
Anti-Tumor Functions of Prelatent Antithrombin on Glioblastoma Multiforme Cells

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Media
2.2. Native and Prelatent AT Purification
2.3. EP and Hepsin Inhibition by Prelatent AT and Complex Formation
2.4. Wound Healing Assay
2.5. Matrigel Invasion Assay
2.6. Cell Proliferation Assay
2.7. Real Time-PCR and Immunoblotting of Selected Cancer Signaling Proteins
2.8. Statistics
3. Results
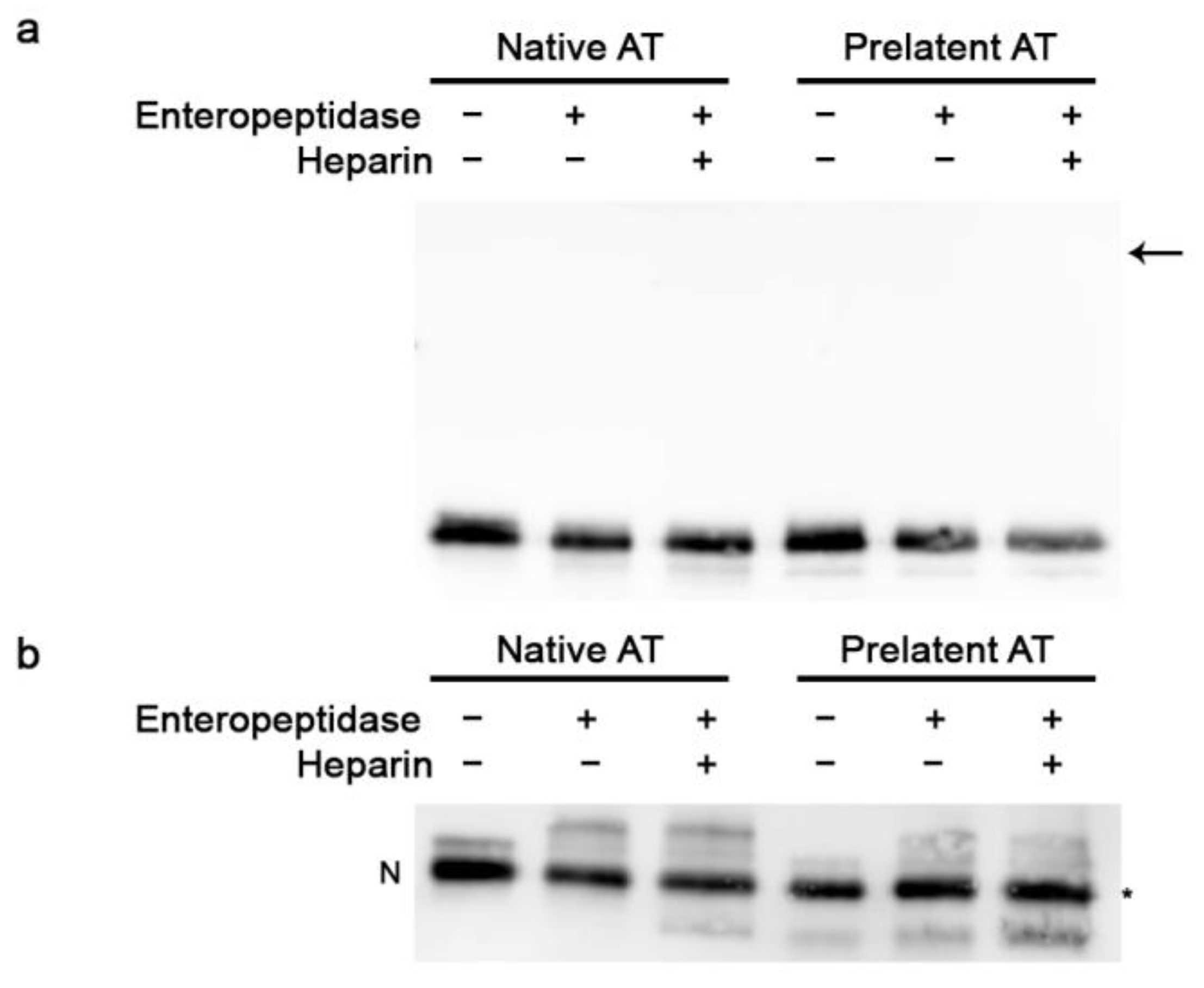
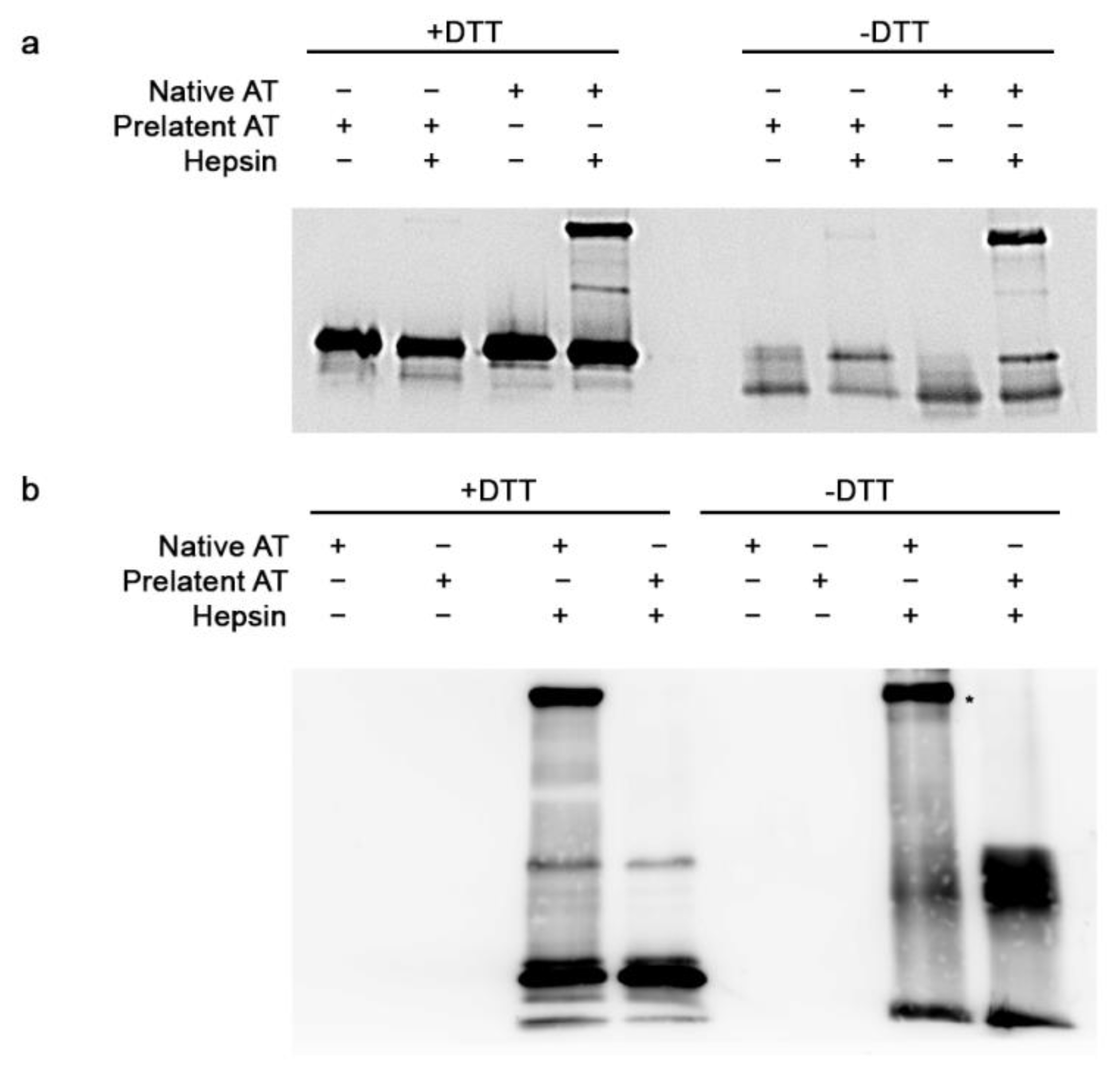
3.1. Inhibitory Effect of Prelatent AT on EP and Hepsin
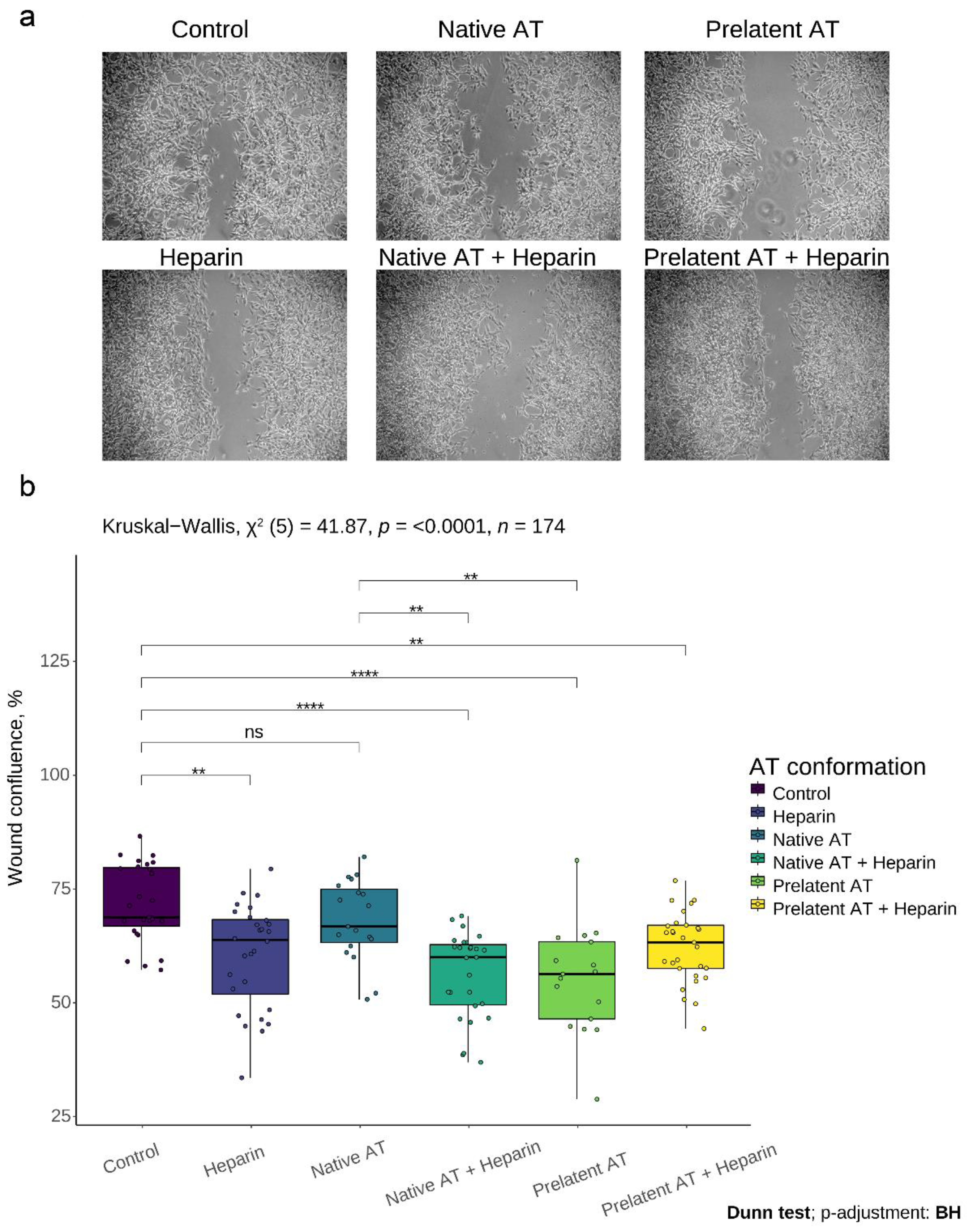
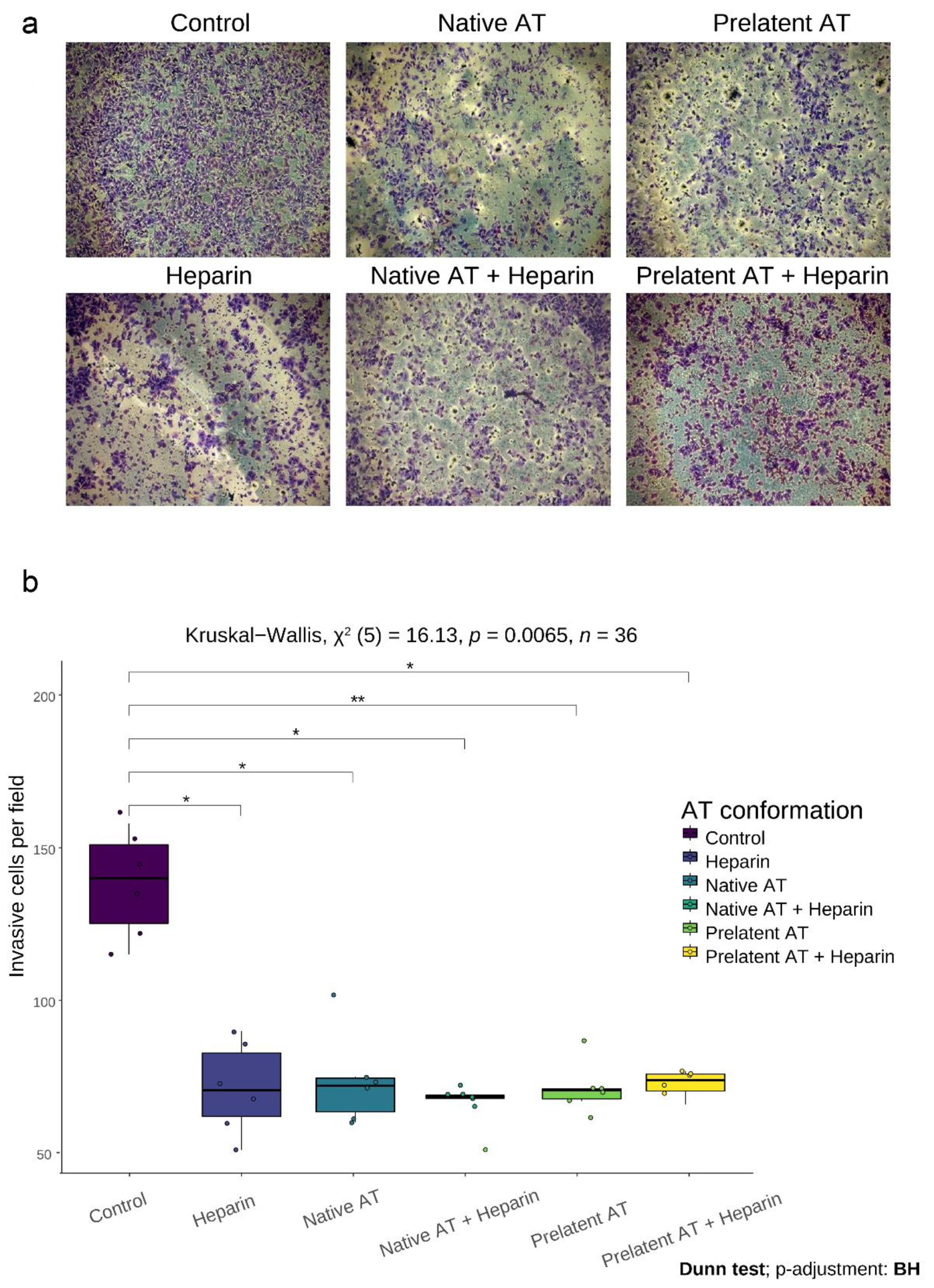
3.2. Prelatent AT Reduces Migration and Invasion of U-87 MG Cells
3.3. Prelatent AT Did Not Affect the Proliferation of U-87 MG or U-251 MG Cells
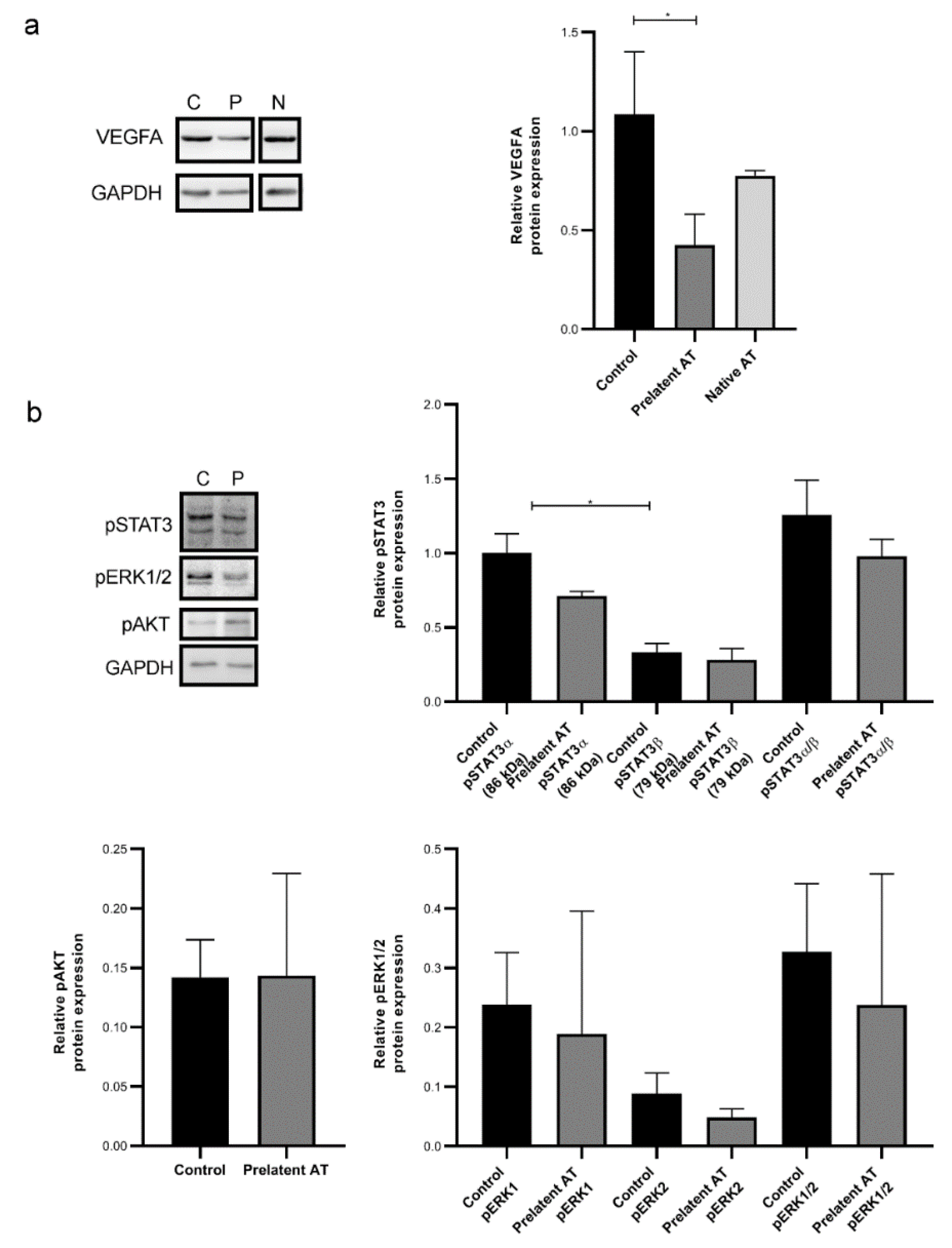
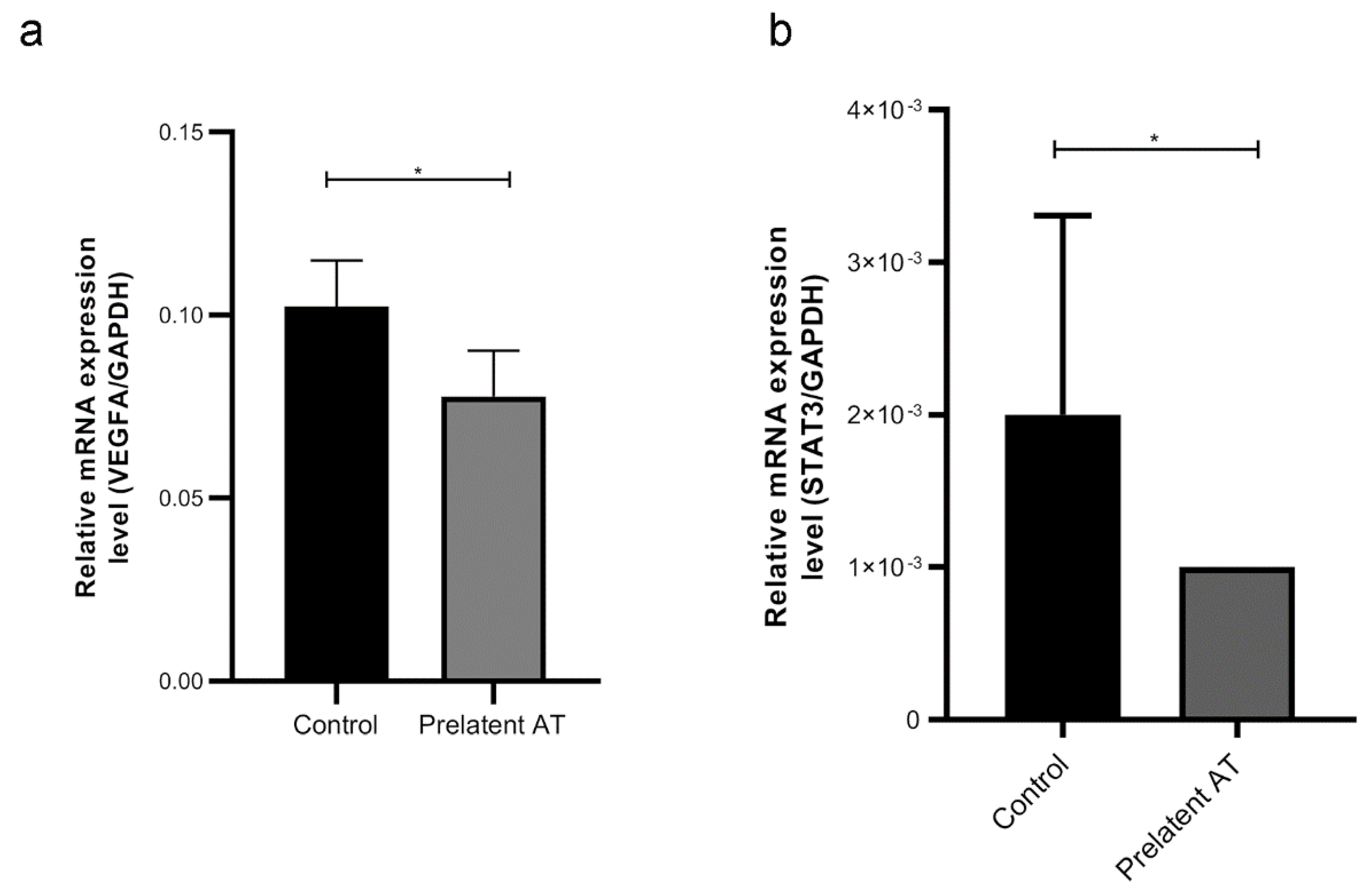
3.4. Prelatent AT Downregulates the Expression or Function of Different Cancer Signaling Molecules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Chuang, Y.-J.; Jin, T.; Swanson, R.; Xiong, Y.; Leung, L.; Olson, S.T. Antiangiogenic Antithrombin Induces Global Changes in the Gene Expression Profile of Endothelial Cells. Cancer Res. 2006, 66, 5047–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, H.; Åkerud, P.; Nordling, K.; Raub-Segall, E.; Claesson-Welsh, L.; Björk, I. A Novel Anti-Angiogenic Form of Antithrombin with Retained Proteinase Binding Ability and Heparin Affinity. J. Biol. Chem. 2001, 276, 11996–12002. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Pirie-Shepherd, S.; Lane, W.S.; Folkman, J. Antiangiogenic Activity of the Cleaved Conformation of the Serpin Antithrombin. Science 1999, 285, 1926–1928. [Google Scholar] [CrossRef]
- Hoffmann, J.N.; Wiedermann, C.J.; Juers, M.; Ostermann, H.; Kienast, J.; Briegel, J.; Strauss, R.; Warren, B.L.; Opal, S.M. Benefit/Risk Profile of High-Dose Antithrombin in Patients with Severe Sepsis Treated with and without Concomitant Heparin. Thromb. Haemost. 2006, 95, 850–856. [Google Scholar] [CrossRef]
- Oelschläger, C.; Römisch, J.; Staubitz, A.; Stauss, H.; Leithäuser, B.; Tillmanns, H.; Hölschermann, H. Antithrombin III Inhibits Nuclear Factor ΚB Activation in Human Monocytes and Vascular Endothelial Cells. Blood 2002, 99, 4015–4020. [Google Scholar] [CrossRef] [Green Version]
- Kienast, J.; Juers, M.; Wiedermann, C.J.; Hoffmann, J.N.; Ostermann, H.; Strauss, R.; Keinecke, H.-O.; Warren, B.L.; Opal, S.M. Treatment Effects of High-Dose Antithrombin without Concomitant Heparin in Patients with Severe Sepsis with or without Disseminated Intravascular Coagulation. J. Thromb. Haemost. 2006, 4, 90–97. [Google Scholar] [CrossRef]
- Guerrero, J.A.; Teruel, R.; Martínez, C.; Arcas, I.; Martínez-Martínez, I.; de la Morena-Barrio, M.E.; Vicente, V.; Corral, J. Protective Role of Antithrombin in Mouse Models of Liver Injury. J. Hepatol. 2012, 57, 980–986. [Google Scholar] [CrossRef]
- Asmal, M.; Seaman, M.; Lin, W.; Chung, R.T.; Letvin, N.L.; Geiben-Lynn, R. Inhibition of HCV by the Serpin Antithrombin III. Virol. J. 2012, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Luengo-Gil, G.; Calvo, M.I.; Martín-Villar, E.; Águila, S.; Bohdan, N.; Antón, A.I.; Espín, S.; Ayala de la Peña, F.; Vicente, V.; Corral, J.; et al. Antithrombin Controls Tumor Migration, Invasion and Angiogenesis by Inhibition of Enteropeptidase. Sci. Rep. 2016, 6, 27544. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Wang, W.; Zhang, Y.; Zhang, B.; Cheng, J.; Teng, P.; Tang, X. Expression Levels and Clinical Significance of Hepsin and HMGB1 Proteins in Cervical Carcinoma. Oncol. Lett. 2017, 14, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhao, J.; Tang, W.; Wang, Y.; Peng, P.; Li, L.; Song, S.; Wu, H.; Li, C.; Yang, C.; et al. High Hepsin Expression Predicts Poor Prognosis in Gastric Cancer. Sci. Rep. 2016, 6, 36902. [Google Scholar] [CrossRef] [Green Version]
- Luengo-Gil, G.; García-Andreo, A.B.; Ortega-Sabater, C.; Bohdan, N.; Espín, S.; Peñas-Martínez, J.; Martínez-Planes, E.; García-Hernández, Á.; Vicente, V.; Quintanilla, M.; et al. Antithrombin Is Incorporated into Exosomes Produced by Antithrombin Non-Expressing Cells. Biochimie 2019, 165, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Whisstock, J.C.; Bottomley, S.P. Molecular Gymnastics: Serpin Structure, Folding and Misfolding. Curr. Opin. Struct. Biol. 2006, 16, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Richard, B.; Swanson, R.; Schedin-Weiss, S.; Ramirez, B.; Izaguirre, G.; Gettins, P.G.W.; Olson, S.T. Characterization of the Conformational Alterations, Reduced Anticoagulant Activity, and Enhanced Antiangiogenic Activity of Prelatent Antithrombin. J. Biol. Chem. 2008, 283, 14417–14429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohdan, N.; Espín, S.; Águila, S.; Teruel-Montoya, R.; Vicente, V.; Corral, J.; Martínez-Martínez, I. Heparanase Activates Antithrombin through the Binding to Its Heparin Binding Site. PLoS ONE 2016, 11, e0157834. [Google Scholar] [CrossRef]
- Mushunje, A.; Evans, G.; Brennan, S.O.; Carrell, R.W.; Zhou, A. Latent Antithrombin and Its Detection, Formation and Turnover in the Circulation. J. Thromb. Haemost. 2004, 2, 2170–2177. [Google Scholar] [CrossRef]
- Gladson, C.L.; Prayson, R.A.; Liu, W.M. The Pathobiology of Glioma Tumors. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 33–50. [Google Scholar] [CrossRef] [Green Version]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT Pathway in Glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/MTOR and Raf/MEK/ERK Pathways in the Treatment of Breast Cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]
- Xie, B.; Zhang, L.; Hu, W.; Fan, M.; Jiang, N.; Duan, Y.; Jing, D.; Xiao, W.; Fragoso, R.C.; Lam, K.S.; et al. Dual Blockage of STAT3 and ERK1/2 Eliminates Radioresistant GBM Cells. Redox Biol. 2019, 24, 101189. [Google Scholar] [CrossRef] [PubMed]
- Aigner, P.; Just, V.; Stoiber, D. STAT3 Isoforms: Alternative Fates in Cancer? Cytokine 2019, 118, 27–34. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 Activity Up-Regulates VEGF Expression and Tumor Angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Johnson, R.S. Hypoxia: A Key Regulator of Angiogenesis in Cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C.E. Identification of Membrane Type-1 Matrix Metalloproteinase as a Target of Hypoxia-Inducible Factor-2 α in von Hippel–Lindau Renal Cell Carcinoma. Oncogene 2005, 24, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-L.; Wang, M.J.; Lee, D.; Liang, C.-C.; Lin, S. Hypoxia-Inducible Factor-1α Regulates Matrix Metalloproteinase-1 Activity in Human Bone Marrow-Derived Mesenchymal Stem Cells. FEBS Lett. 2008, 582, 2615–2619. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Castellano-Sanchez, A.; Kaur, B.; Van Meir, E.G. Genetic and Biologic Progression in Astrocytomas and Their Relation to Angiogenic Dysregulation. Adv. Anat. Pathol. 2002, 9, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor Invasion after Treatment of Glioblastoma with Bevacizumab: Radiographic and Pathologic Correlation in Humans and Mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, R.M.; Torcuator, R.; Jain, R.; Anderson, J.; Doyle, T.; Ellika, S.; Schultz, L.; Mikkelsen, T. Efficacy, Safety and Patterns of Response and Recurrence in Patients with Recurrent High-Grade Gliomas Treated with Bevacizumab plus Irinotecan. J. Neurooncol. 2009, 91, 329. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking Inflammation to Epithelial Cancer-More than a “Gut” Feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 Inhibition Overcomes Temozolomide Resistance in Glioblastoma by Downregulating MGMT Expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.; Ahn, S.H.; Kong, D.-S.; Lee, H.W.; Nam, D.-H. The Role of STAT3 in Glioblastoma Progression through Dual Influences on Tumor Cells and the Immune Microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Cajal, S.R.Y. STAT3 Labels a Subpopulation of Reactive Astrocytes Required for Brain Metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef]
- De Groot, J.; Liang, J.; Kong, L.-Y.; Wei, J.; Piao, Y.; Fuller, G.; Qiao, W.; Heimberger, A.B. Modulating Antiangiogenic Resistance by Inhibiting the Signal Transducer and Activator of Transcription 3 Pathway in Glioblastoma. Oncotarget 2012, 3, 1036. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J. Silibinin Is a Direct Inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef]
- Shingu, T.; Holmes, L.; Henry, V.; Wang, Q.; Latha, K.; Gururaj, A.E.; Gibson, L.A.; Doucette, T.; Lang, F.F.; Rao, G. Suppression of RAF/MEK or PI3K Synergizes Cytotoxicity of Receptor Tyrosine Kinase Inhibitors in Glioma Tumor-Initiating Cells. J. Transl. Med. 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, P.; Nanjaiah, N.D.; Borkotokey, M. Role of MEK-ERK Signaling Mediated Adhesion of Glioma Cells to Extracellular Matrix: Possible Implication on Migration and Proliferation. Ann. Neurosci. 2019, 26, 52–56. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, S.; Wang, Q.; Zheng, J. TMPRSS3 Modulates Ovarian Cancer Cell Proliferation, Invasion and Metastasis. Oncol. Rep. 2016, 35, 81–88. [Google Scholar] [CrossRef]
- Villalba, M.; Exposito, F.; Pajares, M.J.; Sainz, C.; Redrado, M.; Remirez, A.; Wistuba, I.; Behrens, C.; Jantus-Lewintre, E.; Camps, C. TMPRSS4: A Novel Tumor Prognostic Indicator for the Stratification of Stage IA Tumors and a Liquid Biopsy Biomarker for NSCLC Patients. J. Clin. Med. 2019, 8, 2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Lee, K.P.; Park, S.J.; Park, J.H.; Jang, Y.S.; Choi, S.Y.; Jung, J.G.; Jo, K.; Park, D.Y.; Yoon, J.H. TMPRSS4 Promotes Invasion, Migration and Metastasis of Human Tumor Cells by Facilitating an Epithelial–Mesenchymal Transition. Oncogene 2008, 27, 2635–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Du, L.; Li, C.; Wu, W. Human Chorionic Gonadotropin β Induces Cell Motility via ERK1/2 and MMP-2 Activation in Human Glioblastoma U87MG Cells. J. Neurooncol. 2013, 111, 237–244. [Google Scholar] [CrossRef]
- Selvasaravanan, K.D.; Wiederspohn, N.; Hadzalic, A.; Strobel, H.; Payer, C.; Schuster, A.; Karpel-Massler, G.; Siegelin, M.D.; Halatsch, M.-E.; Debatin, K.-M. The Limitations of Targeting MEK Signalling in Glioblastoma Therapy. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Perry, J.R. Thromboembolic Disease in Patients with High-Grade Glioma. Neuro Oncol. 2012, 14, iv73–iv80. [Google Scholar] [CrossRef]
- Perry, J.R.; Julian, J.A.; Laperriere, N.J.; Geerts, W.; Agnelli, G.; Rogers, L.R.; Malkin, M.G.; Sawaya, R.; Baker, R.; Falanga, A. PRODIGE: A Randomized Placebo-controlled Trial of Dalteparin Low-molecular-weight Heparin Thromboprophylaxis in Patients with Newly Diagnosed Malignant Glioma. J. Thromb. Haemost. 2010, 8, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Hentschel, B.; Simon, M.; Schnell, O.; Heese, O.; Tatagiba, M.; Krex, D.; Reithmeier, T.; Kowoll, A.; Weller, M.; et al. A Comprehensive Analysis of Vascular Complications in 3889 Glioma Patients from the German Glioma Network. J. Neurol. 2013, 260, 847–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nghiemphu, P.L.; Green, R.M.; Pope, W.B.; Lai, A.; Cloughesy, T.F. Safety of Anticoagulation Use and Bevacizumab in Patients with Glioma. Neuro Oncol. 2008, 10, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Turnšek, T.L. Mesenchymal Stem Cells Differentially Affect the Invasion of Distinct Glioblastoma Cell Lines. Oncotarget 2017, 8, 25482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lei, B.; Xiang, W.; Wang, H.; Feng, W.; Liu, Y.; Qi, S. Differences in Protein Expression between the U251 and U87 Cell Lines. Turk. Neurosurg. 2017, 27, 894–903. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Vmax405 (mOD/min) |
|---|---|
| Enteropeptidase | 3.357 ± 0.311 |
| Native antithrombin + Enteropeptidase | 2.714 ± 0.679 |
| Prelatent antithrombin + Enteropeptidase | 2.976 ± 1.209 |
| Native antithrombin + Enteropeptidase + Heparin | 0.536 ± 0.258 |
| Prelatent antithrombin + Enteropeptidase + Heparin | 2.571 ± 0.619 |
| Condition | Vmax405 (mOD/min) |
|---|---|
| Hepsin | 3.67 × 106 ± 4.69 × 105 |
| Native antithrombin + Hepsin | 3.21 × 106 ± 1.81 × 105 |
| Native antithrombin+ Heparin + Hepsin | 2.38 × 106 ± 1.91 × 105 |
| Prelatent antithrombin+ Hepsin | 3.65 × 106 ± 1.15 × 105 |
| Prelatent antithrombin+ Hepsin + Native antithrombin | 3.15 × 106 ± 2.86 × 105 |
| * Prelatent antithrombin+ Hepsin + Native antithrombin | 1.19 × 106 ± 3.15 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peñas-Martínez, J.; Luengo-Gil, G.; Espín, S.; Bohdan, N.; Ortega-Sabater, C.; Ródenas, M.C.; Zaragoza-Huesca, D.; López-Andreo, M.J.; Plasencia, C.; Vicente, V.; et al. Anti-Tumor Functions of Prelatent Antithrombin on Glioblastoma Multiforme Cells. Biomedicines 2021, 9, 523. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050523
Peñas-Martínez J, Luengo-Gil G, Espín S, Bohdan N, Ortega-Sabater C, Ródenas MC, Zaragoza-Huesca D, López-Andreo MJ, Plasencia C, Vicente V, et al. Anti-Tumor Functions of Prelatent Antithrombin on Glioblastoma Multiforme Cells. Biomedicines. 2021; 9(5):523. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050523
Chicago/Turabian StylePeñas-Martínez, Julia, Ginés Luengo-Gil, Salvador Espín, Nataliya Bohdan, Carmen Ortega-Sabater, Maria Carmen Ródenas, David Zaragoza-Huesca, María José López-Andreo, Carme Plasencia, Vicente Vicente, and et al. 2021. "Anti-Tumor Functions of Prelatent Antithrombin on Glioblastoma Multiforme Cells" Biomedicines 9, no. 5: 523. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050523