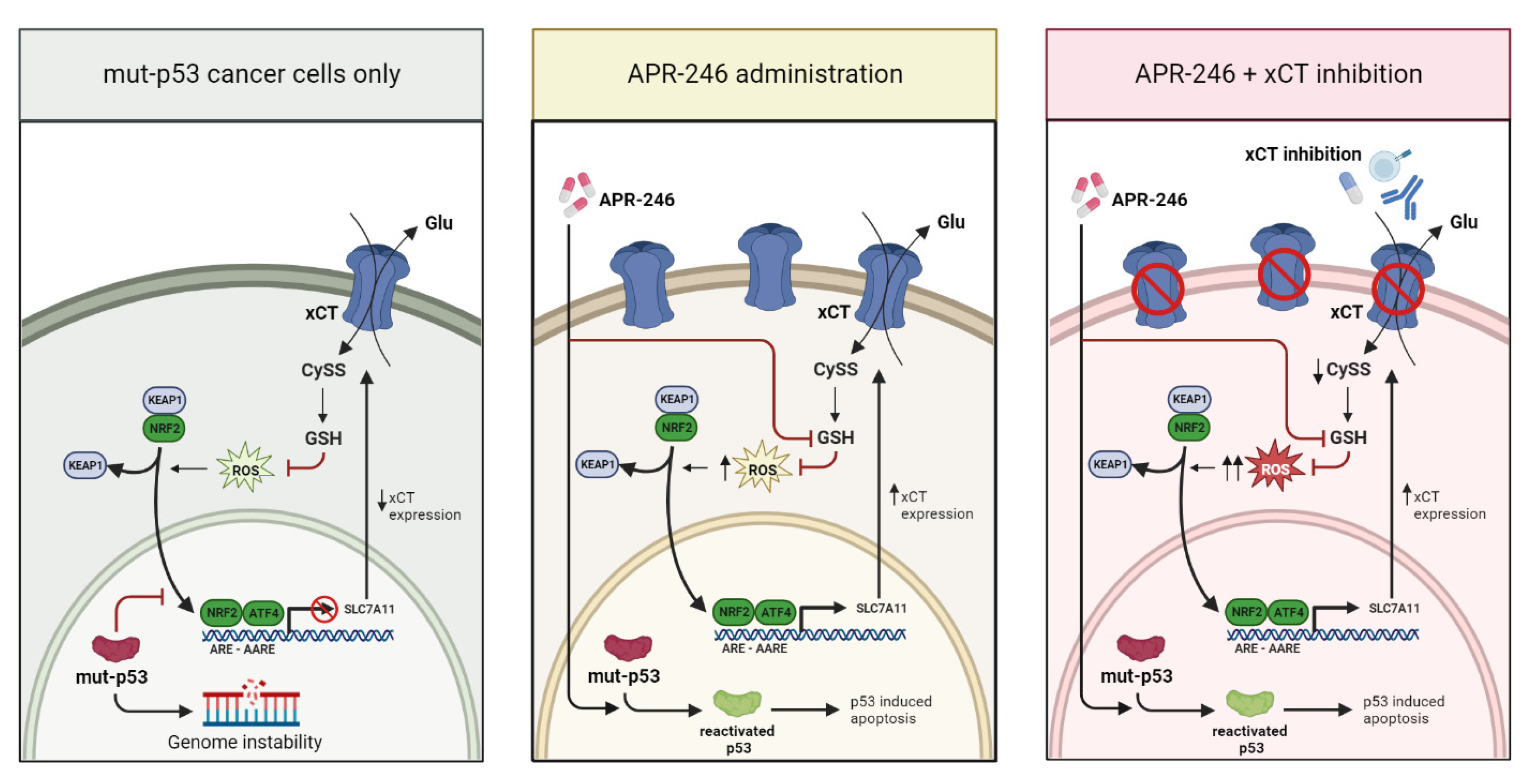
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ayala de la Pena, F.; Andres, R.; Garcia-Saenz, J.A.; Manso, L.; Margeli, M.; Dalmau, E.; Pernas, S.; Prat, A.; Servitja, S.; Ciruelos, E. SEOM clinical guidelines in early stage breast cancer (2018). Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2019, 21, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglino, E.; Conti, L.; Cavallo, F. Breast cancer stem cell antigens as targets for immunotherapy. Semin. Immunol. 2020, 47, 101386. [Google Scholar] [CrossRef] [PubMed]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016, 76, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Ruiu, R.; Rolih, V.; Bolli, E.; Barutello, G.; Riccardo, F.; Quaglino, E.; Merighi, I.F.; Pericle, F.; Donofrio, G.; Cavallo, F.; et al. Fighting breast cancer stem cells through the immune-targeting of the xCT cystine-glutamate antiporter. Cancer Immunol. Immunother. 2018, 68, 131–141. [Google Scholar] [CrossRef]
- Dornier, E.; Rabas, N.; Mitchell, L.; Novo, D.; Dhayade, S.; Marco, S.; Mackay, G.; Sumpton, D.; Pallares, M.; Nixon, C.; et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 2017, 8, 2255. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e629. [Google Scholar] [CrossRef]
- Linares, V.; Alonso, V.; Domingo, J.L. Oxidative stress as a mechanism underlying sulfasalazine-induced toxicity. Expert. Opin. Drug Saf. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Bolli, E.; O’Rourke, J.P.; Conti, L.; Lanzardo, S.; Rolih, V.; Christen, J.M.; Barutello, G.; Forni, M.; Pericle, F.; Cavallo, F. A Virus-Like-Particle immunotherapy targeting Epitope-Specific anti-xCT expressed on cancer stem cell inhibits the progression of metastatic cancer in vivo. Oncoimmunology 2017, 7, e1408746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donofrio, G.; Tebaldi, G.; Lanzardo, S.; Ruiu, R.; Bolli, E.; Ballatore, A.; Rolih, V.; Macchi, F.; Conti, L.; Cavallo, F. Bovine herpesvirus 4-based vector delivering the full length xCT DNA efficiently protects mice from mammary cancer metastases by targeting cancer stem cells. Oncoimmunology 2018, 7, e1494108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolih, V.; Caldeira, J.; Bolli, E.; Salameh, A.; Conti, L.; Barutello, G.; Riccardo, F.; Magri, J.; Lamolinara, A.; Parra, K.; et al. Development of a VLP-Based Vaccine Displaying an xCT Extracellular Domain for the Treatment of Metastatic Breast Cancer. Cancers 2020, 12, 1492. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Bolli, E.; Di Lorenzo, A.; Franceschi, V.; Macchi, F.; Riccardo, F.; Ruiu, R.; Russo, L.; Quaglino, E.; Donofrio, G.; et al. Immunotargeting of the xCT cystine/glutamate antiporter potentiates the efficacy of Her2-targeted immunotherapies in breast cancer. Cancer Immunol. Res. 2020, 8, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- Magri, J.; Gasparetto, A.; Conti, L.; Calautti, E.; Cossu, C.; Ruiu, R.; Barutello, G.; Cavallo, F. Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells 2021, 10, 108. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 in breast cancer: Potential as a therapeutic target and biomarker. Breast Cancer Res. Treat. 2018, 170, 213–219. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahme, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myelodysplasies (GFM). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, K.M.; Corrales Benitez, M.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1(Met) (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Sang, H.; Pisarev, V.M.; Chavez, J.; Robinson, S.; Guo, Y.; Hatcher, L.; Munger, C.; Talmadge, C.B.; Solheim, J.C.; Singh, R.K.; et al. Murine mammary adenocarcinoma cells transfected with p53 and/or Flt3L induce antitumor immune responses. Cancer Gene Ther. 2005, 12, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; de Giovanni, C.; Lollini, P.L.; Nicoletti, G.; Prodi, G. TS/A: A new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clin. Exp. Metastasis 1983, 1, 373–380. [Google Scholar] [CrossRef]
- Lee, K.; Wang, T.; Paszczynski, A.J.; Daoud, S.S. Expression proteomics to p53 mutation reactivation with PRIMA-1 in breast cancer cells. Biochem. Biophys. Res. Commun. 2006, 349, 1117–1124. [Google Scholar] [CrossRef]
- Geninatti Crich, S.; Cadenazzi, M.; Lanzardo, S.; Conti, L.; Ruiu, R.; Alberti, D.; Cavallo, F.; Cutrin, J.C.; Aime, S. Targeting ferritin receptors for the selective delivery of imaging and therapeutic agents to breast cancer cells. Nanoscale 2015, 7, 6527–6533. [Google Scholar] [CrossRef]
- Conti, L.; Lanzardo, S.; Arigoni, M.; Antonazzo, R.; Radaelli, E.; Cantarella, D.; Calogero, R.A.; Cavallo, F. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013, 27, 4731–4744. [Google Scholar] [CrossRef] [PubMed]
- Ruiu, R.; Barutello, G.; Arigoni, M.; Riccardo, F.; Conti, L.; Peppino, G.; Annaratone, L.; Marchio, C.; Mengozzi, G.; Calogero, R.A.; et al. Identification of TENM4 as a Novel Cancer Stem Cell-Associated Molecule and Potential Target in Triple Negative Breast Cancer. Cancers 2021, 13, 894. [Google Scholar] [CrossRef] [PubMed]
- Regis, G.; Icardi, L.; Conti, L.; Chiarle, R.; Piva, R.; Giovarelli, M.; Poli, V.; Novelli, F. IL-6, but not IFN-gamma, triggers apoptosis and inhibits in vivo growth of human malignant T cells on STAT3 silencing. Leukemia 2009, 23, 2102–2108. [Google Scholar] [CrossRef] [PubMed]
- Barutello, G.; Curcio, C.; Spadaro, M.; Arigoni, M.; Trovato, R.; Bolli, E.; Zheng, Y.; Ria, F.; Quaglino, E.; Iezzi, M.; et al. Antitumor immunization of mothers delays tumor development in cancer-prone offspring. Oncoimmunology 2015, 4, e1005500. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Mima, K.; Nakagawa, S.; Ishimoto, T.; Miyake, K.; et al. Cystine/glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellular carcinoma. Oncol. Rep. 2013, 29, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar]
- Lee, J.R.; Roh, J.L.; Lee, S.M.; Park, Y.; Cho, K.J.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Overexpression of cysteine-glutamate transporter and CD44 for prediction of recurrence and survival in patients with oral cavity squamous cell carcinoma. Head Neck 2018, 40, 2340–2346. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Y.; Ling, J.; Zhuo, W.; Yu, Z.; Luo, Y.; Zhu, Y. Overexpression of SLC7A11: A novel oncogene and an indicator of unfavorable prognosis for liver carcinoma. Future Oncol. 2018, 14, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: Use of sulfasalazine. Cancer Chemother. Pharmacol. 2009, 64, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padro, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ‘t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, F.; Matsunaga, N.; Hamamura, K.; Suzuki, K.; Nakao, T.; Okazaki, H.; Kutsukake, M.; Fukumori, S.; Tsuji, Y.; To, H. Administering xCT Inhibitors Based on Circadian Clock Improves Antitumor Effects. Cancer Res. 2017, 77, 6603–6613. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Kuwata, K.; Sugimoto, T.; Igarashi, K.; Osaki, M.; Okada, F.; Fujii, J.; Bannai, S.; Sato, H. Enhanced expression of cystine/glutamate transporter in the lung caused by the oxidative-stress-inducing agent paraquat. Free. Radic. Biol. Med. 2012, 53, 2197–2203. [Google Scholar] [CrossRef]
- Chung, W.J.; Sontheimer, H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB. J. Neurochem. 2009, 110, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Doxsee, D.W.; Gout, P.W.; Kurita, T.; Lo, M.; Buckley, A.R.; Wang, Y.; Xue, H.; Karp, C.M.; Cutz, J.C.; Cunha, G.R.; et al. Sulfasalazine-induced cystine starvation: Potential use for prostate cancer therapy. Prostate 2007, 67, 162–171. [Google Scholar] [CrossRef]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef] [Green Version]
- Shahbandi, A.; Nguyen, H.D.; Jackson, J.G. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 2020, 6, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.I.; Tuszynski, J. The molecular mechanism of action of methylene quinuclidinone and its effects on the structure of p53 mutants. Oncotarget 2018, 9, 37137–37156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1(Met) (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022, 107, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Madden, S.F.; Bykov, V.J.N.; Crown, J.; Wiman, K.G.; Duffy, M.J. The Mutant p53-Targeting Compound APR-246 Induces ROS-Modulating Genes in Breast Cancer Cells. Transl. Oncol. 2018, 11, 1343–1349. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e178. [Google Scholar] [CrossRef] [Green Version]
- Tallerico, R.; Conti, L.; Lanzardo, S.; Sottile, R.; Garofalo, C.; Wagner, A.K.; Johansson, M.H.; Cristiani, C.M.; Karre, K.; Carbone, E.; et al. NK cells control breast cancer and related cancer stem cell hematological spread. Oncoimmunology 2017, 6, e1284718. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, L.; Gomez-Casal, R.; Wang, X.; Hayashi, R.; Appella, E.; Kopelovich, L.; DeLeo, A.B. Targeting cancer stem cells with p53 modulators. Oncotarget 2016, 7, 45079–45093. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | p53 Status | APR-246 * | APR-246 + SAS * |
|---|---|---|---|
| 4T1 | Null | 24.4 | 10.8 |
| TS/A | mutated (R270H) | 11.6 | 4.2 |
| MCF7 | wild type | 26.6 | 16.6 |
| MDA-MB-231 | mutated (R280K) | 9.2 | 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barutello, G.; Di Lorenzo, A.; Gasparetto, A.; Galiazzi, C.; Bolli, E.; Conti, L.; Cavallo, F. Immunotherapy against the Cystine/Glutamate Antiporter xCT Improves the Efficacy of APR-246 in Preclinical Breast Cancer Models. Biomedicines 2022, 10, 2843. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112843
Barutello G, Di Lorenzo A, Gasparetto A, Galiazzi C, Bolli E, Conti L, Cavallo F. Immunotherapy against the Cystine/Glutamate Antiporter xCT Improves the Efficacy of APR-246 in Preclinical Breast Cancer Models. Biomedicines. 2022; 10(11):2843. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112843
Chicago/Turabian StyleBarutello, Giuseppina, Antonino Di Lorenzo, Alessandro Gasparetto, Chiara Galiazzi, Elisabetta Bolli, Laura Conti, and Federica Cavallo. 2022. "Immunotherapy against the Cystine/Glutamate Antiporter xCT Improves the Efficacy of APR-246 in Preclinical Breast Cancer Models" Biomedicines 10, no. 11: 2843. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112843