Reactive Metamizole Metabolites Enhance the Toxicity of Hemin on the ATP Pool in HL60 Cells by Inhibition of Glycolysis

 ,
,  and
and
Abstract
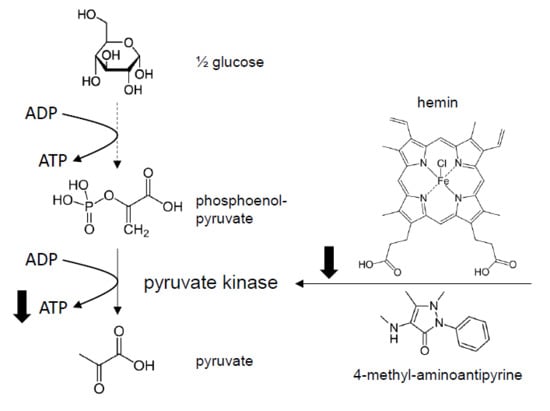
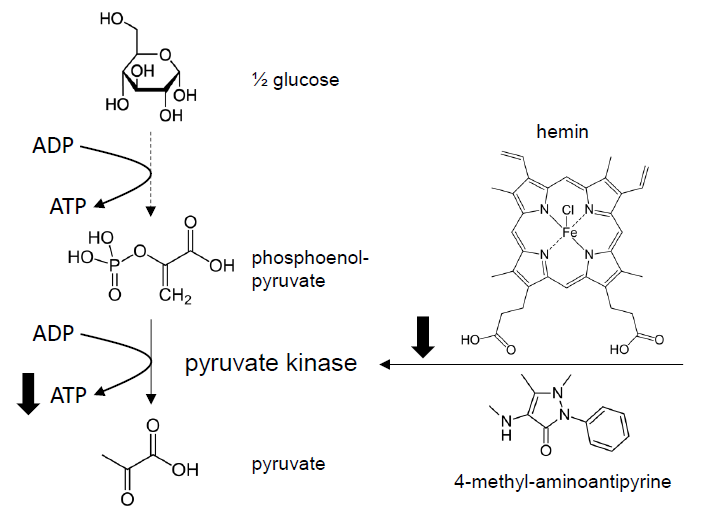
:
1. Introduction
2. Experimental Section
2.1. Chemicals and Cell Culture Reagents
2.2. Time Course of Cellular ATP Depletion
2.3. Time Course of Loss of Membrane Integrity
2.4. Cellular Oxygen Consumption Rate (OCR)
2.5. Mitochondrial Superoxide Generation
2.6. Total Cellular ROS Production
2.7. Mitochondrial DNA Copy Number
2.8. Mitochondrial Membrane Potential
2.9. Cytotoxicity by MAA/Hemin for HL60 Cells Cultured with Galactose
2.10. Effect of MAA/hemin on Glycolysis
2.11. GSH Content of HL60 Cells
2.12. Quantitative Real-Time PCR
2.13. Western Blot Analysis
2.14. Statistics
3. Results
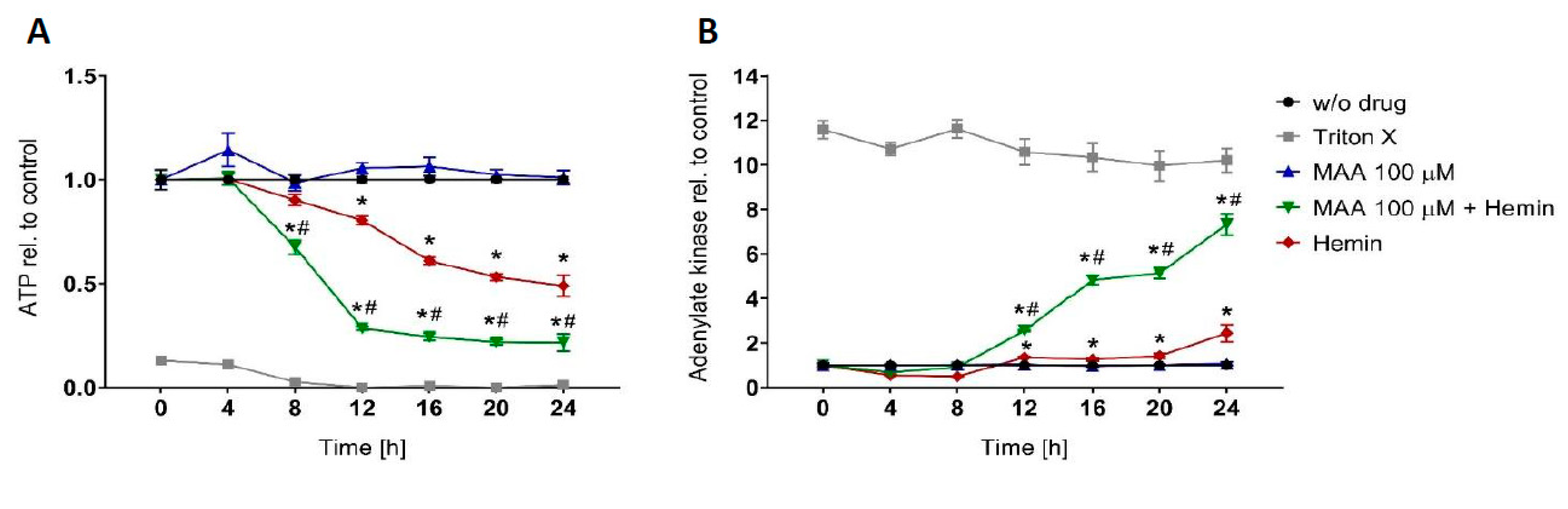
3.1. Time Course of MAA/hemin Toxicity in HL60 Cells
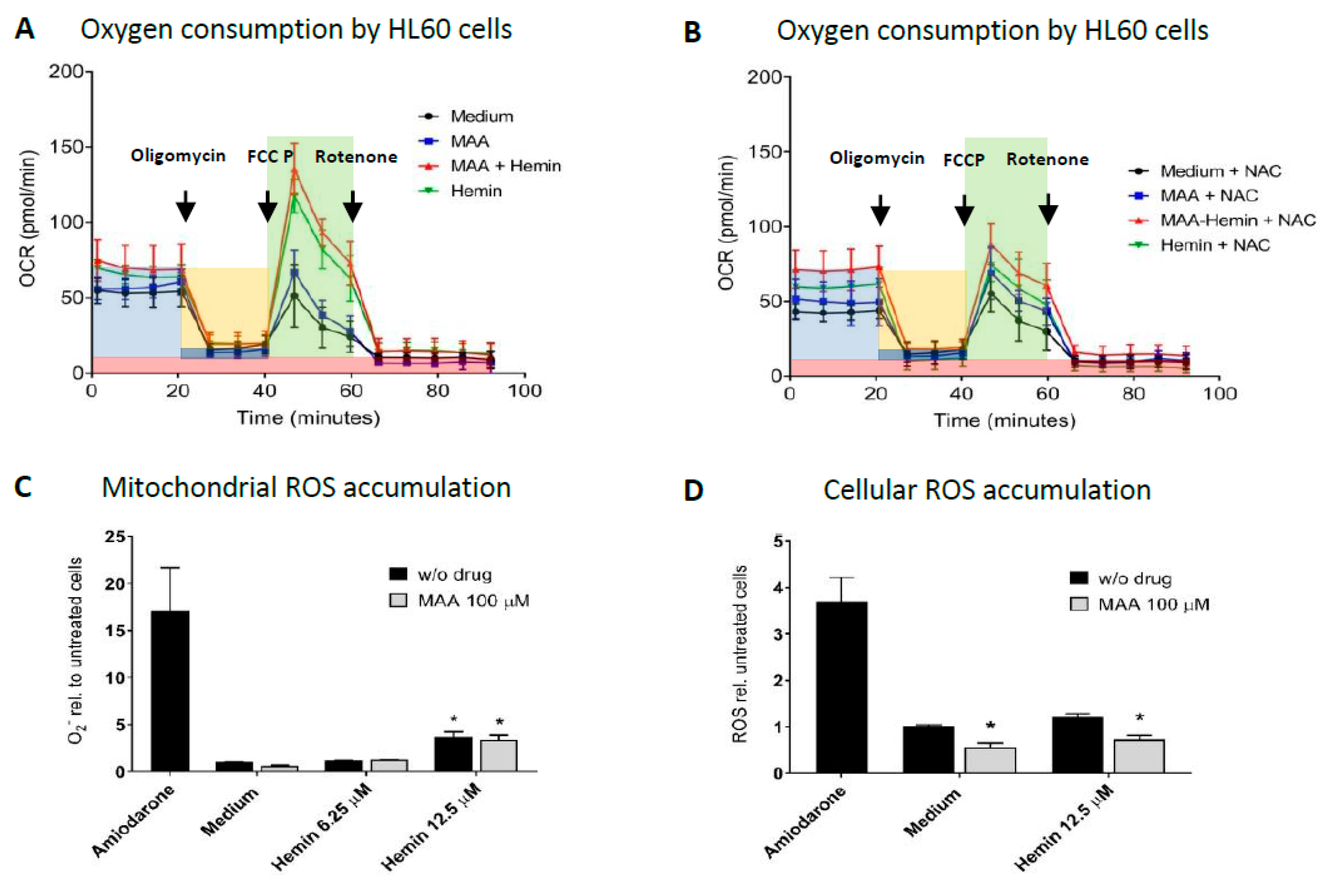
3.2. Mitochondrial Involvement in MAA/hemin Toxicity
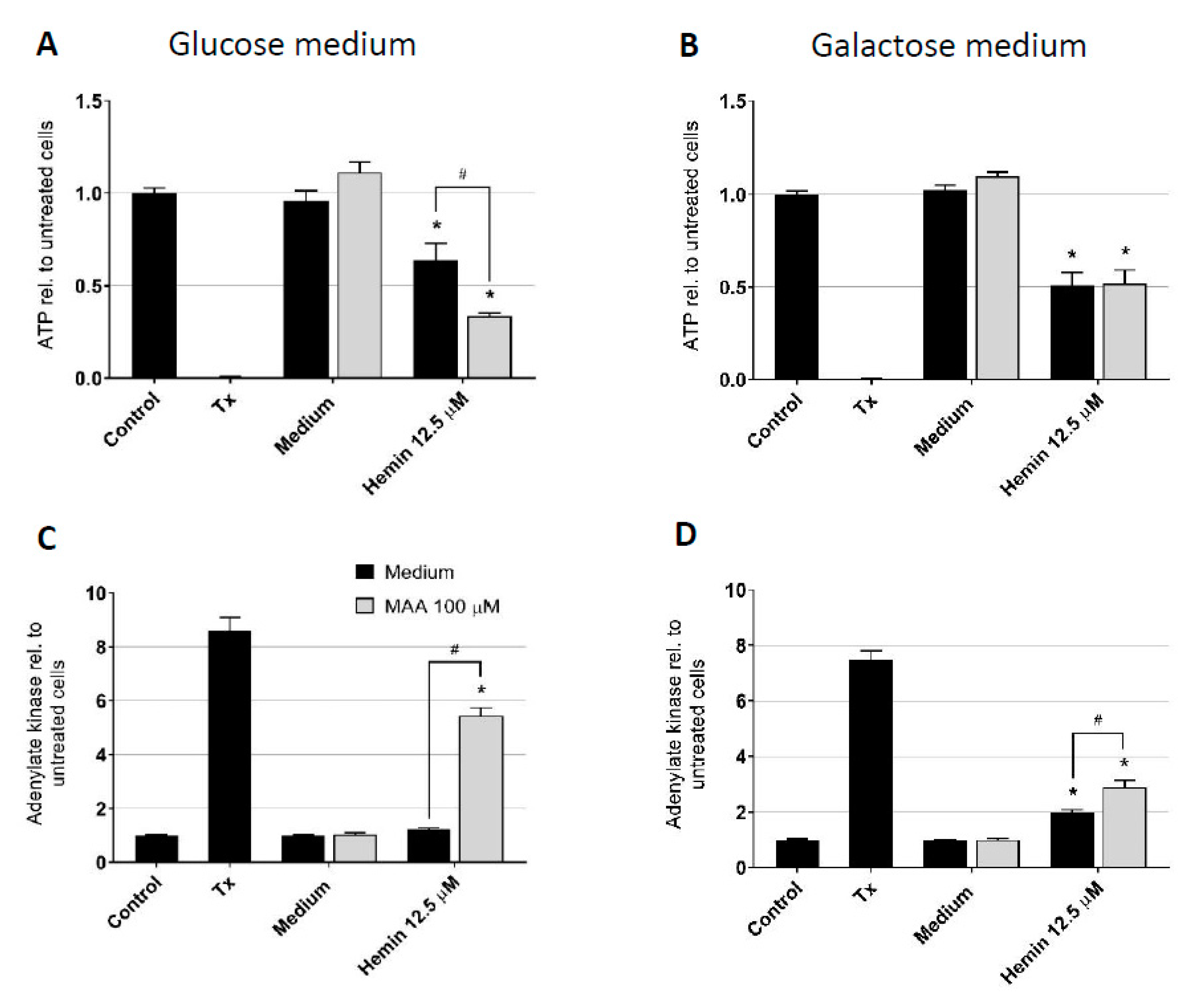
3.3. Impact of Culture Conditions on MAA/Hemin Toxicity
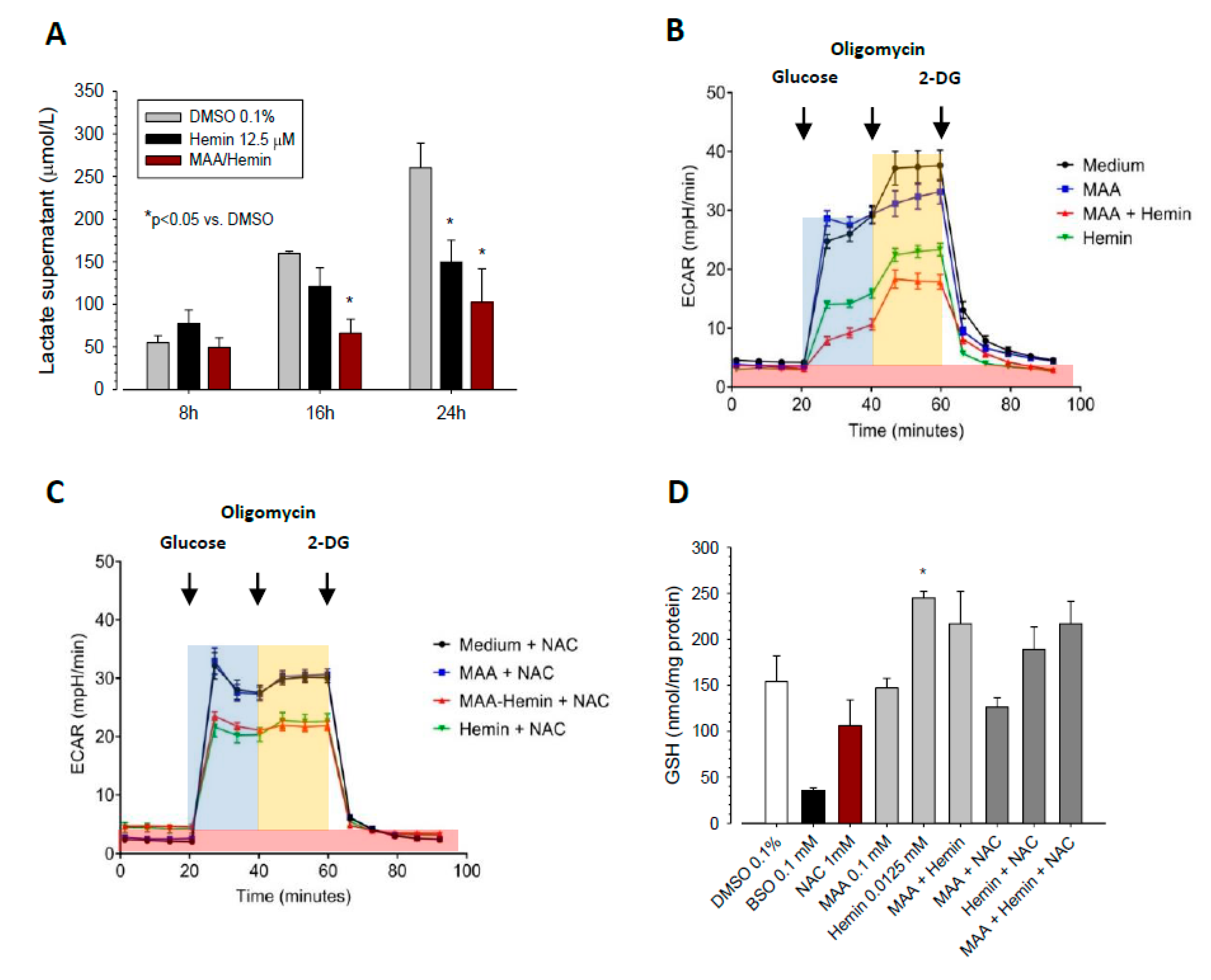
3.4. Effect of MAA/Hemin on Glycolysis
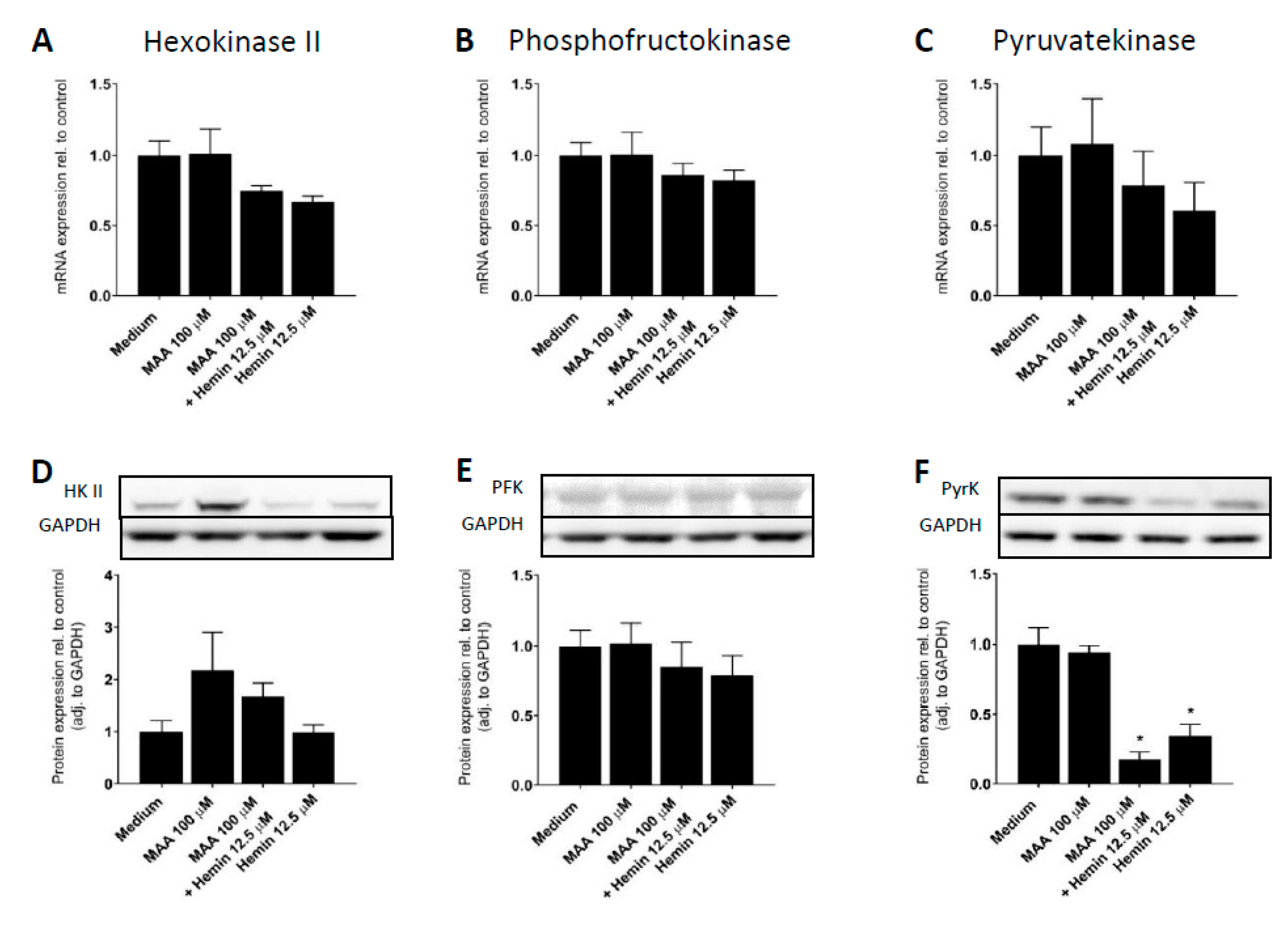
3.5. Molecular Mechanism of the Inhibition of Glycolysis by MAA/Hemin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gomez-Jimenez, J.; Franco-Patino, R.; Chargoy-Vera, J.; Olivares-Sosa, R. Clinical efficacy of mild analgesics in pain following gynaecological or dental surgery: Report on multicentre studies. Br. J. Clin. Pharmacol. 1980, 10, S355–S358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas, M.E.; Gay-Escoda, C.; Bagan, J.V.; Santamaria, J.; Penarrocha, M.; Donado, M.; Puerta, J.L.; Garcia-Magaz, I.; Ruiz, J.; Ortiz, P. Oral metamizol (1 g and 2 g) versus ibuprofen and placebo in the treatment of lower third molar surgery pain: Randomised double-blind multi-centre study. Cooperative Study Group. Eur. J. Clin. Pharmacol. 1998, 53, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Ergün, H.; Frattarelli, D.A.C.; Aranda, J.V. Characterization of the role of physicochemical factors on the hydrolysis of dipyrone. J. Pharm. Biomed. Anal. 2004, 35, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Zylber-Katz, E.; Rosenkranz, B. Clinical pharmacokinetics of dipyrone and its metabolites. Clin. Pharmacokinet. 1995, 28, 216–234. [Google Scholar] [CrossRef] [PubMed]
- Zylber-Katz, E.; Granit, L.; Levy, M. Formation and excretion of dipyrone metabolites in man. Eur. J. Clin. Pharmacol. 1992, 42, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.C.; Schmidt, R.; Brenneis, C.; Michaelis, M.; Geisslinger, G.; Scholich, K. Inhibition of cyclooxygenases by dipyrone. Br. J. Pharmacol. 2007, 151, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Andrade, S.E.; Martinez, C.; Walker, A.M. Comparative safety evaluation of non-narcotic analgesics. J. Clin. Epidemiol. 1998, 51, 1357–1365. [Google Scholar] [CrossRef]
- Garcia-Martin, E.; Esguevillas, G.; Blanca-Lopez, N.; Garcia-Menaya, J.; Blanca, M.; Amo, G.; Canto, G.; Martinez, C.; Cordobes, C.; Agundez, J.A. Genetic determinants of metamizole metabolism modify the risk of developing anaphylaxis. Pharm. Genom. 2015, 25, 462–464. [Google Scholar] [CrossRef]
- Blanca-Lopez, N.; Perez-Sanchez, N.; Agundez, J.A.; Garcia-Martin, E.; Torres, M.J.; Cornejo-Garcia, J.A.; Perkins, J.R.; Miranda, M.A.; Andreu, I.; Mayorga, C.; et al. Allergic Reactions to Metamizole: Immediate and Delayed Responses. Int. Arch. Allergy Immunol. 2016, 169, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Borja, J.M.; Galindo, P.A.; Gomez, E.; Feo, F. Delayed skin reactions to metamizol. Allergy 2003, 58, 84–85. [Google Scholar] [CrossRef]
- Arellano, F.; Sacristan, J.A. Metamizole: Reassessment of its therapeutic role. Eur. J. Clin. Pharmacol. 1990, 38, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Hedenmalm, K.; Spigset, O. Agranulocytosis and other blood dyscrasias associated with dipyrone (metamizole). Eur. J. Clin. Pharmacol. 2002, 58, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Blaser, L.S.; Tramonti, A.; Egger, P.; Haschke, M.; Krahenbuhl, S.; Ratz Bravo, A.E. Hematological safety of metamizole: Retrospective analysis of WHO and Swiss spontaneous safety reports. Eur. J. Clin. Pharmacol. 2015, 71, 209–217. [Google Scholar] [CrossRef]
- Huber, M.; Andersohn, F.; Sarganas, G.; Bronder, E.; Klimpel, A.; Thomae, M.; Konzen, C.; Kreutz, R.; Garbe, E. Metamizole-induced agranulocytosis revisited: Results from the prospective Berlin Case-Control Surveillance Study. Eur. J. Clin. Pharmacol. 2015, 71, 219–227. [Google Scholar] [CrossRef]
- Ibanez, L.; Vidal, X.; Ballarin, E.; Laporte, J.R. Agranulocytosis associated with dipyrone (metamizol). Eur. J. Clin. Pharmacol. 2005, 60, 821–829. [Google Scholar] [CrossRef]
- Levy, M.I.A.; International Agranulocytosis and Aplastic Anemia Study. Risks of agranulocytosis and aplastic anemia. A first report of their relation to drug use with special reference to analgesics. The International Agranulocytosis and Aplastic Anemia Study. JAMA 1986, 256, 1749–1757. [Google Scholar]
- Kowalski, M.L.; Woszczek, G.; Bienkiewicz, B.; Mis, M. Association of pyrazolone drug hypersensitivity with HLA-DQ and DR antigens. Clin. Exp. Allergy 1998, 28, 1153–1158. [Google Scholar] [CrossRef]
- Herdeg, C.; Hilt, F.; Buchtemann, A.; Bianchi, L.; Klein, R. Allergic cholestatic hepatitis and exanthema induced by metamizole: Verification by lymphocyte transformation test. Liver 2002, 22, 507–513. [Google Scholar] [CrossRef]
- Krisai, P.; Rudin, D.; Grunig, D.; Scherer, K.; Pichler, W.; Terracciano, L.; Krahenbuhl, S. Acute Liver Failure in a Patient Treated With Metamizole. Front. Pharmacol. 2019, 10, 996. [Google Scholar] [CrossRef]
- Vlahov, V.; Bacracheva, N.; Tontcheva, D.; Naumova, E.; Mavrudieva, M.; Ilieva, P.; Michailova, A. Genetic factors and risk of agranulocytosis from metamizol. Pharmacogenetics 1996, 6, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Blaser, L.; Hassna, H.; Hofmann, S.; Holbro, A.; Haschke, M.; Ratz Bravo, A.; Zeller, A.; Krahenbuhl, S.; Taegtmeyer, A. Leucopenia associated with metamizole: A case-control study. Swiss Med Wkly. 2017, 147, w14438. [Google Scholar]
- Rudin, D.; Spoendlin, J.; Cismaru, A.L.; Liakoni, E.; Bonadies, N.; Amstutz, U.; Meier, C.R.; Krähenbühl, S.; Haschke, M. Metamizole-associated neutropenia: Comparison of patients with neutropenia and metamizole-tolerant patients. Eur. J. Int. Med. 2019, 68, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Rudin, D.; Lanzilotto, A.; Bachmann, F.; Housecroft, C.E.; Constable, E.C.; Drewe, J.; Haschke, M.; Krahenbuhl, S. Non-immunological toxicological mechanisms of metamizole-associated neutropenia in HL60 cells. Biochem. Pharmacol. 2019, 163, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, P.; Drapier, J.C.; Oliveira, L.; Retmanska, H.; Sochanowicz, B.; Kruszewski, M. Intracellular iron status as a hallmark of mammalian cell susceptibility to oxidative stress: A study of L5178Y mouse lymphoma cell lines differentially sensitive to H2O2. Blood 2000, 95, 2960–2966. [Google Scholar] [CrossRef] [PubMed]
- Rudin, D.; Roos, N.J.; Duthaler, U.; Krahenbuhl, S. Toxicity of metamizole on differentiating HL60 cells and human neutrophil granulocytes. Toxicology 2019, 426, 152254. [Google Scholar] [CrossRef] [PubMed]
- Kummer, O.; Haschke, M.; Tuchscherer, D.; Lampert, M.; Martius, F.; Krahenbuhl, S. Agranulocytosis in a patient treated with metamizole and clopidogrel. Praxis 2006, 95, 1743–1745. [Google Scholar] [CrossRef] [PubMed]
- Asmardi, G.; Jamali, F. Pharmacokinetics of dipyrone in man; role of the administration route. Eur. J. Drug Metab. Pharm. 1985, 10, 121–125. [Google Scholar] [CrossRef]
- Heinemeyer, G.; Gramm, H.J.; Roots, I.; Dennhardt, R.; Simgen, W. The kinetics of metamizol and its metabolites in critical-care patients with acute renal dysfunction. Eur. J. Clin. Pharmacol. 1993, 45, 445–450. [Google Scholar] [CrossRef]
- Aich, A.; Freundlich, M.; Vekilov, P.G. The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 2015, 55, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Waldhauser, K.M.; Torok, M.; Ha, H.R.; Thomet, U.; Konrad, D.; Brecht, K.; Follath, F.; Krahenbuhl, S. Hepatocellular toxicity and pharmacological effect of amiodarone and amiodarone derivatives. J. Pharmacol. Exp. Ther. 2006, 319, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 97, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Fanti, P.; Rossi, R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat. Protoc. 2013, 8, 1660. [Google Scholar] [CrossRef] [PubMed]
- Roos, N.J.; Duthaler, U.; Bouitbir, J.; Krahenbuhl, S. The uricosuric benzbromarone disturbs the mitochondrial redox homeostasis and activates the NRF2 signaling pathway in HepG2 cells. Free Radic. Biol. Med. 2020, 152, 216–226. [Google Scholar] [CrossRef]
- Felser, A.; Blum, K.; Lindinger, P.W.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of hepatocellular toxicity associated with dronedarone--a comparison to amiodarone. Toxicol. Sci. Off. J. Soc. Toxicol. 2013, 131, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.; Gomes, A.; Lima, J.L.; Fernandes, E. Singlet oxygen scavenging activity of non-steroidal anti-inflammatory drugs. Redox Rep. Commun. Free Radic. Res. 2008, 13, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef]
- Schaffer, K.; Taylor, C.T. The impact of hypoxia on bacterial infection. FEBS J. 2015, 282, 2260–2266. [Google Scholar] [CrossRef]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Johnson, M.S.; Hardy, R.W.; Ballinger, S.W.; Darley-Usmar, V.M. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab. Investig. 2013, 93, 690–700. [Google Scholar] [CrossRef]
- Bonkowsky, J.L.; Frazer, J.K.; Buchi, K.F.; Byington, C.L. Metamizole use by Latino immigrants: A common and potentially harmful home remedy. Pediatrics 2002, 109, e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, A.; Kietz, S.; Bahlmann, H.; Jafarzade, G.; Lode, H.N.; Heckmann, M. GM-CSF as successful salvage therapy of metamizole (dipyrone)-induced agranulocytosis with Fournier’s gangrene and severe septic shock in an adolescent. Clin. Case Rep. 2016, 4, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wijk, R.; van Solinge, W.W. The energy-less red blood cell is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef] [PubMed]
- Martinov, M.V.; Plotnikov, A.G.; Vitvitsky, V.M.; Ataullakhanov, F.I. Deficiencies of glycolytic enzymes as a possible cause of hemolytic anemia. Biochim. Et Biophys. Acta Gen. Subj. 2000, 1474, 75–87. [Google Scholar] [CrossRef]
- Valentine, W.N.; Paglia, D.E. The primary cause of hemolysis in enzymopathies of anaerobic glycolysis: A viewpoint. Blood Cells 1980, 6, 819–829. [Google Scholar]
- Alves-Filho, J.C.; Palsson-McDermott, E.M. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front. Immunol. 2016, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.P.; Dean, R.T. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem. J. 1986, 234, 399–403. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No N-Acetylcysteine (NAC) | ||
|---|---|---|
| Glucose-Dependent Glycolysis | Maximal Glycolysis | |
| Control (DMSO 0.1%) | 21.0 ± 2.3 | 31.5 ± 4.9 |
| MAA 100 µM | 22.5 ± 2.6 | 25.9 ± 2.6 |
| Hemin 12.5 µM | 10.9 ± 0.4 * | 19.1 ± 0.8 * |
| MAA + hemin | 4.9 ± 1.5 *,+ | 13.5 ± 1.1 *,+ |
| Plus N-acetylcysteine (NAC) 1 mM | ||
| Glucose-Dependent Glycolysis | Maximal Glycolysis | |
| Control (DMSO 0.1%) | 25.9 ± 1.9 | 26.6 ± 1.1 |
| MAA 100 µM | 25.9 ± 3.4 | 27.0 ± 1.5 |
| Hemin 12.5 µM | 17.3 ± 2.6 | 19.1 ± 1.1 * |
| MAA + hemin | 18.4 ± 1.5 | 17.6 ± 0.4 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudin, D.; Schmutz, M.; Roos, N.J.; Bouitbir, J.; Krähenbühl, S. Reactive Metamizole Metabolites Enhance the Toxicity of Hemin on the ATP Pool in HL60 Cells by Inhibition of Glycolysis. Biomedicines 2020, 8, 212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070212
Rudin D, Schmutz M, Roos NJ, Bouitbir J, Krähenbühl S. Reactive Metamizole Metabolites Enhance the Toxicity of Hemin on the ATP Pool in HL60 Cells by Inhibition of Glycolysis. Biomedicines. 2020; 8(7):212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070212
Chicago/Turabian StyleRudin, Deborah, Maurice Schmutz, Noëmi Johanna Roos, Jamal Bouitbir, and Stephan Krähenbühl. 2020. "Reactive Metamizole Metabolites Enhance the Toxicity of Hemin on the ATP Pool in HL60 Cells by Inhibition of Glycolysis" Biomedicines 8, no. 7: 212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070212