
Creation of an iPSC-Based Skeletal Muscle Model of McArdle Disease Harbouring the Mutation c.2392T>C (p.Trp798Arg) in the PYGM Gene

Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Samples
iPSC Lines
2.2. iPSC Culture
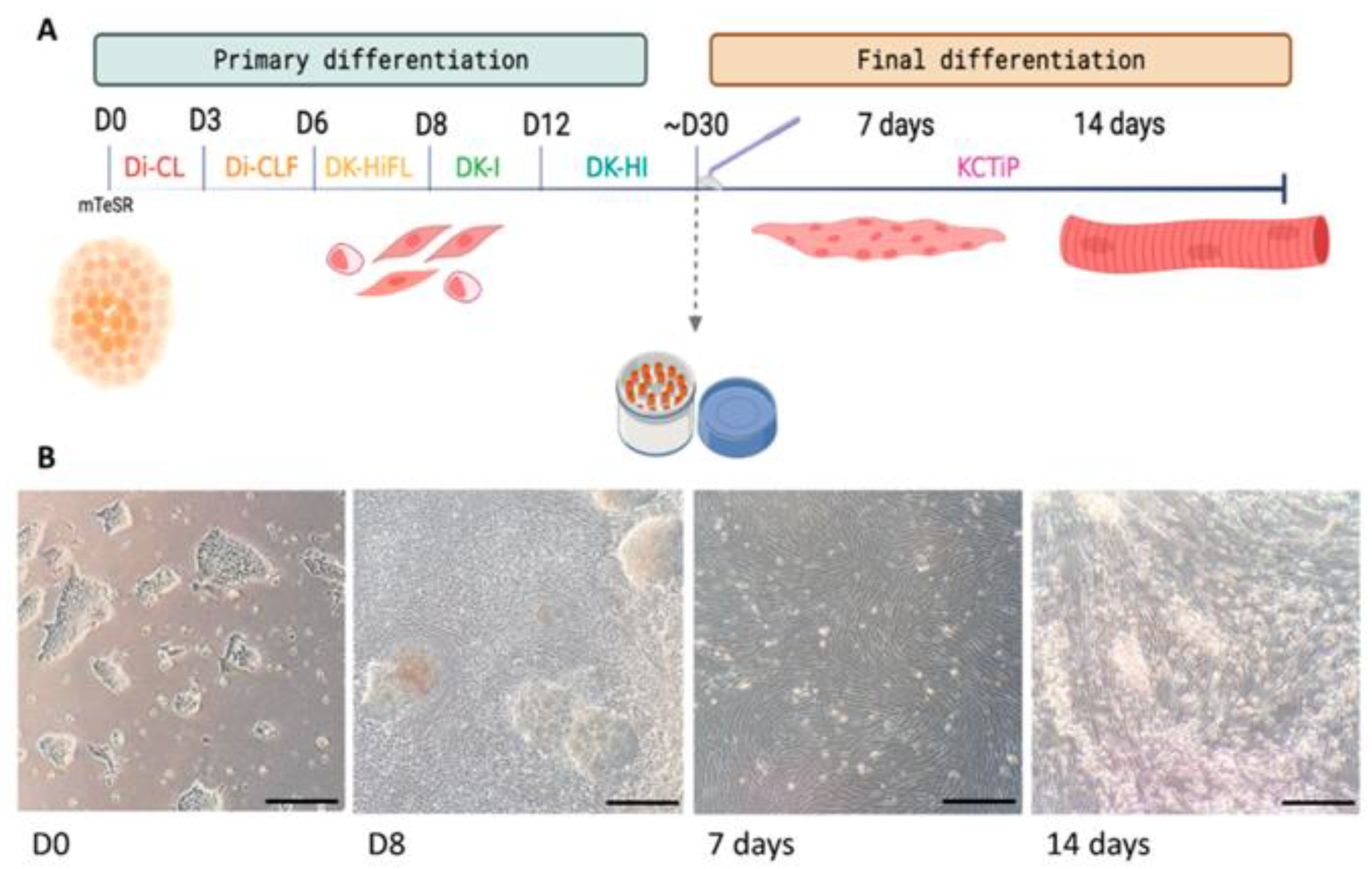
2.3. Skeletal Muscle Differentiation of iPSCs
2.3.1. Primary Differentiation
2.3.2. Expansion and Cryopreservation of Myogenic Progenitors
2.3.3. Final Differentiation
2.4. Characterization of Myogenic Cells
2.4.1. RT-qPCR
2.4.2. Immunofluorescence of Myogenic Cells
2.5. Validation of the Human iPSC-Based Model of McArdle Disease
2.5.1. PYGM Gene Expression Analyses
2.5.2. Western Blot
2.5.3. PAS Staining
3. Results
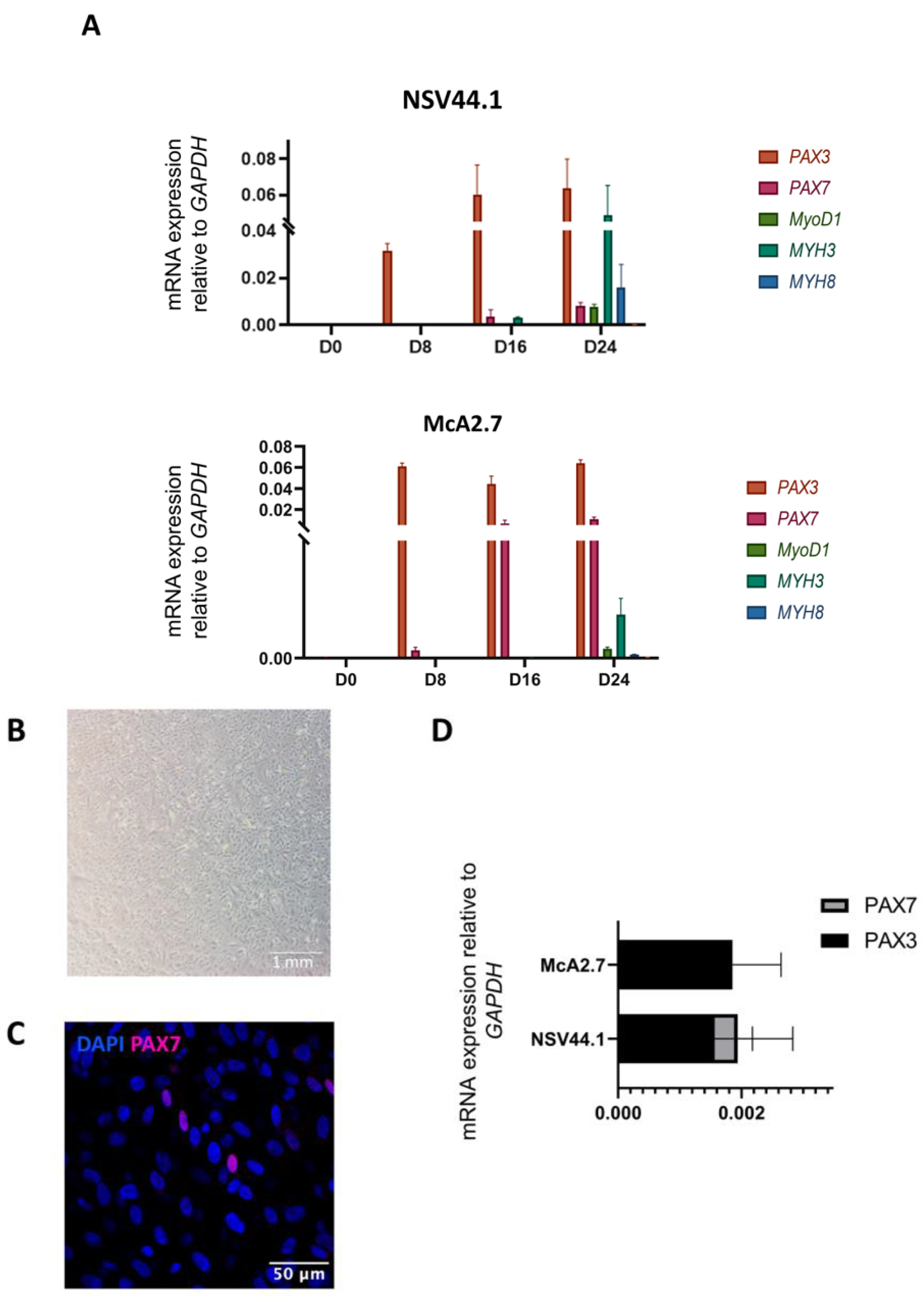
3.1. Generation of a Human iPSC-Based Skeletal Muscle Model of McArdle Disease



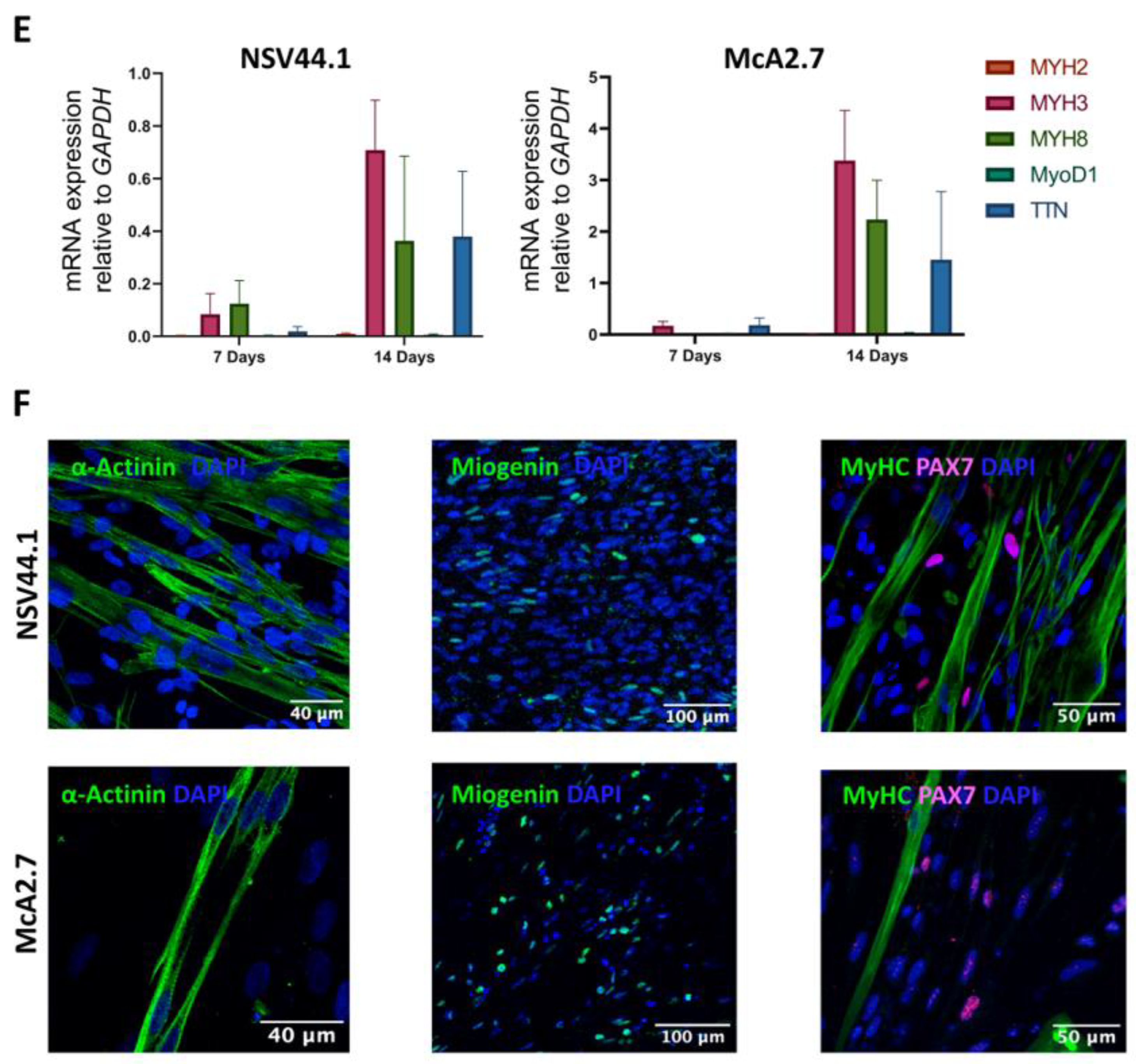
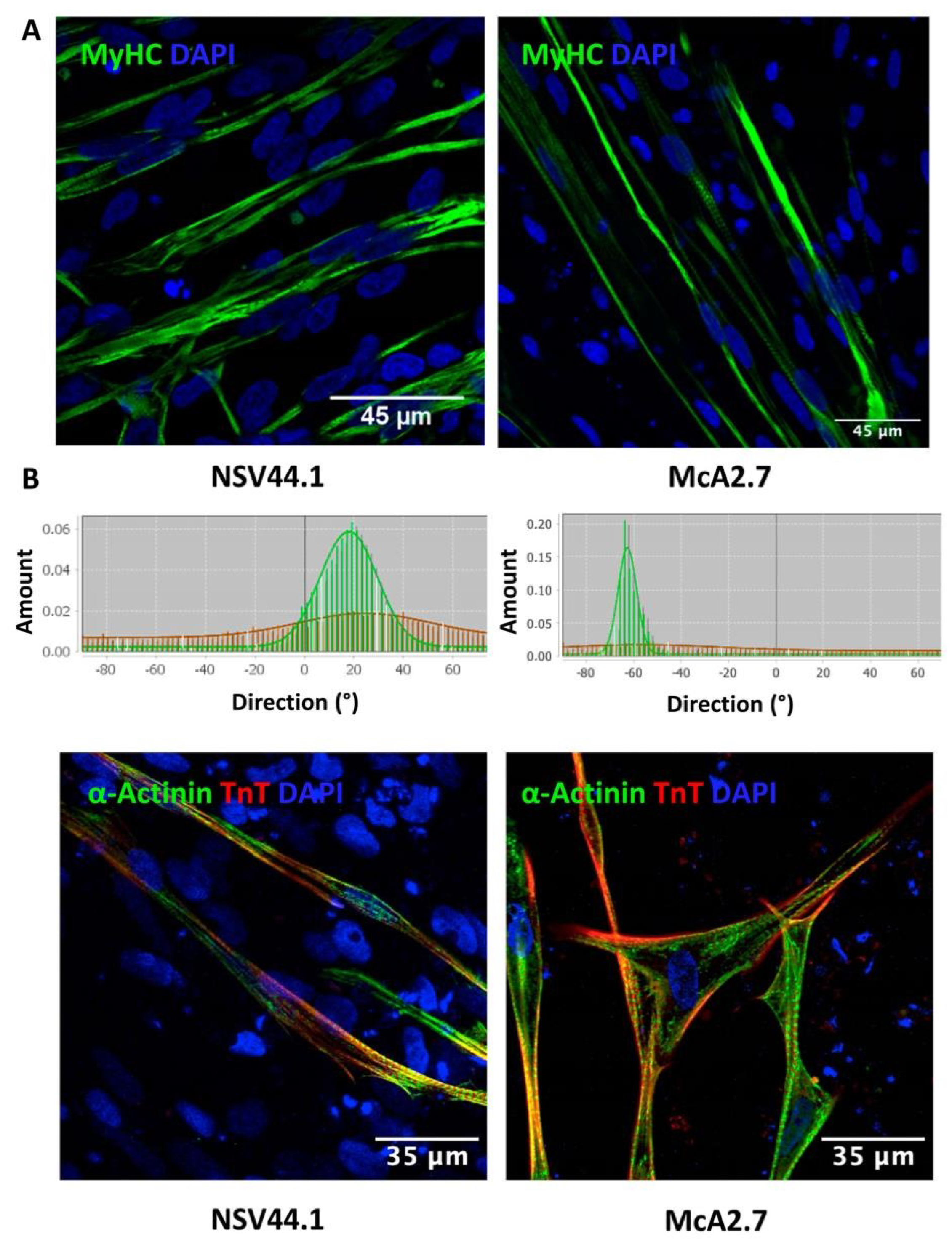
Maturation of the Myofibres
3.2. Validation of the McArdle Disease Skeletal Muscle Model
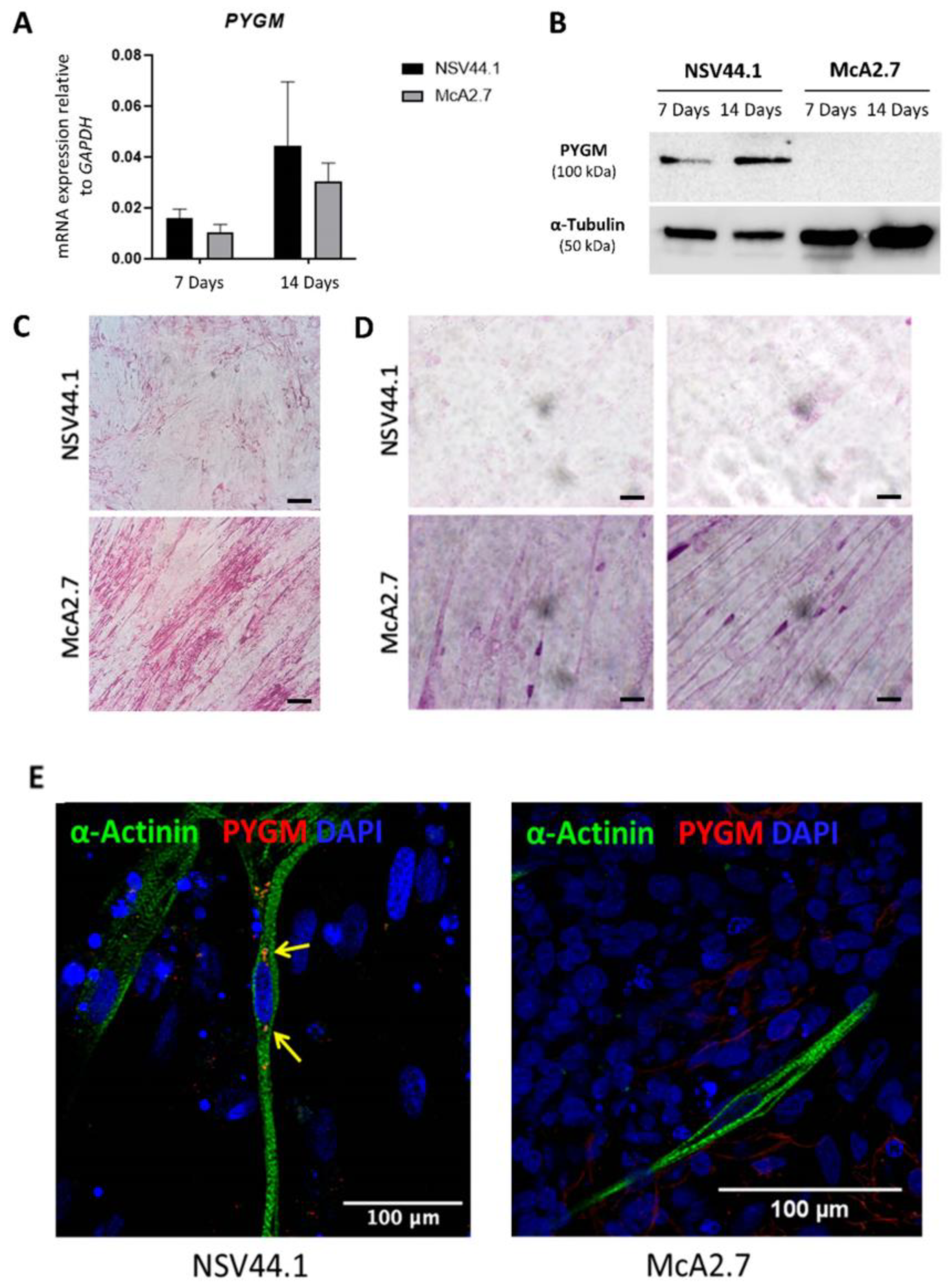
3.2.1. Expression of PYGM in iPSC-Derived Skeletal Muscle
3.2.2. Immunodetection Analysis
3.2.3. PAS Staining
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Özen, H. Glycogen storage diseases: New perspectives. World J. Gastroenterol. 2007, 13, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- Santalla, A.; Nogales-Gadea, G.; Ørtenblad, N.; Brull, A.; de Luna, N.; Pinós, T.; Lucia, A. McArdle Disease: A Unique Study Model in Sports Medicine. Sport. Med. 2014, 44, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Gadea, G.; Brull, A.; Santalla, A.; Andreu, A.L.; Arenas, J.; Martín, M.A.; Lucia, A.; de Luna, N.; Pinós, T. McArdle Disease: Update of Reported Mutations and Polymorphisms in the PYGM Gene. Hum. Mutat. 2015, 36, 669–678. [Google Scholar] [CrossRef]
- Santalla, A.; Nogales-Gadea, G.; Encinar, A.B.; Vieitez, I.; González-Quintana, A.; Serrano-Lorenzo, P.; Consuegra, I.G.; Asensio, S.; Ballester-Lopez, A.; Pintos-Morell, G.; et al. Genotypic and phenotypic features of all Spanish patients with McArdle disease: A 2016 update. BMC Genom. 2017, 18, 819. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, R.; Martinuzzi, A.; Schoser, B. Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V). Cochrane Database Syst. Rev. 2014, 2014, CD003458. [Google Scholar] [CrossRef]
- Lucia, A.; Quinlivan, R.; Wakelin, A.; Martín, M.A.; Andreu, A.L. The “McArdle paradox”: Exercise is a good advice for the exercise intolerant. Br. J. Sports Med. 2013, 47, 728–729. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- del Ortuño-Costela, M.C.; Cerrada, V.; Moreno-Izquierdo, A.; García-Consuegra, I.; Laberthonnière, C.; Delourme, M.; Garesse, R.; Arenas, J.; Fuster García, C.; García García, G.; et al. Generation of the First Human In Vitro Model for McArdle Disease Based on iPSC Technology. Int. J. Mol. Sci. 2022, 23, 13964. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Díaz, F.; Canals, I.; Hansen, M.G.; Rufián-Vázquez, L.; Ehinger, J.K.; Elmér, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial dysfunction and calcium dysregulation in leigh syndrome induced pluripotent stem cell derived neurons. Int. J. Mol. Sci. 2020, 21, 3191. [Google Scholar] [CrossRef]
- Cerrada, V.; García-López, M.; Alvarez-Galeano, S.; Moreno-Izquierdo, A.; Lucia, A.; Rabasa Pérez, M.; Arenas, J.; Gallardo, M.E. Generation of the iPSC line IISHDOi007-A from peripheral blood mononuclear cells from a patient with McArdle disease harbouring the mutation c.2392 T > C; p.Trp798Arg. Stem Cell Res. 2020, 49, 49. [Google Scholar] [CrossRef]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Zimmerman, J.F.; Rao, J.; Sieiro, D.; McNamara, H.M.; Cherrier, T.; Rodríguez-DelaRosa, A.; Hick-Colin, A.; Bousson, F.; Fugier-Schmucker, C.; et al. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell-derived skeletal muscle in vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2022960118. [Google Scholar] [CrossRef] [PubMed]
- Schaart, G.; Hesselink, R.P.; Keizer, H.A.; Van Kranenburg, G.; Drost, M.R.; Hesselink, M.K.C. A modified PAS stain combined with immunofluorescence for quantitative analyses of glycogen in muscle sections. Histochem. Cell Biol. 2004, 122, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Wu, J.; Ortiz-Vitali, J.L.; Li, Y.; Darabi, R. Directed differentiation of human pluripotent stem cells toward skeletal myogenic progenitors and their purification using surface markers. Cells 2021, 10, 2746. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Villarreal-Salazar, M.; Brull, A.; Nogales-Gadea, G.; Andreu, A.L.; Martín, M.A.; Arenas, J.; Santalla, A.; Lucia, A.; Vissing, J.; Krag, T.O.; et al. Preclinical Research in McArdle Disease: A Review of Research Models and Therapeutic Strategies. Genes 2022, 13, 74. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef]
- del Carmen Ortuño-Costela, M.; García-López, M.; Cerrada, V.; Gallardo, M.E. iPSCs: A powerful tool for skeletal muscle tissue engineering. J. Cell Mol. Med. 2019, 23, 3784–3794. [Google Scholar] [CrossRef]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef]
- Jarocha, D.; Stangel-Wojcikiewicz, K.; Basta, A.; Majka, M. Efficient myoblast expansion for regenerative medicine use. Int. J. Mol. Med. 2014, 34, 83–91. [Google Scholar] [CrossRef]
- Melendez, J.; Sieiro, D.; Salgado, D.; Morin, V.; Dejardin, M.J.; Zhou, C.; Mullen, A.C.; Marcelle, C. TGFβ signalling acts as a molecular brake of myoblast fusion. Nat. Commun. 2021, 12, 749. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Tranchant, C.; Vilquin, J.T.; Labouret, P.; Warter, J.M.; Poindron, P. Stimulating effects of prednisolone on acetylcholine receptor expression and myogenesis in primary culture of newborn rat muscle cells. J. Neurol. Sci. 1989, 92, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Mondragon-Gonzalez, R.; Xu, B.; Magli, A.; Kim, H.; Lainé, J.; Kiley, J.; McKee, H.; Rinaldi, F.; Aho, J.; et al. Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. Elife 2019, 8, e47970. [Google Scholar] [CrossRef] [PubMed]
- Mzaaleyrat, K.; Badja, C.; Broucqsault, N.; Chevalier, R.; Laberthonnière, C.; Dion, C.; Baldasseroni, L.; El-Yazidi, C.; Thomas, M.; Bachelier, R.; et al. Multilineage Differentiation for Formation of Innervated Skeletal Muscle Fibers from Healthy and Diseased Human Pluripotent Stem Cells. Cells 2020, 9, 1531. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.L.; Ang, L.T.; Loh, K.M. A critical look: Challenges in differentiating human pluripotent stem cells into desired cell types and organoids. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e368. [Google Scholar] [CrossRef]
- García-Consuegra, I.; Asensio-Peña, S.; Ballester-Lopez, A.; Francisco-Velilla, R.; Pinos, T.; Pintos-Morell, G.; Coll-Cantí, J.; González-Quintana, A.; Andreu, A.L.; Arenas, J.; et al. Missense mutations have unexpected consequences: The McArdle disease paradigm. Hum. Mutat. 2018, 39, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Zierz, S. McArdle Disease: Clinical, biochemical and molecular genetic analysis of 58 patients. Neuromuscul. Disord. 2017, 27, S204–S205. [Google Scholar] [CrossRef]
- Rae, D.E.; Noakes, T.D.; San Juan, A.F.; Pérez, M.; Nogales-Gadea, G.; Ruiz, J.R.; Morán, M.; Martín, M.A.; Andreu, A.L.; Arenas, J.; et al. Excessive skeletal muscle recruitment during strenuous exercise in McArdle patients. Eur. J. Appl. Physiol. 2010, 110, 1047–1055. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration | Company and Reference | |
|---|---|---|
| DMEM/F12 | ThermoFisher, USA, 11320-082 | |
| FBS | 18% | Sigma Aldrich, USA, F7524-500ML |
| Insulin | 1% | Sigma Aldrich, USA, I9278 |
| P/S | 0.2% | Sigma Aldrich, USA, P0781 |
| Human recombinant EGF | 20 ng/mL | Stemcell technologies, Canada, 78006.1 |
| Human bFGF-2 | 20 ng/mL | Miltenyi, Germany, 130-093-839 |
| Human recombinant HGF | 10 ng/mL | Stemcell technologies, Canada, 78019.1 |
| Dexamethasone | 10 µM | Sigma Aldrich, USA, D4902 |
| Target | Forward/Reverse Primer (5′-3′) |
|---|---|
| MYOD1 | GACGGCATGATGGACTACAG/AGGCAGTCTAGGCTCGACAC |
| PAX3 | TACAGGTCTGGTTTAGCAAC/GATCTGACACAGCTTGTGGA |
| PAX7 | CAGACAGGTGGCGACTCC/CGCGGCTAATCGAACTCAC |
| MYH2 | GGAGCTGGTGGAGGGGCCAA/TGCTCCATGGCACCAGGAGTTT |
| MYH3 | GCTTGTGGGCGGAGGTCTGG/AGGGCTGGTTCTGAGCCTCGAT |
| MYH8 | TCCACCAAGAACCCAGAGAGTGG/TGGGCCTCAATCCGCTCCTT |
| TITIN | CCGAAATGCATCAGTCAGCG/CCTTGCAAGCTTGTGTCACC |
| Antibody | Dilution | Company and RRID |
|---|---|---|
| Rabbit anti PAX7 | 1:50 | Abcam, Cambridge, UK, (ab187339) RRID: AB_2813893 |
| Mouse anti Myogenin | 1:25 | Abcam, UK (ab212668) RRID: AB_262133 |
| Mouse anti α-Actinin | 1:500 | Sigma Aldrich, USA, (A7811) RRID: AB_476766 |
| Mouse anti MyHC | 1:250 | Sigma Aldrich, USA, (M4276) RRID: AB_477190 |
| Rabbit anti PYGM | 1:50 | Sigma Aldrich, USA, (HPA056003) RRID: AB_2683006 |
| Goat anti Mouse IgG (H + L), Alexa Fluor 488 | 1:300 | ThermoFisher, USA, (A-11029) RRID: AB_2534088 |
| Goat anti Rabbit IgG, Alexa Fluor 568 | 1:1000 | Invitrogen, USA, (A-11011) RRID: AB_143157 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerrada, V.; García-Consuegra, I.; Arenas, J.; Gallardo, M.E. Creation of an iPSC-Based Skeletal Muscle Model of McArdle Disease Harbouring the Mutation c.2392T>C (p.Trp798Arg) in the PYGM Gene. Biomedicines 2023, 11, 2434. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092434
Cerrada V, García-Consuegra I, Arenas J, Gallardo ME. Creation of an iPSC-Based Skeletal Muscle Model of McArdle Disease Harbouring the Mutation c.2392T>C (p.Trp798Arg) in the PYGM Gene. Biomedicines. 2023; 11(9):2434. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092434
Chicago/Turabian StyleCerrada, Victoria, Inés García-Consuegra, Joaquín Arenas, and M. Esther Gallardo. 2023. "Creation of an iPSC-Based Skeletal Muscle Model of McArdle Disease Harbouring the Mutation c.2392T>C (p.Trp798Arg) in the PYGM Gene" Biomedicines 11, no. 9: 2434. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092434