Comparison of Circulating Tumour DNA and Extracellular Vesicle DNA by Low-Pass Whole-Genome Sequencing Reveals Molecular Drivers of Disease in a Breast Cancer Patient

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Patient Samples
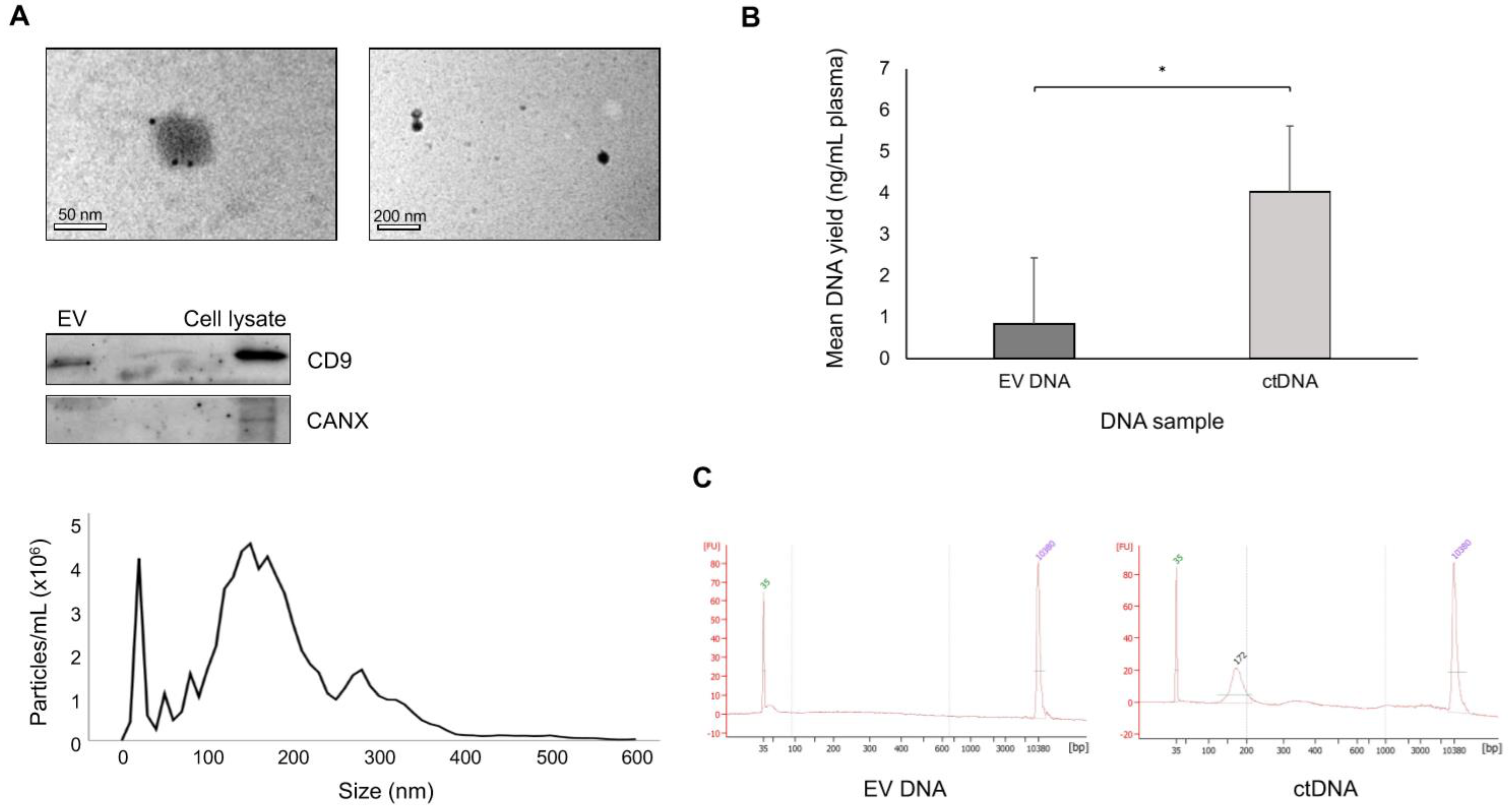
2.2. EV Isolation
2.3. Immuno-Electron Microscopy
2.4. Nanoparticle Tracking Analysis
2.5. Western Blot
2.6. DNA Extraction
2.7. Low-Pass Whole Genome Sequencing
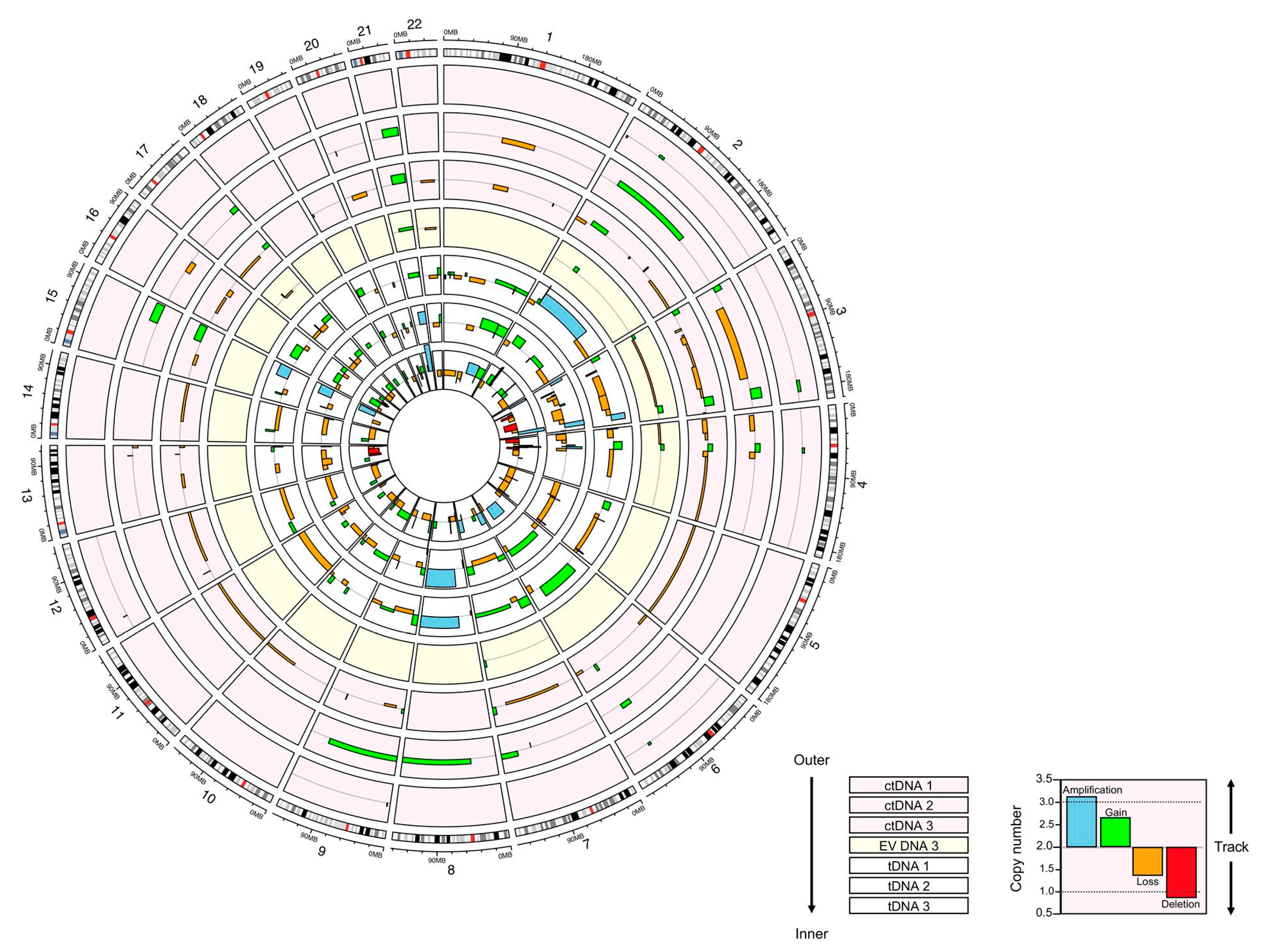
2.8. Data Analysis
3. Results
3.1. Patient Characteristics
3.2. EV DNA vs. ctDNA Yield
3.3. Copy Number Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Non-invasive detection of genome-wide somatic copy number alterations by liquid biopsies. Mol. Oncol. 2016, 10, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Stover, D.G.; Parsons, H.A.; Ha, G.; Freeman, S.S.; Barry, W.T.; Guo, H.; Choudhury, A.D.; Gydush, G.; Reed, S.C.; Rhoades, J.; et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J. Clin. Oncol. 2018, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 154. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [Green Version]
- San Lucas, F.A.; Allenson, K.; Bernard, V.; Castillo, J.; Kim, D.U.; Ellis, K.; Ehli, E.A.; Davies, G.E.; Petersen, J.L.; Li, D.; et al. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann. Oncol. 2016, 27, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Yoon, H.; Park, S.; Kim, J.S.; Ahn, Y.-H.; Kwon, K.; Lee, D.; Kim, K.H. Urinary exosomal and cell-free DNA detects somatic mutation and copy number alteration in urothelial carcinoma of bladder. Sci. Rep. 2018, 8, 14707. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Fang, L.; Bai, J.; Wang, Z. Characteristics and Changes of DNA in Extracellular Vesicles. DNA Cell Biol. 2020, 39, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, X.; Guo, S.; Wang, Y.; Chen, L.; Liu, Y.; Jia, M.; An, J.; Tao, K.; Xing, J. Next generation sequencing-based analysis of mitochondrial DNA characteristics in plasma extracellular vesicles of patients with hepatocellular carcinoma. Oncol. Lett. 2020, 20, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Kalra, H.; Adda, C.G.; Liem, M.; Ang, C.S.; Mechler, A.; Simpson, R.J.; Hulett, M.D.; Mathivanan, S. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 2013, 13, 3354–3364. [Google Scholar] [CrossRef]
- Kurywchak, P.; Tavormina, J.; Kalluri, R. The emerging roles of exosomes in the modulation of immune responses in cancer. Genome Med. 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Scheinin, I.; Sie, D.; Bengtsson, H.; van de Wiel, M.A.; Olshen, A.B.; van Thuijl, H.F.; van Essen, H.F.; Eijk, P.P.; Rustenburg, F.; Meijer, G.A.; et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014, 24, 2022–2032. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Akashi-Tanaka, S.; Watanabe, C.; Takamaru, T.; Kuwayama, T.; Ikeda, M.; Ohyama, H.; Mori, M.; Yoshida, R.; Hashimoto, R.; Terumasa, S.; et al. BRCAness predicts resistance to taxane-containing regimens in triple negative breast cancer during neoadjuvant chemotherapy. Clin. Breast Cancer 2015, 15, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.A.; Schmidt, M.K.; van Beers, E.H.; Cornelissen, S.; Holtkamp, M.; Froklage, F.E.; de Vries, E.G.E.; et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann. Oncol. 2011, 22, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.; Bernard, V.; San Lucas, F.A.; Allenson, K.; Capello, M.; Kim, D.U.; Gascoyne, P.; Mulu, F.C.; Stephens, B.M.; Huang, J.; et al. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Monlong, J.; Bourque, G.; Girard, S.L.; Meloche, C.; Cossette, P.; Rouleau, G. Human copy number variants are enriched in regions of low mappability. Nucl. Acids Res. 2018, 46, 7236–7249. [Google Scholar] [CrossRef]
- Luo, Z.; Fan, X.; Su, Y.; Huang, Y.S. Accurity: Accurate tumor purity and ploidy inference from tumor-normal WGS data by jointly modelling somatic copy number alterations and heterozygous germline single-nucleotide-variants. Bioinformatics 2018, 34, 2004–2011. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.-D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659. [Google Scholar]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Swystun, L.L.; Mukherjee, S.; Liaw, P.C. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J. Thromb. Haemost. 2011, 9, 2313–2321. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruhen, O.; Mirzai, B.; Clark, M.E.; Nguyen, B.; Salomon, C.; Erber, W.; Meehan, K. Comparison of Circulating Tumour DNA and Extracellular Vesicle DNA by Low-Pass Whole-Genome Sequencing Reveals Molecular Drivers of Disease in a Breast Cancer Patient. Biomedicines 2021, 9, 14. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010014
Ruhen O, Mirzai B, Clark ME, Nguyen B, Salomon C, Erber W, Meehan K. Comparison of Circulating Tumour DNA and Extracellular Vesicle DNA by Low-Pass Whole-Genome Sequencing Reveals Molecular Drivers of Disease in a Breast Cancer Patient. Biomedicines. 2021; 9(1):14. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010014
Chicago/Turabian StyleRuhen, Olivia, Bob Mirzai, Michael E. Clark, Bella Nguyen, Carlos Salomon, Wendy Erber, and Katie Meehan. 2021. "Comparison of Circulating Tumour DNA and Extracellular Vesicle DNA by Low-Pass Whole-Genome Sequencing Reveals Molecular Drivers of Disease in a Breast Cancer Patient" Biomedicines 9, no. 1: 14. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010014