
Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment



Abstract
:1. Introduction
2. The Role of Androgen Receptor (AR) in AR-Dependent Castration-Resistant Prostate Cancer (CRPC)
2.1. Secondary AR Alterations
2.1.1. AR Mutation
2.1.2. AR Amplification
2.1.3. AR Splice Variants
2.2. AR Bypass/Crosstalk Mechanisms
2.2.1. Growth Factors
2.2.2. Cytokines
2.2.3. Phosphatidylinositol-3 Kinase (PI3K)/AKT Pathway
2.2.4. Wnt Pathway
2.2.5. Glucocorticoid Receptor Upregulation
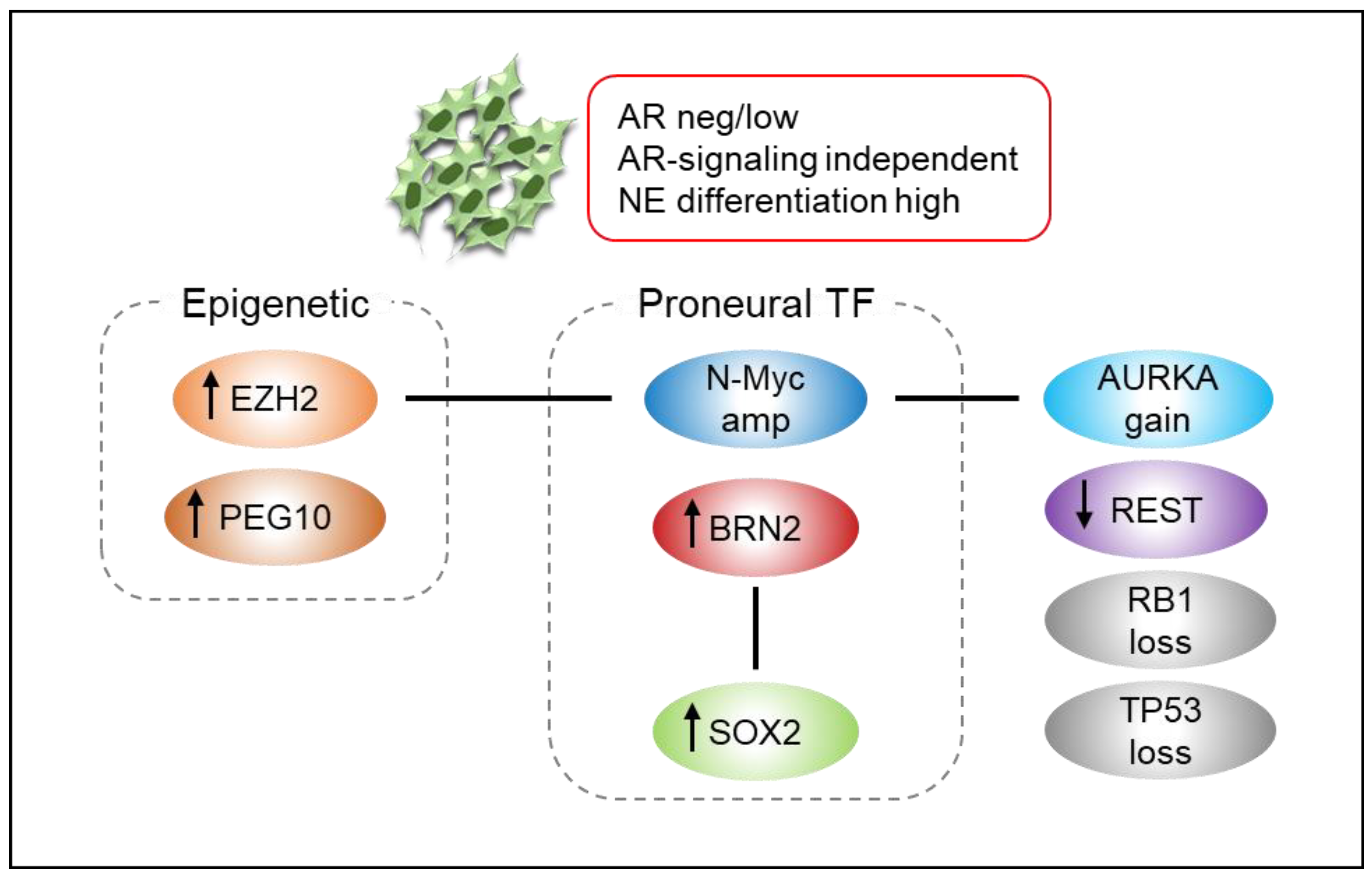
3. Neuroendocrine Prostate Cancer (NEPC)
3.1. Definition
3.2. Mechanisms
3.3. Diagnosis
3.3.1. Serum Markers
Chromogranin A (CgA)
Neuron-Specific Enolase (NSE)
Progastrin-Releasing Peptide (Pro-GRP)
3.3.2. Radiological Characteristics
3.3.3. Histological Characteristics
3.4. Treatment
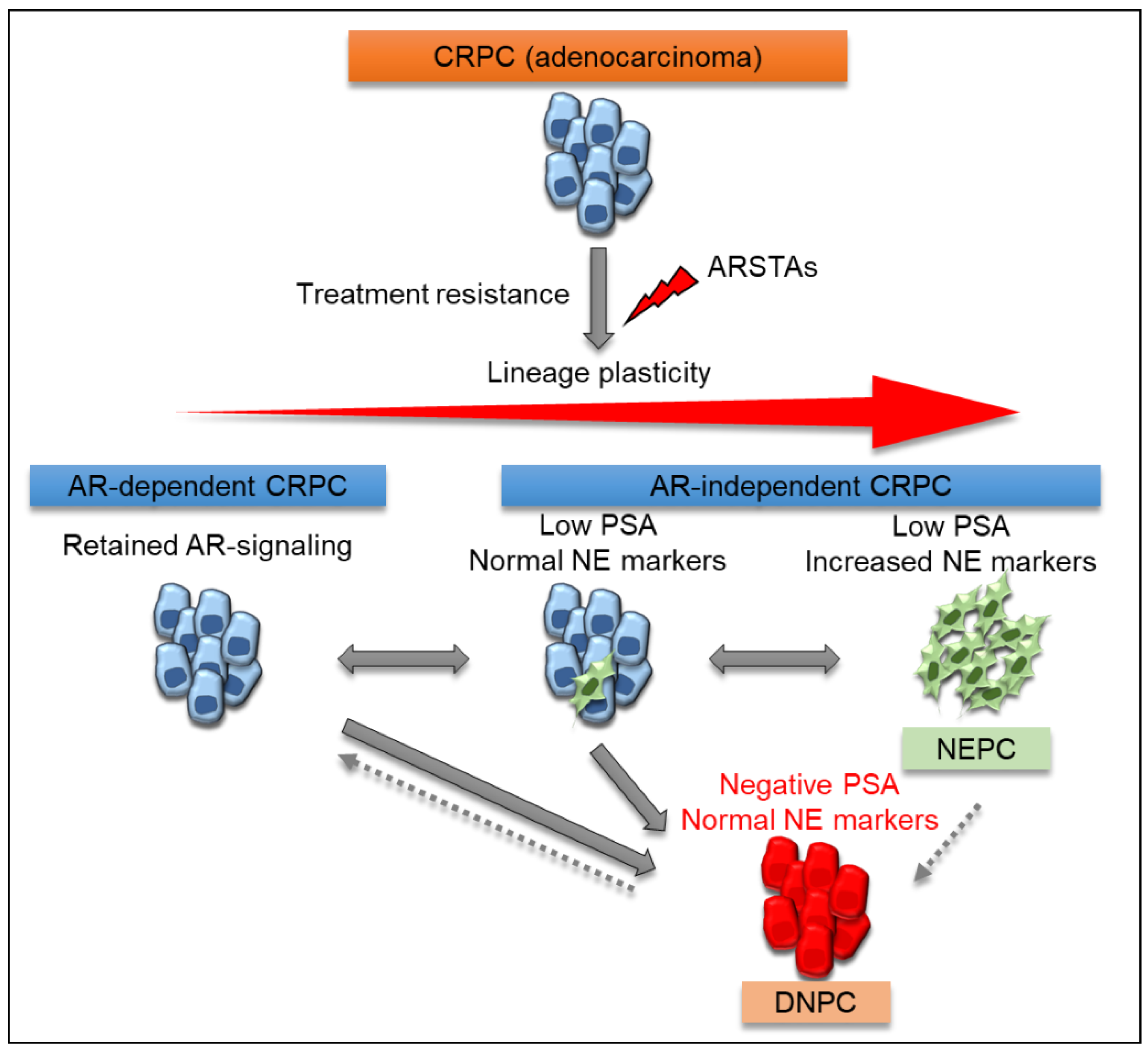
4. Prostate-Specific Antigen (PSA)-Null and NE-Null Double-Negative CRPC (DNPC)
4.1. DNPC Position
4.2. Potential Mechanisms
4.3. Potential Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, S.; Sowalsky, A.G.; Voznesensky, O.S.; Mostaghel, E.A.; Nelson, P.S.; Cai, C.; Balk, S.P. Rapid Induction of Androgen Receptor Splice Variants by Androgen Deprivation in Prostate Cancer. Clin. Cancer Res. 2014, 20, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.P.; Arnold, J.T.; et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef]
- Iwamoto, H.; Izumi, K.; Shimada, T.; Kano, H.; Kadomoto, S.; Makino, T.; Naito, R.; Yaegashi, H.; Shigehara, K.; Kadono, Y.; et al. Androgen receptor signaling-targeted therapy and taxane chemotherapy induce visceral metastasis in castration-resistant prostate cancer. Prostate 2021, 81, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef] [PubMed]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The Androgen Receptor Gene Mutations Database The androgen receptor gene mutations database: 2012 Update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Gao, A.C. Mechanisms of persistent activation of the androgen receptor in CRPC: Recent advances and future perspectives. World J. Urol. 2011, 30, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steketee, K.; Timmerman, L.; Der Made, A.C.Z.-V.; Doesburg, P.; Brinkmann, A.O.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; A Watson, P.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Lawrence, M.G.; Takayama, K.; Choo, N.; Risbridger, G.P.; Takahashi, S.; Inoue, S. Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 581515. [Google Scholar] [CrossRef] [PubMed]
- Linja, M.J.; Savinainen, K.J.; Saramäki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar] [PubMed]
- Koivisto, P.A.; Helin, H.J. Androgen receptor gene amplification increases tissue PSA protein expression in hormone-refractory prostate carcinoma. J. Pathol. 1999, 189, 219–223. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Huang, H.; Tindall, N.J. Androgen receptor signaling in androgen-refractory prostate cancer. J. Natl. Cancer Inst. 2001, 93, 1687–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, C.W.; Johnson, R.T., Jr.; Mohler, J.L.; French, F.S.; Wilson, E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001, 61, 2892–2898. [Google Scholar] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Altuwaijri, S.; Deng, F.; Chen, L.; Lal, P.; Bhanot, U.K.; Korets, R.; Wenske, S.; Lilja, H.G.; Chang, C.; et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 2009, 175, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A Novel Androgen Receptor Splice Variant Is Up-regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-Independent Androgen Receptor Variants Derived from Splicing of Cryptic Exons Signify Hormone-Refractory Prostate Cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, M.; Lu, C.; Chen, Y.; Paller, C.J.; Carducci, M.A.; Eisenberger, M.A.; Luo, J.; Antonarakis, E.S. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann. Oncol. 2015, 26, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2018, 129, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ledet, E.; Li, D.; Dotiwala, A.; Steinberger, A.; Feibus, A.; Li, J.; Qi, Y.; Silberstein, J.; Lee, B.; et al. A Whole Blood Assay for AR-V7 and AR v567es in Patients with Prostate Cancer. J. Urol. 2016, 196, 1758–1763. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Sprenger, C.; Sun, S.; Epilepsia, K.S.; Haugk, K.; Zhang, X.; Coleman, I.; Nelson, P.S.; Plymate, S. AR Variant ARv567es Induces Carcinogenesis in a Novel Transgenic Mouse Model of Prostate Cancer. Neoplasia 2013, 15, 1009-IN3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.; Plymate, S.R. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J. Cell. Biochem. 2006, 99, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Camarco, D.P.; Strock, C.J.; Johnston, P.A.; Trask, O.J. High Content Positional Biosensor Assay to Screen for Compounds that Prevent or Disrupt Androgen Receptor and Transcription Intermediary Factor 2 Protein-Protein Interactions. Adv. Struct. Saf. Stud. 2017, 1683, 211–227. [Google Scholar] [CrossRef]
- Saraon, P.; Jarvi, K.; Diamandis, E.P. Molecular Alterations during Progression of Prostate Cancer to Androgen Independence. Clin. Chem. 2011, 57, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr. Relat. Cancer 2009, 16, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (Review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandarlapaty, S. Negative Feedback and Adaptive Resistance to the Targeted Therapy of Cancer. Cancer Discov. 2012, 2, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic Targeting of PI3K/AKT Pathway and Androgen Receptor Axis Significantly Delays Castration-Resistant Prostate Cancer Progression In Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [Green Version]
- Truica, C.I.; Byers, S.; Gelmann, E.P. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000, 60, 4709–4713. [Google Scholar] [PubMed]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/beta-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Buttigliero, C.; Tucci, M.; Bertaglia, V.; Vignani, F.; Bironzo, P.; Di Maio, M.; Scagliotti, G.V. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 2015, 41, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Griend, N.J.V.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.M.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2017, 24, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.-M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed Morphologic Classification of Prostate Cancer with Neuroendocrine Differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.-M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.J.; et al. Platinum-Based Chemotherapy for Variant Castrate-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Yao, J.L.; di Sant’Agnese, P.A.; Yang, Q.; Bourne, P.A.; Na, Y. Immunohistochemical characterization of neuroendocrine cells in prostate cancer. Prostate 2006, 66, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.Y.; di Sant’Agnese, P.A.; Moynes, R.A.; Hiipakka, R.A.; Liao, S.; Cockett, A.T.; Abrahamsson, P.A. The androgen receptor status of neuroendocrine cells in human benign and malignant prostatic tissue. Cancer Res. 1993, 53, 1967–1970. [Google Scholar] [PubMed]
- Ellis, L.; Loda, M. LSD1: A single target to combat lineage plasticity in lethal prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4530–4531. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, C.E. The Role of Splicing Regulators in the Emergence of Treatment-induced Neuroendocrine Prostate Cancer: The Next Generation of Drug Targets? Eur. Urol. 2019, 76, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Zoubeidi, A.; A Selth, L. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr. Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; Macdonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Colleen, M.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; Macdonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef]
- Zhang, X.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lucas, J.M.; Lam, H.-M.; Dumpit, R.; Corey, E.; Chéry, L.; et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4698–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor–Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Cates, J.M.; Morrissey, C.; You, C.; Grabowska, M.M.; Zhang, J.; DeGraff, D.J.; Strand, D.W.; Franco, O.E.; Lin-Tsai, O.; et al. SOX2 expression in the developing, adult, as well as, diseased prostate. Prostate Cancer Prostatic Dis. 2014, 17, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.-F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of Pluripotency Networks by the Rb Tumor Suppressor Restricts Reprogramming and Tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Lin, C.-P.; Ho, J.J.; He, X.; Okada, N.; Bu, P.; Zhong, Y.; Kim, S.Y.; Bennett, M.J.; Chen, C.; et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 2011, 13, 1353–1360. [Google Scholar] [CrossRef]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Xiao, Y.; Hu, Z.; Chen, Y.; Liu, N.; Hu, G. PEG10 directly regulated by E2Fs might have a role in the development of hepatocellular carcinoma. FEBS Lett. 2008, 582, 2793–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thaper, D.; Bidnur, S.; Toren, P.; Akamatsu, S.; Bishop, J.L.; Colins, C.; Vahid, S.; Zoubeidi, A. PEG10 is associated with treatment-induced neuroendocrine prostate cancer. J. Mol. Endocrinol. 2019, 63, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosquera, J.M.; Beltran, H.; Park, K.; Macdonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN Gene Amplifications Are Harbingers of Lethal TreatmentRelated Neuroendocrine Prostate Cancer. Neoplasia 2013, 15, 1-IN4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxby, H.; Mikropoulos, C.; Boussios, S. An Update on the Prognostic and Predictive Serum Biomarkers in Metastatic Prostate Cancer. Diagnostics 2020, 10, 549. [Google Scholar] [CrossRef]
- Sargos, P.; Ferretti, L.; Gross-Goupil, M.; Orre, M.; Cornelis, F.; De Figueiredo, B.H.; Houédé, N.; Merino, C.; Roubaud, G.; Dallaudiére, B.; et al. Characterization of prostate neuroendocrine cancers and therapeutic management: A literature review. Prostate Cancer Prostatic Dis. 2014, 17, 220–226. [Google Scholar] [CrossRef]
- Kamiya, N.; Akakura, K.; Suzuki, H.; Isshiki, S.; Komiya, A.; Ueda, T.; Ito, H. Pretreatment serum level of neuron specific enolase (NSE) as a prognostic factor in metastatic prostate cancer patients treated with endocrine therapy. Eur. Urol. 2003, 44, 309–314. [Google Scholar] [CrossRef]
- Heck, M.M.; Thaler, M.A.; Schmid, S.C.; Seitz, A.-K.; Tauber, R.; Kübler, H.; Maurer, T.; Thalgott, M.; Hatzichristodoulou, G.; Höppner, M.; et al. Chromogranin A and neurone-specific enolase serum levels as predictors of treatment outcome in patients with metastatic castration-resistant prostate cancer undergoing abiraterone therapy. BJU Int. 2017, 119, 30–37. [Google Scholar] [CrossRef]
- Molina, R.; Filella, X.; Augé, J.M. ProGRP: A new biomarker for small cell lung cancer. Clin. Biochem. 2004, 37, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Taal, B.G.; Vincent, A.; van Velthuysen, M.-L.F.; Baas, P.; Buning-Kager, J.C.; Linders, T.C.; Bonfrer, J.M. Choice of tumour markers in patients with neuroendocrine tumours is dependent on the histological grade. A marker study of Chromogranin A, Neuron specific enolase, Progastrin-releasing peptide and cytokeratin fragments. Eur. J. Cancer 2012, 48, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Taal, B.G.; Bonfrer, J.M.G.; Vincent, A.; Van Velthuysen, M.L.; Baas, P. An elevated progastrin-releasing peptide level in patients with well-differentiated neuroendocrine tumours indicates a primary tumour in the lung and predicts a shorter survival. Ann. Oncol. 2011, 22, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Martucci, F.; Pascale, M.; Valli, M.C.; Pesce, G.A.; Froesch, P.; Giovanella, L.; Richetti, A.; Treglia, G. Impact of 18F-FDG PET/CT in Staging Patients With Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Front. Med. 2020, 6, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratt, D.E.; Gavane, S.; Tarlinton, L.; Fareedy, S.B.; Doran, M.G.; Zelefsky, M.J.; Osborne, J.R. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate 2014, 74, 1153–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, S.; Van Der Lely, A.; Hofland, L. New somatostatin analogs: Will they fulfil old promises? Eur. J. Endocrinol. 2002, 146, 701–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocr. 1999, 20, 157–198. [Google Scholar] [CrossRef]
- Rai, U.; Thrimawithana, T.R.; Valery, C.; Young, S.A. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol. Ther. 2015, 152, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Grozinsky-Glasberg, S.; Shimon, I.; Korbonits, M.A.; Grossman, A.B. Somatostatin analogues in the control of neuroendocrine tumours: Efficacy and mechanisms. Endocr. Relat. Cancer 2008, 15, 701–720. [Google Scholar] [CrossRef] [Green Version]
- Spieth, M.E.; Lin, Y.G.; Nguyen, T.T. Diagnosing and Treating Small-Cell Carcinomas of Prostatic Origin. Clin. Nucl. Med. 2002, 27, 11–17. [Google Scholar] [CrossRef]
- Mori, H.; Nakajima, K.; Kadomoto, S.; Mizokami, A.; Ikeda, H.; Wakabayashi, H.; Kinuya, S. Imaging Somatostatin Receptor Activity in Neuroendocrine-differentiated Prostate Cancer. Intern. Med. 2018, 57, 3123–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usmani, S.; Orevi, M.; Stefanelli, A.; Zaniboni, A.; Gofrit, O.N.; Bnà, C.; Illuminati, S.; Lojacono, G.; Noventa, S.; Savelli, G. Neuroendocrine differentiation in castration resistant prostate cancer. Nuclear medicine radiopharmaceuticals and imaging techniques: A narrative review. Crit. Rev. Oncol. 2019, 138, 29–37. [Google Scholar] [CrossRef]
- Gomes-Porras, M.; Cárdenas-Salas, J.; Álvarez-Escolá, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binderup, T.; Knigge, U.; Loft, A.; Mortensen, J.; Pfeifer, A.; Federspiel, B.; Hansen, C.P.; Højgaard, L.; Kjaer, A. Functional Imaging of Neuroendocrine Tumors: A Head-to-Head Comparison of Somatostatin Receptor Scintigraphy, 123I-MIBG Scintigraphy, and 18F-FDG PET. J. Nucl. Med. 2010, 51, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garin, E.; Le Jeune, F.; Devillers, A.; Cuggia, M.; De Lajarte-Thirouard, A.-S.; Bouriel, C.; Boucher, E.; Raoul, J.-L. Predictive Value of 18F-FDG PET and Somatostatin Receptor Scintigraphy in Patients with Metastatic Endocrine Tumors. J. Nucl. Med. 2009, 50, 858–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainio, M.; Visakorpi, T.; Tolonen, T.; Ilvesaro, J.; Bova, G.S. Expression of neuroendocrine differentiation markers in lethal metastatic castration-resistant prostate cancer. Pathol. Res. Pract. 2018, 214, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.; Zhang, T.; Small, E.J.; Armstrong, A.J. Neuroendocrine Prostate Cancer: Subtypes, Biology, and Clinical Outcomes. J. Natl. Compr. Cancer Netw. 2014, 12, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Winkler, H.; Westhead, E. Th molecular organization of adrenal chromaffin granules. Neuroscience 1980, 5, 1803–1823. [Google Scholar] [CrossRef]
- Nilsson, O.; Jakobsen, A.-M.L.; Kölby, L.; Bernhardt, P.; Forssell-Aronsson, E.; Ahlman, H. Importance of Vesicle Proteins in the Diagnosis and Treatment of Neuroendocrine Tumors. Ann. N. Y. Acad. Sci. 2004, 1014, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Portela-Gomes, G.M.; Lukinius, A.; Grimelius, L. Synaptic Vesicle Protein 2, A New Neuroendocrine Cell Marker. Am. J. Pathol. 2000, 157, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Rindi, G.; Paolotti, D.; Fiocca, R.; Wiedenmann, B.; Henry, J.-P.; Solcia, E. Vesicular monoamine transporter 2 as a marker of gastric enterochromaffin-like cell tumors. Virchows Arch. 2000, 436, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–418. [Google Scholar] [CrossRef]
- Tahara, S.; Sanno, N.; Teramoto, A.; Osamura, R.Y. Expression of Rab3, a Ras-related GTP-binding protein, in human non-tumorous pituitaries and pituitary adenomas. Mod. Pathol. 1999, 12, 627–634. [Google Scholar]
- Regazzi, R.; Wollheim, C.; Lang, J.; Theler, J.; Rossetto, O.; Montecucco, C.; Sadoul, K.; Weller, U.; Palmer, M.; Thorens, B. VAMP-2 and cellubrevin are expressed in pancreatic beta-cells and are essential for Ca(2+)-but not for GTP gamma S-induced insulin secretion. EMBO J. 1995, 14, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.E.; Fritz, B.A.; Wong, S.M.; Lowe, A.W. Identification of a vesicle-associated membrane protein (VAMP)-like mem-brane protein in zymogen granules of the rat exocrine pancreas. J. Biol. Chem. 1994, 269, 5328–5335. [Google Scholar] [CrossRef]
- Tischler, A.S.; Mobtaker, H.; Mann, K.; Nunnemacher, G.; Jason, W.J.; Dayal, Y.; Delellis, R.A.; Adelman, L.; Wolfe, H.J. An-ti-lymphocyte antibody Leu-7 (HNK-1) recognizes a constituent of neuroendocrine granule matrix. J. Histochem. Cytochem. 1986, 34, 1213–1216. [Google Scholar] [CrossRef]
- Lipinski, M.; Braham, K.; Caillaud, J.M.; Carlu, C.; Tursz, T. HNK-1 antibody detects an antigen expressed on neuroectodermal cells. J. Exp. Med. 1983, 158, 1775–1780. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fléchon, A.; Pouessel, D.; Ferlay, C.; Perol, D.; Beuzeboc, P.; Gravis, G.; Joly, F.; Oudard, S.; Deplanque, G.; Zanetta, S.; et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate Cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Geni-to-Urinary Tumor Group (GETUG) P01. Trial. Ann. Oncol. 2011, 22, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ohtsubo, K.; Mouri, H.; Yamashita, K.; Yasumoto, K.; Izumi, K.; Zen, Y.; Watanabe, H.; Yano, S. Combined chemotherapy with carboplatin plus irinotecan showed favorable efficacy in a patient with relapsed small cell carcinoma of the prostate complicated with meningeal carcinomatosis. Int. J. Clin. Oncol. 2009, 14, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Ishidoya, S.; Ito, A.; Endoh, M.; Shimazui, T.; Arai, Y. Experience of the treatment with gemcitabine, docetaxel, and carboplatin (GDC) chemotherapy for patients with small-cell carcinoma of the prostate. Int. J. Urol. 2006, 13, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.N.; Daliani, D.D.; Thall, P.F.; Tu, S.-M.; Wang, X.; Reyes, A.; Troncoso, P.; Logothetis, C.J. Results of a Phase II Study with Doxorubicin, Etoposide, and Cisplatin in Patients with Fully Characterized Small-Cell Carcinoma of the Prostate. J. Clin. Oncol. 2002, 20, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, F.; Nakai, Y.; Tomizawa, M.; Owari, T.; Miyake, M.; Inoue, T.; Anai, S.; Tanaka, N.; Fujimoto, K. Amrubicin is ef-fective against small cell carcinoma of the prostate as a second-line chemotherapeutic agent: A case report. IJU Case Rep. 2019, 2, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, L.; Nientiedt, C.; Winkler, E.C.; Berger, A.K.; Kratochwil, C.; Kaiser, A.; Becker, A.-S.; Jäger, D.; Hohenfellner, M.; Hüttenbrink, C.; et al. Clinical characteristics, treatment outcomes and potential novel therapeutic options for patients with neuroendocrine carcinoma of the prostate. Oncotarget 2019, 10, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC di-agnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [Green Version]
- Spetsieris, N.; Boukovala, M.; Patsakis, G.; Alafis, I.; Efstathiou, E. Neuroendocrine and Aggressive-Variant Prostate Cancer. Cancers 2020, 12, 3792. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A phase II trial of the Aurora kinase a inhibitor alisertib for patients with castration-resistant and neuroendocrine prostate cancer: Efficacy and bi-omarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, E.-C.; Rice, M.A.; Bermudez, A.; Marques, F.J.G.; Aslan, M.; Liu, S.; Ghoochani, A.; Zhang, C.A.; Chen, Y.-S.; Zlitni, A.; et al. Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP. Proc. Natl. Acad. Sci. USA 2020, 117, 2032–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Epstein, J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J Surg. Pathol. 2008, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Han, H.H.; Wang, Y.; Zhang, B.; Zhou, B.; Cheng, Y.; Rumandla, A.; Gurrapu, S.; Chakraborty, G.; Su, J.; et al. The Polycomb repressor Complex 1 drives dou-ble-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell. 2019, 36, 139–155.e10. [Google Scholar] [CrossRef]
- Izumi, K.; Mizokami, A. Suppressive Role of Androgen/Androgen Receptor Signaling via Chemokines on Prostate Cancer Cells. J. Clin. Med. 2019, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for andro-gen-deprivation therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nat. Cell Biol. 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M. Inducible FGFR-1 Activation Leads to Irreversible Prostate Adenocarcinoma and an Epithelial-to-Mesenchymal Transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten Loss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, H.; Chatani, Y.; Kohno, M.; Kawakita, M.; Ogawa, O. Constitutive activation of the 41- and 43-kDa mitogen-activated protein (MAP) kinases in the progression of prostate cancer to an androgen-independent state. Int. J. Urol. 2005, 12, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, G.K.; Mutton, L.N.; Khalili, M.; McMullin, R.P.; Hicks, J.L.; Bianchi-Frias, D.; Horn, L.A.; Kulac, I.; Moubarek, M.S.; Nelson, P.S.; et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2015, 76, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.M. A Positive Step toward Understanding Double-Negative Metastatic Prostate Cancer. Cancer Cell 2019, 36, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2018, 110, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, H.; Izumi, K.; Mizokami, A. Is the C-C Motif Ligand 2–C-C Chemokine Receptor 2 Axis a Promising Target for Cancer Therapy and Diagnosis? Int. J. Mol. Sci. 2020, 21, 9328. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated with the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Buttigliero, C.; Pisano, C.; Di Stefano, R.F.; Tabbò, F.; Turco, F.; Vignani, F.; Scagliotti, G.V.; Di Maio, M.; Tucci, M. Bipolar androgen therapy in prostate cancer: Current evidences and future perspectives. Crit. Rev. Oncol. 2020, 152, 102994. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Song, X.-L.; Wang, C.; Yu, Y.-Z.; Wang, J.-Q.; Chen, Z.-S.; Zhao, S.-C. The role of androgen therapy in prostate cancer: From testosterone replacement therapy to bipolar androgen therapy. Drug Discov. Today 2021. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Mirochnik, Y.; Veliceasa, D.; Williams, L.; Maxwell, K.; Yemelyanov, A.; Budunova, I.; Volpert, O.V. Androgen Receptor Drives Cellular Senescence. PLoS ONE 2012, 7, e31052. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| AR-Dependent CRPC | Ref. | NEPC | Ref. | DNPC | Ref. | |
|---|---|---|---|---|---|---|
| Prevalence | ARSTA pre-approval era; 88.4% | [1] | ARSTA pre-approval era; 6.3% | [1] | ARSTA pre-approval era; 5.4% | [1] |
| ARSTA post-approval era; 63.3% | ARSTA post-approval era; 13.3% | ARSTA post-approval era; 23.3% | ||||
| Prognosis | 15–32 months | [12] | 7–16 months | [11,125,126] | No data reported | |
| Serum markers |
|
|
| |||
|
|
| ||||
| Histological features |
| [9] |
| [9] |
| [9] |
|
|
| ||||
| Clinical features |
| [11] |
| [11] |
| [11] |
| Molecular features |
| [2,3,4,41,45,51,54,55,56] |
| [8,68,69,70,71] |
| [1,132,140] |
|
|
| ||||
|
|
| ||||
|
|
| ||||
| ||||||
| ||||||
| Treatments |
| [12,118] |
| [61,118,119,120,121,122,123,124,127] |
| [1,142,144,146,147,148] |
|
|
| ||||
|
| |||||
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makino, T.; Izumi, K.; Mizokami, A. Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment. Biomedicines 2021, 9, 414. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040414
Makino T, Izumi K, Mizokami A. Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment. Biomedicines. 2021; 9(4):414. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040414
Chicago/Turabian StyleMakino, Tomoyuki, Kouji Izumi, and Atsushi Mizokami. 2021. "Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment" Biomedicines 9, no. 4: 414. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040414