
STAT3 Signalling via the IL-6ST/gp130 Cytokine Receptor Promotes Epithelial Integrity and Intestinal Barrier Function during DSS-Induced Colitis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DSS-Induced Colitis
2.3. Tissue Collection
2.4. In Vivo Intestinal Permeability Assay
2.5. Gene Expression Analysis
2.6. Western Blot
2.7. Histology and Immunohistochemistry
2.8. Multiplex ELISA
2.9. Statistical Analysis
3. Results
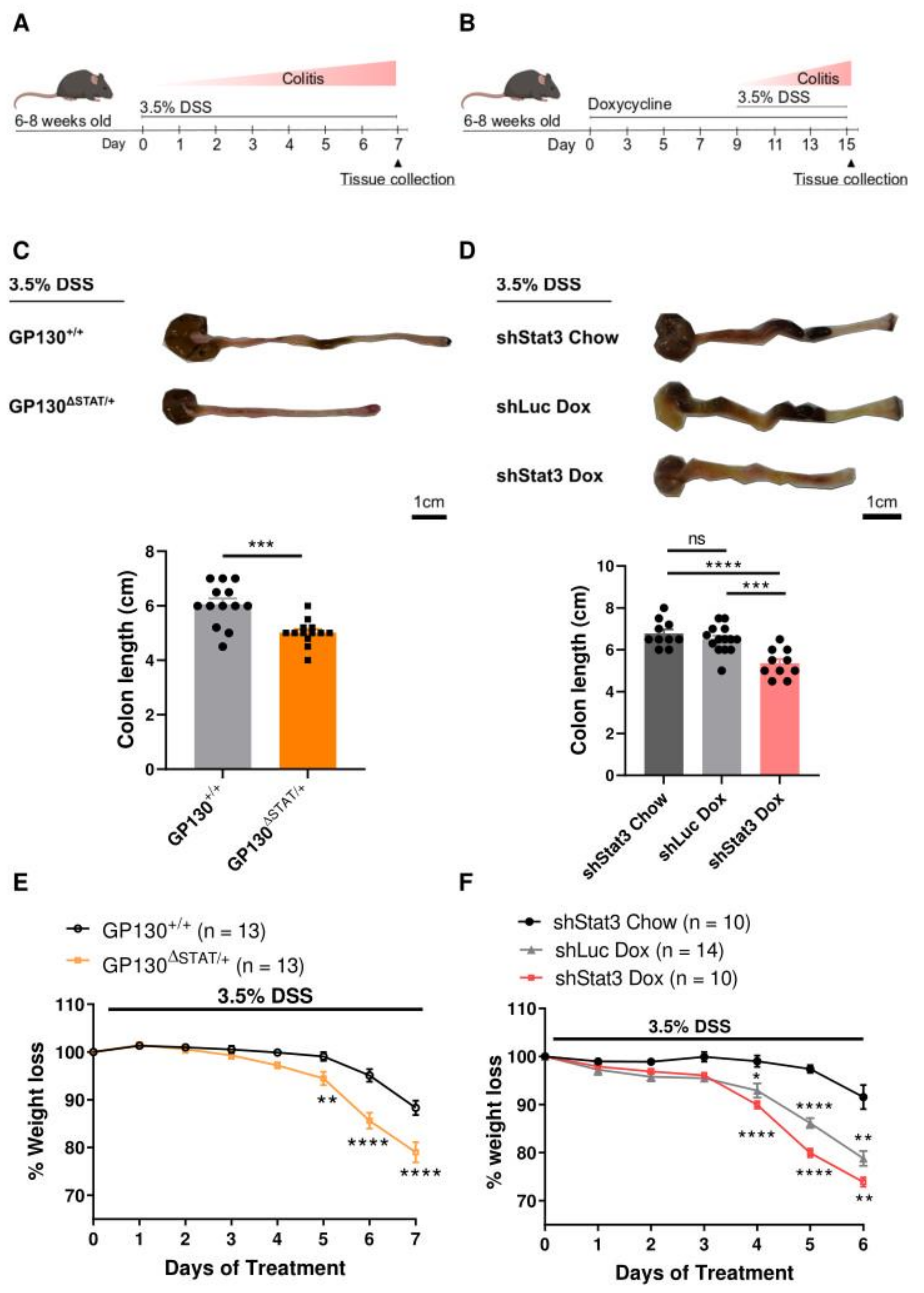
3.1. Mice with Reduced STAT3 Activity Are Highly Susceptible to DSS-Induced Colitis In Vivo
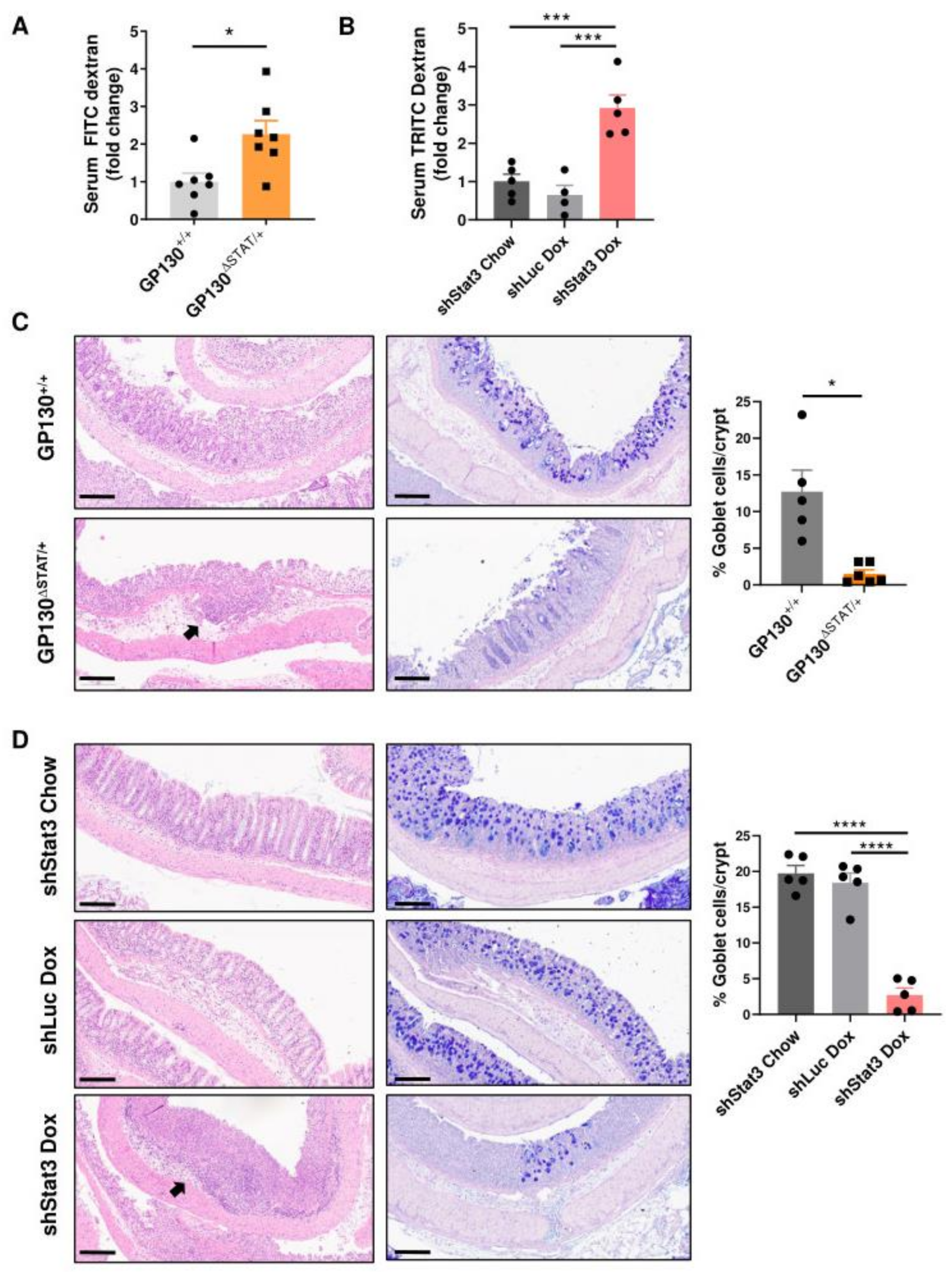
3.2. Reduction in STAT3 Activity Decreases Intestinal Barrier Function and Compromises Barrier Integrity
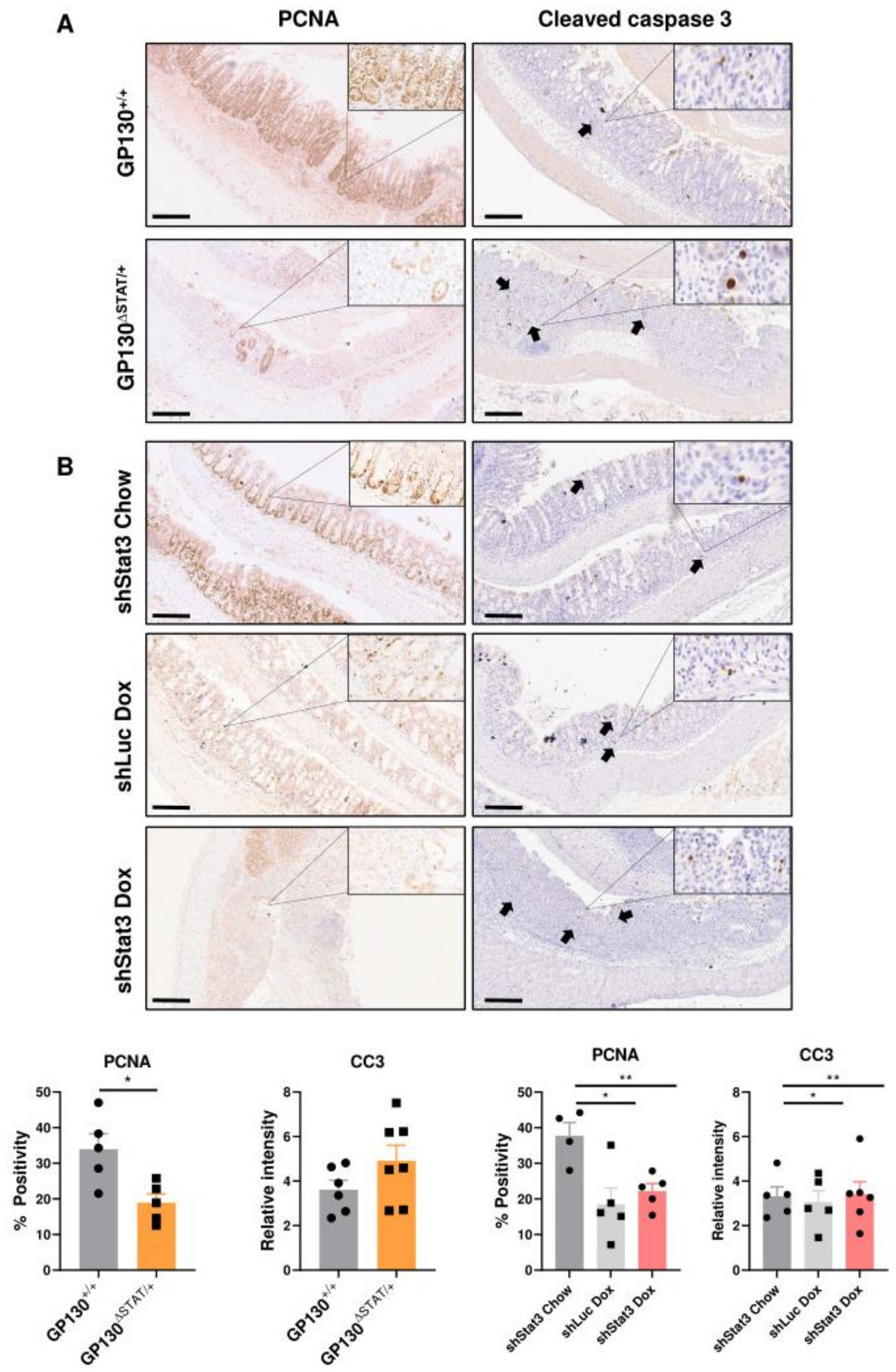
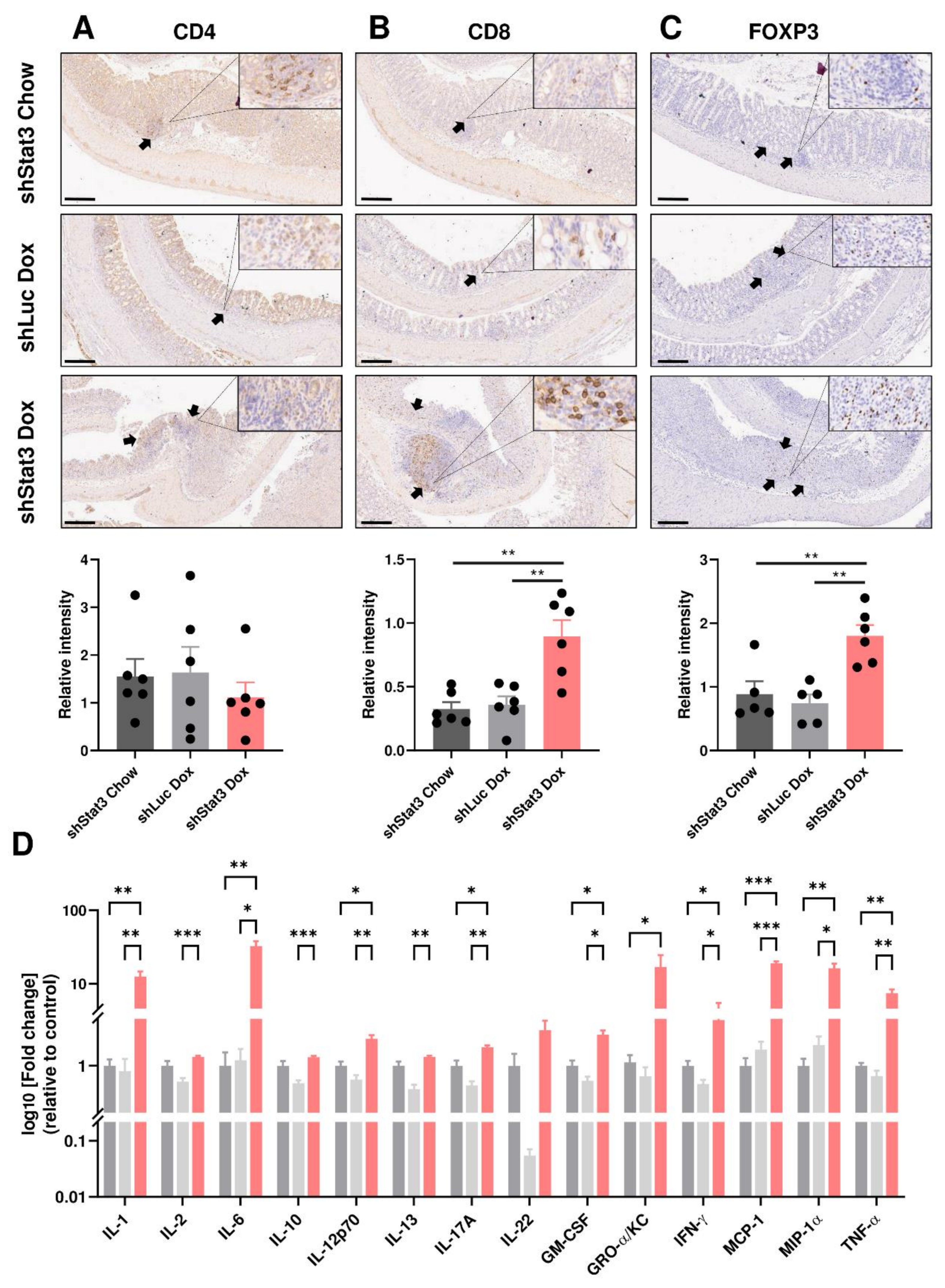
3.3. Reduction in STAT3 Activity Decreases Epithelial Proliferation while Exacerbating Inflammatory Responses
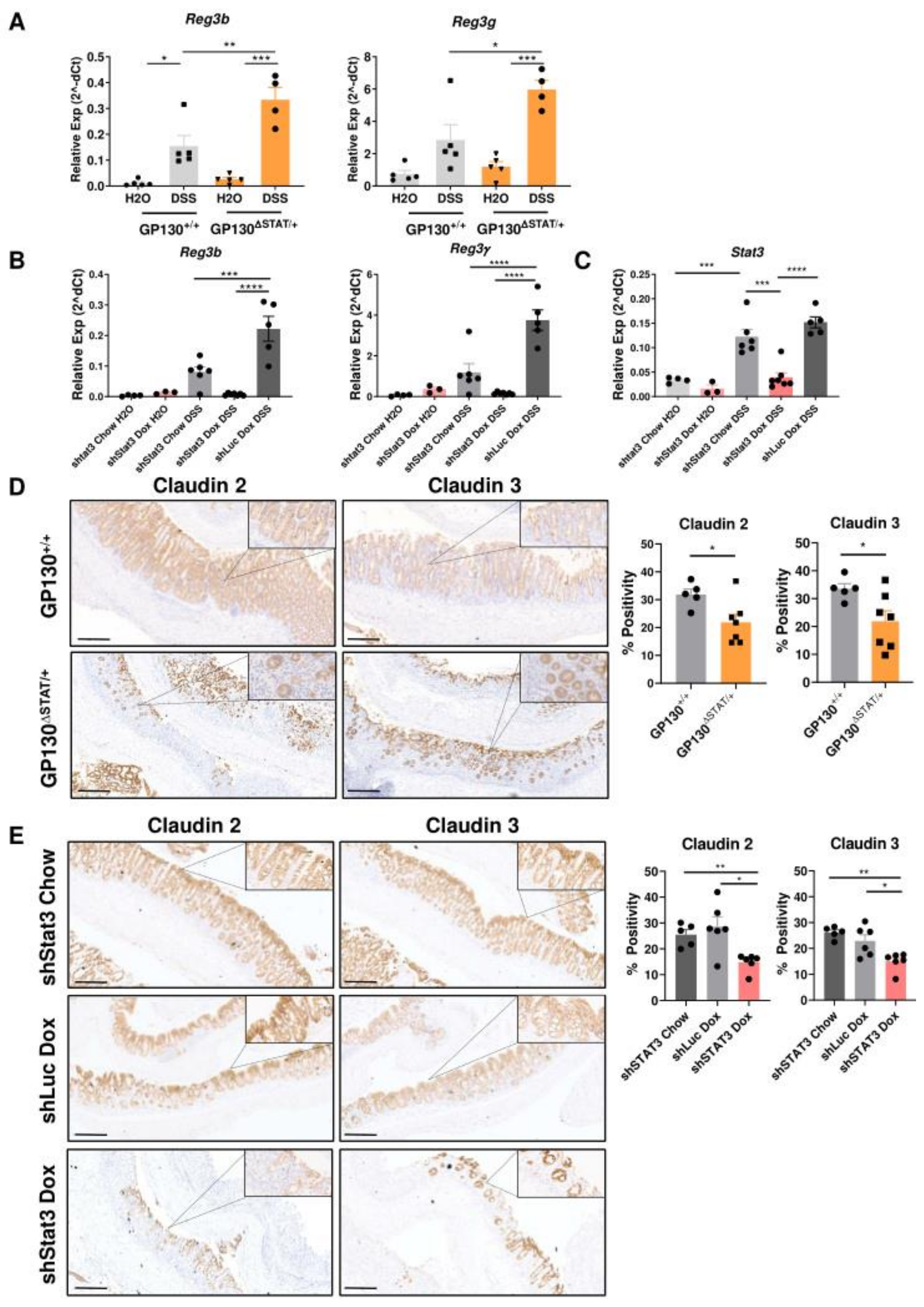
3.4. Reduction in STAT3 Activity Compromises Expression of Antimicrobial Genes and Tight Junction Proteins
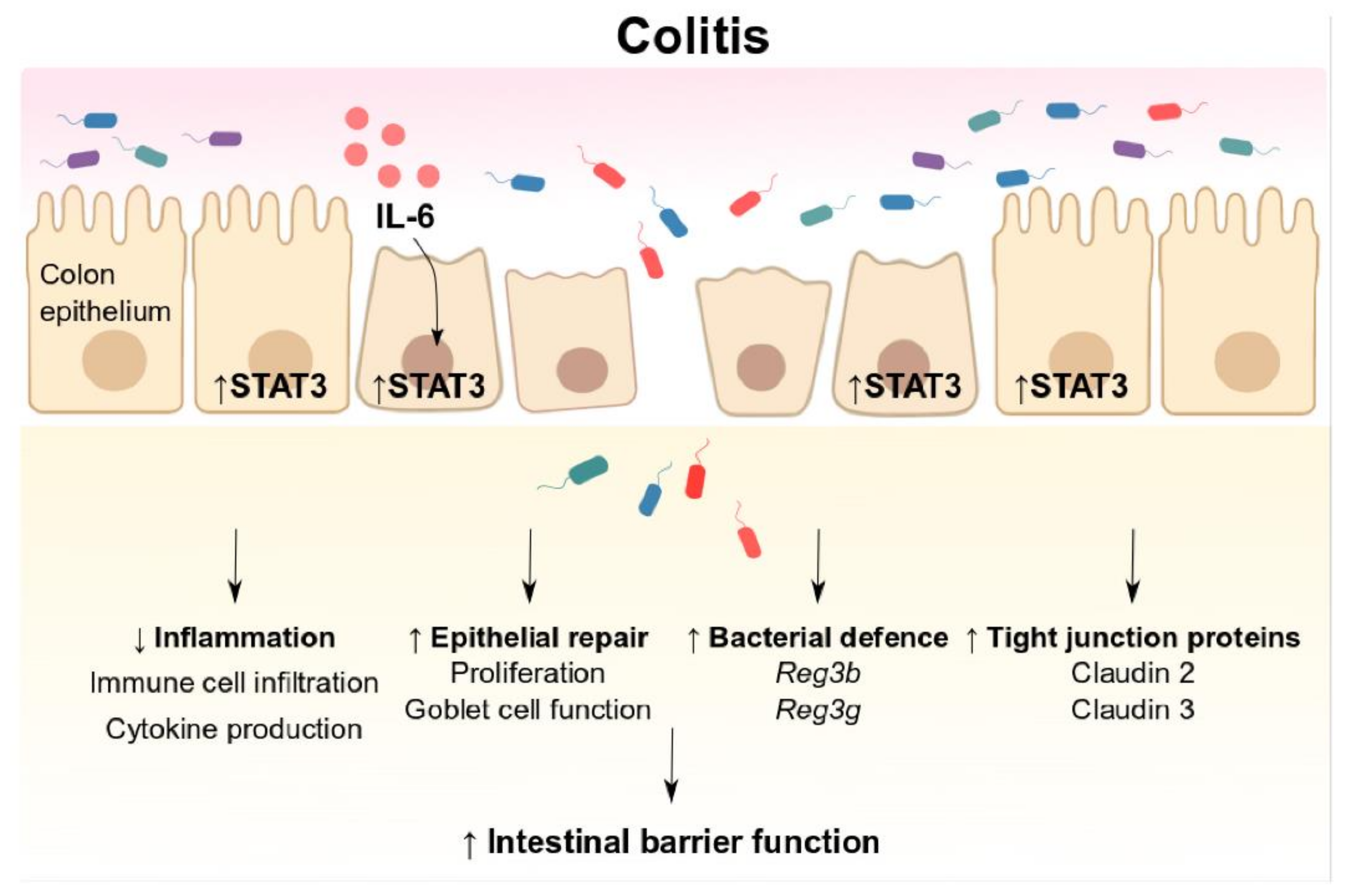
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.M.; Putoczki, T.L.; Ernst, M. STAT3-Activating Cytokines: A Therapeutic Opportunity for Inflammatory Bowel Disease? J. Interferon Cytokine Res. 2015, 35, 340–350. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.; Tromm, A.; Griga, T.; Fricke, H.; Folwaczny, C.; Hocke, M.; Eitner, K.; Marx, M.; Duerig, N.; Epplen, J.T. A polymorphism in the IL11 gene is associated with ulcerative colitis. Genes Immun. 2002, 3, 494–496. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.G.; Zheng, X.B.; Wu, Z.G.; Dai, S.X.; Wan, Z.; Zou, Y. Association of the interleukin-22 genetic polymorphisms with ulcerative colitis. Diagn. Pathol. 2014, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S.; et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Förster, I.; Akira, S. Enhanced Th1 Activity and Development of Chronic Enterocolitis in Mice Devoid of Stat3 in Macrophages and Neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Kweon, M.-N.; Kuwata, H.; Schreiber, R.D.; Kiyono, H.; Takeda, K.; Akira, S. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J. Clin. Investig. 2003, 111, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.-W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.-A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Wittkopf, N.; Pickert, G.; Billmeier, U.; Mahapatro, M.; Wirtz, S.; Martini, E.; Leppkes, M.; Neurath, M.F.; Becker, C. Activation of Intestinal Epithelial Stat3 Orchestrates Tissue Defense during Gastrointestinal Infection. PLoS ONE 2015, 10, e0118401. [Google Scholar] [CrossRef]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef]
- Alorro, M.G.; Pierce, T.P.; Eissmann, M.F.; Dijkstra, C.; Dickins, R.A.; Ernst, M.; Buchert, M.; Masson, F. Generation of an inducible mouse model to reversibly silence Stat3. Genesis 2017, 55, e23023. [Google Scholar] [CrossRef]
- Moschen, A.R.; Gerner, R.R.; Wang, J.; Klepsch, V.; Adolph, T.E.; Reider, S.J.; Hackl, H.; Pfister, A.; Schilling, J.; Moser, P.L.; et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host Microbe 2016, 19, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.B.; Tebbutt, N.C.; Buchert, M.; Putoczki, T.L.; Doggett, K.; Bao, S.; Johnstone, C.N.; Masson, F.; Hollande, F.; Burgess, A.W.; et al. Glycoprotein A33 deficiency: A new mouse model of impaired intestinal epithelial barrier function and inflammatory disease. Dis. Models Mech. 2015, 8, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.; Inglese, M.; Waring, P.; Campbell, I.K.; Bao, S.; Clay, F.J.; Alexander, W.S.; Wicks, I.P.; Tarlinton, D.M.; Novak, U.; et al. Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J. Exp. Med. 2001, 194, 189–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, N.A.; Jenkins, B.J.; Quinn, J.M.W.; Nakamura, A.; Glatt, M.; Gillespie, M.T.; Ernst, M.; Martin, T.J. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J. Clin. Investig. 2004, 113, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10−/− mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Choi, S.-M.; McAleer, J.P.; Zheng, M.; Pociask, D.A.; Kaplan, M.H.; Qin, S.; Reinhart, T.A.; Kolls, J.K. Innate Stat3-mediated induction of the antimicrobial protein Reg3γ is required for host defense against MRSA pneumonia. J. Exp. Med. 2013, 210, 551–561. [Google Scholar] [CrossRef]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Han, J.; Li, L.; Wang, Y.; Li, Y.; Zhang, S. Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front. Immunol. 2019, 10, 4–6. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 2011, 286, 31263–31271. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.-Y.; Zhang, B.; He, W.-Q.; Zha, J.-M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017, 21, 671–681.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mumm, J.B.; Herbst, R.; Kolbeck, R.; Wang, Y. IL-22 Increases Permeability of Intestinal Epithelial Tight Junctions by Enhancing Claudin-2 Expression. J. Immunol. 2017, 199, 3316–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koss, K.; Satsangi, J.; Welsh, K.I.; Jewell, D.P. Is interleukin-6 important in inflammatory bowel disease? Genes Immun. 2000, 1, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, K.A.; Schulz, H.M.; Regner, E.H.; Severs, E.L.; Hendrickson, J.D.; Mehta, G.; Whitney, A.K.; Ir, D.; Ohri, N.; Robertson, C.E.; et al. Bacteroidales recruit IL-6-producing intraepithelial lymphocytes in the colon to promote barrier integrity. Mucosal Immunol. 2018, 11, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Sun, X.; He, S.; Lv, C.; Sun, X.; Wang, J.; Zheng, W.; Wang, D. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 2893–2898. [Google Scholar] [CrossRef] [Green Version]
- Däbritz, J.; Judd, L.M.; Chalinor, H.V.; Menheniott, T.R.; Giraud, A.S. Altered gp130 signalling ameliorates experimental colitis via myeloid cell-specific STAT3 activation and myeloid-derived suppressor cells. Sci. Rep. 2016, 6, 20584. [Google Scholar] [CrossRef]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [Green Version]
- van Beelen Granlund, A.; Østvik, A.E.; Brenna, Ø.; Torp, S.H.; Gustafsson, B.I.; Sandvik, A.K. REG gene expression in inflamed and healthy colon mucosa explored by in situ hybridisation. Cell Tissue Res. 2013, 352, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Goto, R.; Fujimoto, M.; Okada, N.; Hardt, W.-D. The Bactericidal Lectin RegIII Prolongs Gut Colonization and Enteropathy in the Streptomycin Mouse Model for Salmonella Diarrhea. Cell Host Microbe 2017, 21, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fouts, D.E.; Stärkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Bidu, C.; Escoula, Q.; Bellenger, S.; Spor, A.; Galan, M.; Geissler, A.; Bouchot, A.; Dardevet, D.; Morio, B.; Cani, P.D.; et al. The Transplantation of ω3 PUFA–Altered Gut Microbiota of fat-1 Mice to Wild-Type Littermates Prevents Obesity and Associated Metabolic Disorders. Diabetes 2018, 67, 1512–1523. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagómez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Mingrino, R.; Kaukinen, K.; Hayes, K.L.; Powell, R.M.; MacDonald, T.T.; Collins, J.E. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab. Investig. 2005, 85, 1139–1162. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Kumar, B.; Chen, Z.; Chen, X.; Müller, D.; Lele, S.M.; Washington, M.K.; Batra, S.K.; Dhawan, P.; Singh, A.B. Loss of claudin-3 expression induces IL6/gp130/Stat3 signaling to promote colon cancer malignancy by hyperactivating Wnt/β-catenin signaling. Oncogene 2017, 36, 6592–6604. [Google Scholar] [CrossRef]
- Turgeon, N.; Blais, M.; Gagné, J.-M.; Tardif, V.; Boudreau, F.; Perreault, N.; Asselin, C. HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE 2013, 8, e73785. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-L.; Chassard, C.; Hausmann, M.; von Itzstein, M.; Hennet, T. Sialic acid catabolism drives intestinal inflammation and microbial dysbiosis in mice. Nat. Commun. 2015, 6, 8141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onoda, T.; Ono, T.; Dhar, D.K.; Yamanoi, A.; Fujii, T.; Nagasue, N. Doxycycline inhibits cell proliferation and invasive potential: Combination therapy with cyclooxygenase-2 inhibitor in human colorectal cancer cells. J. Lab. Clin. Med. 2004, 143, 207–216. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Huynh, J.; Alorro, M.G.; Li, X.; Ernst, M.; Chand, A.L. STAT3 Signalling via the IL-6ST/gp130 Cytokine Receptor Promotes Epithelial Integrity and Intestinal Barrier Function during DSS-Induced Colitis. Biomedicines 2021, 9, 187. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020187
Pang L, Huynh J, Alorro MG, Li X, Ernst M, Chand AL. STAT3 Signalling via the IL-6ST/gp130 Cytokine Receptor Promotes Epithelial Integrity and Intestinal Barrier Function during DSS-Induced Colitis. Biomedicines. 2021; 9(2):187. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020187
Chicago/Turabian StylePang, Lokman, Jennifer Huynh, Mariah G. Alorro, Xia Li, Matthias Ernst, and Ashwini L. Chand. 2021. "STAT3 Signalling via the IL-6ST/gp130 Cytokine Receptor Promotes Epithelial Integrity and Intestinal Barrier Function during DSS-Induced Colitis" Biomedicines 9, no. 2: 187. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020187