
How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis?

 ,
,
Abstract
:1. Introduction
2. Adenosine as a Neuromodulator
3. Adenosine Receptors

{kind=link}
{kind=link}
| Adenosine Receptor Subtype | Central Nervous System | Peripheral Organs/Tissues and Non-neuronal Cells |
|---|---|---|
| A1 | Widely distributed with highest levels in the cerebral cortex, hippocampus, cerebellum, thalamus, brain stem and dorsal horn of the spinal cord [35,36,37,38] | Widely distributed, including mononuclear cells in the blood, heart, kidney, adipose tissue [34,35,39,40] |
| A2A | Highly concentrated in dorsal and ventral striatum (on striatopallidal medium spiny neurons (MSNs)). Additionally expressed in the globus pallidus (external), nucleus accumbens, olfactory tubercle [17,18,41]. Expressed in lower levels in the hippocampus, thalamus, cerebellum, cerebral cortex [38,42,43,44] and spinal cord motor neurons [36,41,45]. | Spleen, thymus, blood vessels, heart, lung, immune cells, platelets, glial cells [34,35,46] |
| A2B | Widely distributed (low density) [34,35] | Widely distributed (very low density). Higher levels in the cecum, colon, bladder, macrophages, mast cells [34,35] |
| A3 | Widely distributed (low density) [34,35] | Widely distributed (low density). Higher levels in mast cells, eosinophils [34,35,47] |
- Identification of A2AR-specific expression in the medium spiny neurons (MSNs, also known as spiny projection neurons), projecting through the striatum to GPe [50].
- Demonstration of A2A antagonist efficacy in functional animal models for PD [53].
- Discovery of physiological significance of A2ARs in the MSN and establishing the mechanism of action for A2AR antagonism in PD therapy [43].
4. Pathophysiology in Adenosine Levels and A2A Receptor Density in ALS
5. Pharmacology of Adenosine A2A Receptor Blockade on ALS Animal Models
- Disease stage (pre-symptomatic phase, onset of symptomatic phase and end stage);
- The ALS model used and the mechanisms underlying the motor neuron disease.
6. Uric Acid as a Proposed Biomarker in Patients with ALS
- What is the sequence for initiating motor neuron death? Increased level of adenosine and/or via adenosine A2AR activation?
- What causes loss of ADA?
- What are the mechanisms that drive the upregulation/down regulation of A2ARs?
- What are the timings by which the alterations of the adenosinergic system occur during ALS pathogenesis in patients and animal models?
- What are the processes of each event in the entire pathogenesis of ALS from pre-symptomatic, symptomatic and end stages?
- How can plasma UA be utilized as a biomarker for diagnosis and therapy?
- Can A2AR antagonists (and agonists) be useful pharmacotherapy during an ALS patient’s journey, and if so, what is the optimal timing for such therapy?
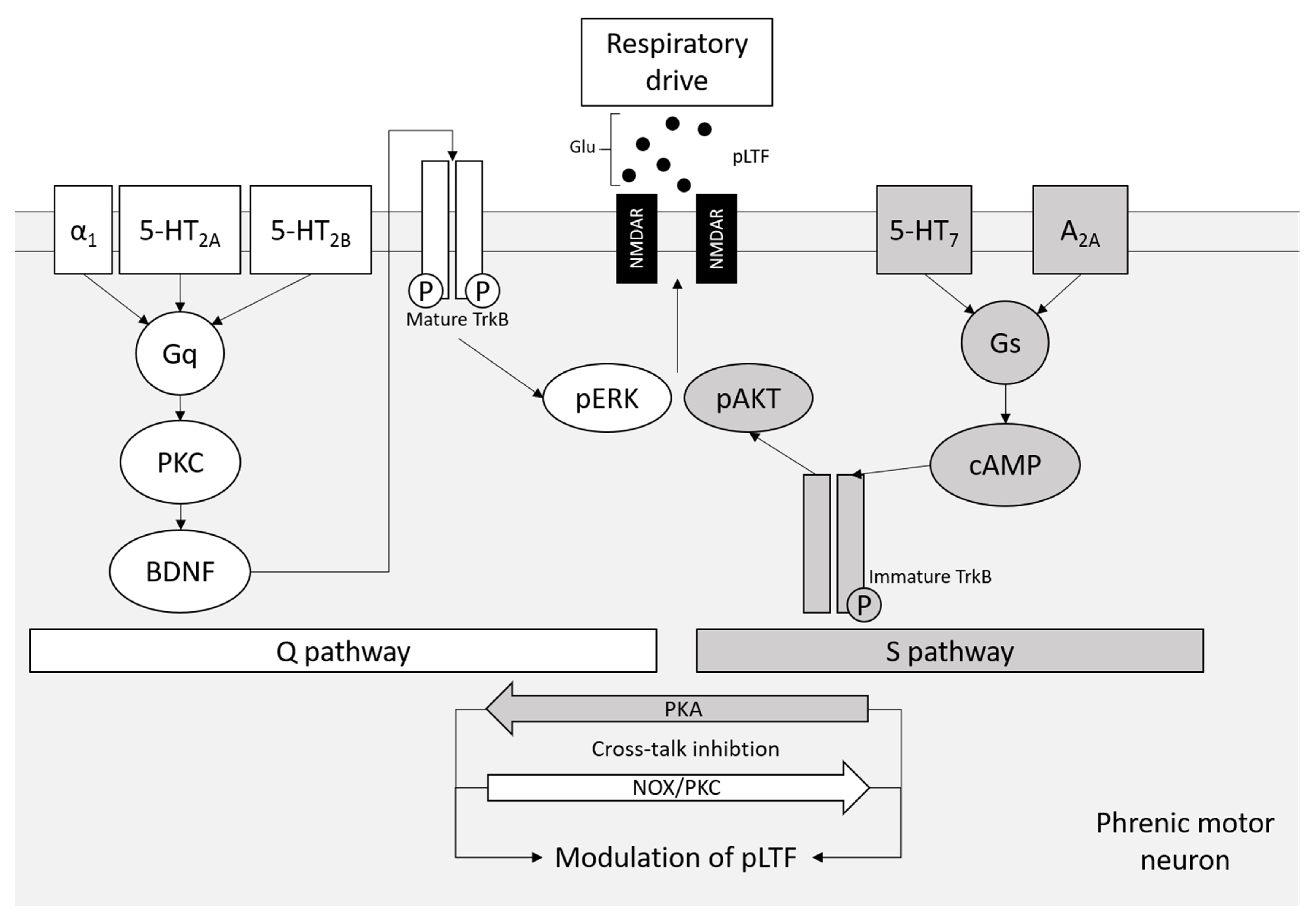
7. A2A Receptors in Respiratory Motor Neurons and tAIH Treatment for ALS
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: Separating the wheat from the chaff. J. Parkinson’s Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Baker, M.R. ALS—Dying forward, backward or outward? Nat. Rev. Neurol. 2014, 10, 660. [Google Scholar] [CrossRef] [Green Version]
- Casas, C.; Manzano, R.; Vaz, R.; Osta, R.; Brites, D. Synaptic failure: Focus in an integrative view of ALS. Brain Plast. 2016, 1, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef]
- Ballarín, M.; Fredholm, B.B.; Ambrosio, S.; Mahy, N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: Inhibition of uptake and metabolism. Acta Physiol. Scand. 1991, 142, 97–103. [Google Scholar] [CrossRef]
- Pedata, F.; Corsi, C.; Melani, A.; Bordoni, F.; Latini, S. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann. N. Y. Acad. Sci. 2001, 939, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Delaney, S.M.; Geiger, J.D. Levels of endogenous adenosine in rat striatum. II. Regulation of basal and N-methyl-D-aspartate-induced levels by inhibitors of adenosine transport and metabolism. J. Pharmacol. Exp. Ther. 1998, 285, 568–572. [Google Scholar]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. Tripartite synapses: Roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 2009, 57, 343–346. [Google Scholar] [CrossRef] [Green Version]
- Mori, A. Mode of action of adenosine A2A receptor antagonists as symptomatic treatment for Parkinson’s disease. Int. Rev. Neurobiol. 2014, 119, 87–116. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Le Moine, C.; Aubert, I.; Burbaud, P.; Fredholm, B.B.; Bloch, B. Cellular distribution of adenosine A2A receptor mRNA in the primate striatum. J. Comp. Neurol. 1998, 399, 229–240. [Google Scholar] [CrossRef]
- Huang, Z.-L.; Zhang, Z.; Qu, W.-M. Roles of adenosine and its receptors in sleep–wake regulation. Int. Rev. Neurobiol. 2014, 119, 349–371. [Google Scholar] [PubMed]
- Ishikawa, T.; Aw, W.; Kaneko, K. Metabolic interactions of purine derivatives with human ABC transporter ABCG2: Genetic testing to assess gout risk. Pharmaceuticals 2013, 6, 1347–1360. [Google Scholar] [CrossRef]
- Klyuch, B.P.; Dale, N.; Wall, M.J. Deletion of ecto-5′-nucleotidase (CD73) reveals direct action potential-dependent adenosine release. J. Neurosci. 2012, 32, 3842–3847. [Google Scholar] [CrossRef] [Green Version]
- Pajski, M.L.; Venton, B.J. Adenosine release evoked by short electrical stimulations in striatal brain slices is primarily activity dependent. ACS Chem. Neurosci. 2010, 1, 775–787. [Google Scholar] [CrossRef]
- Street, S.E.; Walsh, P.L.; Sowa, N.A.; Taylor-Blake, B.; Guillot, T.S.; Vihko, P.; Wightman, R.M.; Zylka, M.J. PAP and NT5E inhibit nociceptive neurotransmission by rapidly hydrolyzing nucleotides to adenosine. Mol. Pain 2011, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Street, S.E.; Kramer, N.J.; Walsh, P.L.; Taylor-Blake, B.; Yadav, M.C.; King, I.F.; Vihko, P.; Wightman, R.M.; Millán, J.L.; Zylka, M.J. Tissue-nonspecific alkaline phosphatase acts redundantly with PAP and NT5E to generate adenosine in the dorsal spinal cord. J. Neurosci. 2013, 33, 11314–11322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonta, C.; Négyessy, L.; Renaud, L.; Barone, P. Areal and subcellular localization of the ubiquitous alkaline phosphatase in the primate cerebral cortex: Evidence for a role in neurotransmission. Cereb. Cortex 2004, 14, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez-Zaera, M.; Díaz-Hernández, J.I.; Hernández-Álvarez, E.; Zimmermann, H.; Díaz-Hernández, M.; Miras-Portugal, M.T. Tissue-nonspecific alkaline phosphatase promotes axonal growth of hippocampal neurons. Mol. Biol. Cell 2011, 22, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xiong, W.; Chu, S.; Sun, C.; Albensi, B.C.; Parkinson, F.E. Inhibition of hippocampal synaptic activity by ATP, hypoxia or oxygen-glucose deprivation does not require CD73. PLoS ONE 2012, 7, e39772. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Venton, B.J. Regional variations of spontaneous, transient adenosine release in brain slices. ACS Chem. Neurosci. 2018, 9, 505–513. [Google Scholar] [CrossRef]
- Carruthers, A.M.; Sellers, L.A.; Jenkins, D.W.; Jarvie, E.M.; Feniuk, W.; Humphrey, P.P. Adenosine A(1) receptor-mediated inhibition of protein kinase A-induced calcitonin gene-related peptide release from rat trigeminal neurons. Mol. Pharmacol. 2001, 59, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.J.; Jang, I.S.; Nabekura, J.; Akaike, N. Adenosine A1 receptor-mediated presynaptic inhibition of GABAergic transmission in immature rat hippocampal CA1 neurons. J. Neurophys. 2003, 89, 1214–1222. [Google Scholar] [CrossRef]
- Fenton, R.A.; Shea, L.G.; Doddi, C.; Dobson, J.G., Jr. Myocardial adenosine A(1)-receptor-mediated adenoprotection involves phospholipase C, PKC-epsilon, and p38 MAPK, but not HSP27. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1671–H1678. [Google Scholar] [CrossRef] [Green Version]
- Josselyn, S.A.; Nguyen, P.V. CREB, synapses and memory disorders: Past progress and future challenges. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Waltereit, R.; Weller, M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 2003, 27, 99–106. [Google Scholar] [CrossRef]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Choca, J.I.; Proudfit, H.K.; Green, R.D. Identification of A1 and A2 adenosine receptors in the rat spinal cord. J. Pharmacol. Exp. Ther. 1987, 242, 905–910. [Google Scholar] [PubMed]
- Rebola, N.; Pinheiro, P.C.; Oliveira, C.R.; Malva, J.O.; Cunha, R.A. Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003, 987, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.; Richardson, P.J.; Freeman, T.C. Tissue distribution of adenosine receptor mRNAs in the rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.B.; Silva, C.; Gonzalez, G.; Holden, J.; Warren, K.G.; Metz, L.M.; Power, C. Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann. Neurol. 2001, 49, 650–658. [Google Scholar] [CrossRef]
- Mayne, M.; Shepel, P.N.; Jiang, Y.; Geiger, J.D.; Power, C. Dysregulation of adenosine A1 receptor-mediated cytokine expression in peripheral blood mononuclear cells from multiple sclerosis patients. Ann. Neurol. 1999, 45, 633–639. [Google Scholar] [CrossRef]
- Kaelin-Lang, A.; Lauterburg, T.; Burgunder, J.M. Expression of adenosine A2a receptors gene in the olfactory bulb and spinal cord of rat and mouse. Neurosci. Lett. 1999, 261, 189–191. [Google Scholar] [CrossRef]
- Svenningsson, P.; Hall, H.; Sedvall, G.; Fredholm, B.B. Distribution of adenosine receptors in the postmortem human brain: An extended autoradiographic study. Synapse 1997, 27, 322–335. [Google Scholar] [CrossRef]
- Mori, A. How do adenosine A(2A) receptors regulate motor function? Parkinsonism Relat. Disord. 2020, 80, S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.S.; Rademakers, R.; Miller, B.L. Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Ann. N. Y. Acad. Sci. 2015, 1338, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Boison, D.; Chen, J.F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- Libert, F.; Parmentier, M.; Lefort, A.; Dinsart, C.; Van Sande, J.; Maenhaut, C.; Simons, M.J.; Dumont, J.E.; Vassart, G. Selective amplification and cloning of four new members of the G protein-coupled receptor family. Science 1989, 244, 569–572. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Aradi, S.D.; Hauser, R.A. Istradefylline—A first generation adenosine A2A antagonist for the treatment of Parkinson’s disease. Exp. Rev. Neurother. 2021, 21, 317–333. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferré, S. Adenosine A2A receptors and basal ganglia physiology. Progr. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef] [Green Version]
- Simola, N.; Morelli, M.; Pinna, A. Adenosine A2A receptor antagonists and Parkinson’s disease: State of the art and future directions. Curr. Pharm. Des. 2008, 14, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Shimada, J.; Suzuki, F.; Nonaka, H.; Ishii, A.; Ichikawa, S. (E)-1,3-dialkyl-7-methyl-8-(3,4,5-trimethoxystyryl)xanthines: Potent and selective adenosine A2 antagonists. J. Med. Chem. 1992, 35, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Jenner, P. Can adenosine A(2A) receptor antagonists modify motor behavior and dyskinesia in experimental models of Parkinson’s disease? Parkinsonism Relat. Disord. 2020, 80, S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Dridi, M.; Hornykiewicz, O.; Bedard, P.J.; Rajput, A.H.; Di Paolo, T. Increased adenosine A2A receptors in the brain of Parkinson’s disease patients with dyskinesias. Brain 2004, 127, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Villar-Menendez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Diaz-Sanchez, S.; Albasanz, J.L.; Ferrer, I.; Martin, M.; Barrachina, M. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol. Dis. 2014, 69, 206–214. [Google Scholar] [CrossRef]
- Mishina, M.; Ishiwata, K.; Naganawa, M.; Kimura, Y.; Kitamura, S.; Suzuki, M.; Hashimoto, M.; Ishibashi, K.; Oda, K.; Sakata, M.; et al. Adenosine A(2A) receptors measured with [C]TMSX PET in the striata of Parkinson’s disease patients. PLoS ONE 2011, 6, e17338. [Google Scholar] [CrossRef]
- Morelli, M.; Di Paolo, T.; Wardas, J.; Calon, F.; Xiao, D.; Schwarzschild, M.A. Role of adenosine A2A receptors in parkinsonian motor impairment and l-DOPA-induced motor complications. Prog. Neurobiol. 2007, 83, 293–309. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Bose, S.K.; Ahmed, I.; Turkheimer, F.E.; Pavese, N.; Brooks, D.J. Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology 2011, 76, 1811–1816. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Hubble, J.P.; Truong, D.D. Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology 2003, 61, 297–303. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Aradi, S.D.; Hauser, R.A.; Rascol, O. The challenge of developing adenosine A(2A) antagonists for Parkinson disease: Istradefylline, preladenant, and tozadenant. Parkinsonism Relat. Disord. 2020, 80, S54–S63. [Google Scholar] [CrossRef]
- Yoshida, Y.; Une, F.; Utatsu, Y.; Nomoto, M.; Furukawa, Y.; Maruyama, Y.; Machigashira, N.; Matsuzaki, T.; Osame, M. Adenosine and neopterin levels in cerebrospinal fluid of patients with neurological disorders. Int. Med. 1999, 38, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, A.M.; Rei, N.; Ribeiro, J.A. Amyotrophic Lateral Sclerosis (ALS) and Adenosine Receptors. Front. Pharmacol. 2018, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of adenosine 2a receptor (A2aR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2015, 267, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenza, R.L.; Armida, M.; Ferrante, A.; Pèzzola, A.; Matteucci, A.; Puopolo, M.; Popoli, P. Effects of chronic caffeine intake in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2013, 91, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Rei, N.; Rombo, D.M.; Ferreira, M.F.; Baqi, Y.; Müller, C.E.; Ribeiro, J.A.; Sebastião, A.M.; Vaz, S.H. Hippocampal synaptic dysfunction in the SOD1(G93A) mouse model of Amyotrophic Lateral Sclerosis: Reversal by adenosine A(2A)R blockade. Neuropharmacology 2020, 171, 108106. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Popoli, P.; Varani, K. A2A adenosine receptors are up-regulated in lymphocytes from amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 406–413. [Google Scholar] [CrossRef]
- Boison, D.; Aronica, E. Comorbidities in neurology: Is adenosine the common link? Neuropharmacology 2015, 97, 18–34. [Google Scholar] [CrossRef] [Green Version]
- Mojsilovic-Petrovic, J.; Jeong, G.B.; Crocker, A.; Arneja, A.; David, S.; Russell, D.S.; Kalb, R.G. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J. Neurosci. 2006, 26, 9250–9263. [Google Scholar] [CrossRef]
- Niccolini, F.; Foltynie, T.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Ju, T.C.; Chen, H.M.; Jang, Y.S.; Lee, L.M.; Lai, H.L.; Tai, H.C.; Fang, J.M.; Lin, Y.L.; Tu, P.H.; et al. Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Liu, Y.-J.; Lai, H.-L.; Chen, H.-M.; Kuo, H.-C.; Liao, Y.-P.; Chern, Y. The D2 dopamine receptor interferes with the protective effect of the A2A adenosine receptor on TDP-43 mislocalization in experimental models of motor neuron degeneration. Front. Neurosci. 2018, 12, 187. [Google Scholar] [CrossRef]
- Yanpallewar, S.U.; Barrick, C.A.; Buckley, H.; Becker, J.; Tessarollo, L. Deletion of the BDNF truncated receptor TrkB.T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39946. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.; Pousinha, P.A.; Correia, A.M.; Gomes, R.; Sebastião, A.M.; Ribeiro, J.A. Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre-symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS ONE 2014, 9, e104081. [Google Scholar] [CrossRef] [PubMed]
- Armida, M.; Matteucci, A.; Pèzzola, A.; Baqi, Y.; Müller, C.E.; Popoli, P.; Potenza, R.L. Modulating P1 adenosine receptors in disease progression of SOD1(G93A) mutant mice. Neurochem. Res. 2019, 44, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.; Sebastião, A.M.; Ribeiro, J.A. Presymptomatic and symptomatic ALS SOD1(G93A) mice differ in adenosine A1 and A2A receptor-mediated tonic modulation of neuromuscular transmission. Purinergic Signal. 2015, 11, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seven, Y.B.; Simon, A.K.; Sajjadi, E.; Zwick, A.; Satriotomo, I.; Mitchell, G.S. Adenosine 2A receptor inhibition protects phrenic motor neurons from cell death induced by protein synthesis inhibition. Exp. Neurol. 2020, 323, 113067. [Google Scholar] [CrossRef]
- Napolitano, F.; Pasqualetti, M.; Usiello, A.; Santini, E.; Pacini, G.; Sciamanna, G.; Errico, F.; Tassone, A.; Di Dato, V.; Martella, G.; et al. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol. Dis. 2010, 38, 434–445. [Google Scholar] [CrossRef] [Green Version]
- Correia-de-Sá, P.; Sebastião, A.M.; Ribeiro, J.A. Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve ending of the rat. Br. J. Pharmacol. 1991, 103, 1614–1620. [Google Scholar] [CrossRef]
- Ferré, S.; Rubio, A.; Fuxe, K. Stimulation of adenosine A2 receptors induces catalepsy. Neurosci. Lett. 1991, 130, 162–164. [Google Scholar] [CrossRef]
- Ramanathan, M.; Pinhal-Enfield, G.; Hao, I.; Leibovich, S.J. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol. Biol. Cell. 2007, 18, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Petimar, J.; O’Reilly, É.; Adami, H.O.; van den Brandt, P.A.; Buring, J.; English, D.R.; Freedman, D.M.; Giles, G.G.; Håkansson, N.; Kurth, T.; et al. Coffee, tea, and caffeine intake and amyotrophic lateral sclerosis mortality in a pooled analysis of eight prospective cohort studies. Eur. J. Neurol. 2019, 26, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Chen, J.-B.; Liu, E.M.; Chern, T.-R.; Yang, C.-W.; Lin, C.-I.; Huang, N.-K.; Lin, Y.-L.; Chern, Y.; Lin, J.-H.; Fang, J.-M. Design and synthesis of novel dual-action compounds targeting the adenosine A2A receptor and adenosine transporter for neuroprotection. Chem. Med. Chem. 2011, 6, 1390–1400. [Google Scholar] [CrossRef]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, N.D.; Sheean, R.K.; Scott, J.W.; Kemp, B.E.; Horne, M.K.; Turner, B.J. Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS ONE 2014, 9, e90449. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef]
- Fabbrini, E.; Serafini, M.; Colic Baric, I.; Hazen, S.L.; Klein, S. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes 2014, 63, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Proctor, P. Similar functions of uric acid and ascorbate in man? Nature 1970, 228, 868. [Google Scholar] [CrossRef] [PubMed]
- Yeum, K.J.; Russell, R.M.; Krinsky, N.I.; Aldini, G. Biomarkers of antioxidant capacity in the hydrophilic and lipophilic compartments of human plasma. Arch. Biochem. Biophys. 2004, 430, 97–103. [Google Scholar] [CrossRef]
- Oh, S.I.; Baek, S.; Park, J.S.; Piao, L.; Oh, K.W.; Kim, S.H. Prognostic role of serum levels of uric acid in amyotrophic lateral sclerosis. J. Clin. Neurol. 2015, 11, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Raknuzzaman, M.; Jannaty, T.; Ali Masum, A.S.M.H.; Sagir, G.; Wahiduzzaman, M.; Islam, M.N. Association of serum uric acid, homocystine and ferritin among amyotrophic lateral sclerosis patients. Int. J. Adv. Med. 2021, 8, 7. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Q.; Ke, Y.; Hao, J.; Lu, L.; Lu, N.; Chen, X. Serum uric acid levels in patients with amyotrophic lateral sclerosis: A meta-analysis. Sci. Rep. 2018, 8, 1100. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Zhang, M.; Quiroz Zárate, A.; Jaffa, M.; Yu, H.; Cudkowicz, M.E.; Wills, A.M. Uric acid levels predict survival in men with amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 1923–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshi, R.; Macklin, E.A.; Hung, A.Y.; Hayes, M.T.; Hyman, B.T.; Wills, A.M.; Gomperts, S.N.; Growdon, J.H.; Ascherio, A.; Scherzer, C.R.; et al. Associations of lower caffeine intake and plasma urate levels with idiopathic Parkinson’s disease in the Harvard biomarkers study. J. Parkinson’s Dis. 2020, 10, 505–510. [Google Scholar] [CrossRef]
- Crotty, G.F.; Maciuca, R.; Macklin, E.A.; Wang, J.; Montalban, M.; Davis, S.S.; Alkabsh, J.I.; Bakshi, R.; Chen, X.; Ascherio, A.; et al. Association of caffeine and related analytes with resistance to Parkinson disease among LRRK2 mutation carriers: A metabolomic study. Neurology 2020, 95, e3428–e3437. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Xu, D.; Hou, X.; Song, X.; Liu, C.; Chen, Y.; Wang, Y.; Li, X. Inverse association between serum uric acid levels and Alzheimer’s disease risk. Mol. Neurobiol. 2016, 53, 2594–2599. [Google Scholar] [CrossRef]
- Paganoni, S.; Schwarzschild, M.A. Urate as a marker of risk and progression of neurodegenerative disease. Neurotherapeutics 2017, 14, 148–153. [Google Scholar] [CrossRef]
- Auinger, P.; Kieburtz, K.; McDermott, M.P. The relationship between uric acid levels and Huntington’s disease progression. Mov. Disord. 2010, 25, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Euser, S.M.; Hofman, A.; Westendorp, R.G.; Breteler, M.M. Serum uric acid and cognitive function and dementia. Brain 2009, 132, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, M.C.; Raman, R.; Schwarzschild, M.A.; Becerra, L.M.; Thomas, R.G.; Peterson, R.C.; Ascherio, A.; Aisen, P.S. Plasma urate and progression of mild cognitive impairment. Neurodegener. Dis. 2009, 6, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Song, S.K.; Sohn, Y.H.; Lee, P.H. Uric acid as a potential disease modifier in patients with multiple system atrophy. Mov. Disord. 2011, 26, 1533–1536. [Google Scholar] [CrossRef]
- Fang, P.; Li, X.; Luo, J.J.; Wang, H.; Yang, X.F. A double-edged sword: Uric acid and neurological disorders. Brain Disord. Ther. 2013, 2, 109. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Burdett, T.C.; Desjardins, C.A.; Logan, R.; Cipriani, S.; Xu, Y.; Schwarzschild, M.A. Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wu, G.; Schwarzschild, M.A. Urate in Parkinson’s disease: More than a biomarker? Curr. Neurol. Neurosci. Rep. 2012, 12, 367–375. [Google Scholar] [CrossRef]
- Bergeron, C. Oxidative stress: Its role in the pathogenesis of amyotrophic lateral sclerosis. J. Neurol. Sci. 1995, 129, 81–84. [Google Scholar] [CrossRef]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of oxidative stress and inflammatory biomarkers in ALS: A pilot study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzo, F.; Mirra, A.; Carrì, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Chen, C.P.; Tseng, C.Y.; Eisenberg, Y.; Firestein, B.L. Astroglia-mediated effects of uric acid to protect spinal cord neurons from glutamate toxicity. Glia 2007, 55, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Meng, F.; Guo, Z.; Hu, Y.; Mai, W.; Zhang, Z.; Zhang, B.; Ge, Q.; Lou, H.; Guo, F.; Chen, J.; et al. CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signaling. Brain 2019, 142, 700–718. [Google Scholar] [CrossRef]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Sonsalla, P.K.; Pedata, F.; Melani, A.; Domenici, M.R.; Popoli, P.; Geiger, J.; Lopes, L.V.; de Mendonça, A. Adenosine A2A receptors and brain injury: Broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog. Neurobiol. 2007, 83, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Pantoni, L.; Bordoni, F.; Gianfriddo, M.; Bianchi, L.; Vannucchi, M.G.; Bertorelli, R.; Monopoli, A.; Pedata, F. The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res. 2003, 959, 243–250. [Google Scholar] [CrossRef]
- Melani, A.; Gianfriddo, M.; Vannucchi, M.G.; Cipriani, S.; Baraldi, P.G.; Giovannini, M.G.; Pedata, F. The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006, 1073–1074, 470–480. [Google Scholar] [CrossRef]
- Melani, A.; Cipriani, S.; Vannucchi, M.G.; Nosi, D.; Donati, C.; Bruni, P.; Giovannini, M.G.; Pedata, F. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain 2009, 132, 1480–1495. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Zhu, W.; Shao, M.; Zhang, F.; Guo, J.; Xu, H.; Jiang, J.; Ma, X.; Xia, X.; Zhi, X.; et al. Ecto-5′-nucleotidase (CD73) attenuates inflammation after spinal cord injury by promoting macrophages/microglia M2 polarization in mice. J. Neuroinflam. 2018, 15, 155. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F.; Schwarzschild, M.A. Do caffeine and more selective adenosine A2A receptor antagonists protect against dopaminergic neurodegeneration in Parkinson’s disease? Parkinsonism Relat. Disord. 2020, 80, S45–S53. [Google Scholar] [CrossRef]
- Iłzecka, J.; Stelmasiak, Z.; Balicka, G. Respiratory function in amyotrophic lateral sclerosis. Neurol. Sci. 2003, 24, 288–289. [Google Scholar] [CrossRef]
- Lyall, R.A.; Donaldson, N.; Polkey, M.I.; Leigh, P.N.; Moxham, J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain 2001, 124, 2000–2013. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Verma, R.; Garg, R.K.; Singh, M.K.; Shukla, R.; Verma, S.K. Assessment of respiratory functions by spirometry and phrenic nerve studies in patients of amyotrophic lateral sclerosis. J. Neurol. Sci. 2011, 306, 76–81. [Google Scholar] [CrossRef]
- Dale-Nagle, E.A.; Hoffman, M.S.; MacFarlane, P.M.; Satriotomo, I.; Lovett-Barr, M.R.; Vinit, S.; Mitchell, G.S. Spinal plasticity following intermittent hypoxia: Implications for spinal injury. Ann. N. Y. Acad. Sci. 2010, 1198, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, C.; Rozovsky, J. Effect of spinal cord injury on the respiratory system. Am. J. Phys. Med. Rehabil. 2003, 82, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Bourke, S.C.; Shaw, P.J.; Gibson, G.J. Respiratory function vs sleep-disordered breathing as predictors of QOL in ALS. Neurology 2001, 57, 2040–2044. [Google Scholar] [CrossRef]
- Lechtzin, N.; Rothstein, J.; Clawson, L.; Diette, G.B.; Wiener, C.M. Amyotrophic lateral sclerosis: Evaluation and treatment of respiratory impairment. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2002, 3, 5–13. [Google Scholar] [CrossRef]
- Dale-Nagle, E.A.; Hoffman, M.S.; MacFarlane, P.M.; Mitchell, G.S. Multiple pathways to long-lasting phrenic motor facilitation. Adv. Exp. Med. Biol. 2010, 669, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Golder, F.J.; Mitchell, G.S. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J. Neurosci. 2005, 25, 2925–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.S.; Johnson, S.M. Neuroplasticity in respiratory motor control. J. Appl. Physiol. 2003, 94, 358–374. [Google Scholar] [CrossRef]
- Xing, T.; Fong, A.Y.; Bautista, T.G.; Pilowsky, P.M. Acute intermittent hypoxia induced neural plasticity in respiratory motor control. Clin. Exp. Pharmacol. Physiol. 2013, 40, 602–609. [Google Scholar] [CrossRef]
- Bocchiaro, C.M.; Feldman, J.L. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc. Natl. Acad. Sci. USA 2004, 101, 4292–4295. [Google Scholar] [CrossRef] [Green Version]
- Devinney, M.J.; Huxtable, A.G.; Nichols, N.L.; Mitchell, G.S. Hypoxia-induced phrenic long-term facilitation: Emergent properties. Ann. N. Y. Acad. Sci. 2013, 1279, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.L.; Mitchell, G.S.; Nattie, E.E. Breathing: Rhythmicity, plasticity, chemosensitivity. Ann. Rev. Neurosci. 2003, 26, 239–266. [Google Scholar] [CrossRef] [Green Version]
- Bach, K.B.; Mitchell, G.S. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Resp. Physiol. 1996, 104, 251–260. [Google Scholar] [CrossRef]
- Baker, T.L.; Fuller, D.D.; Zabka, A.G.; Mitchell, G.S. Respiratory plasticity: Differential actions of continuous and episodic hypoxia and hypercapnia. Resp. Physiol. 2001, 129, 25–35. [Google Scholar] [CrossRef]
- Fuller, D.D.; Bach, K.B.; Baker, T.L.; Kinkead, R.; Mitchell, G.S. Long term facilitation of phrenic motor output. Resp. Physiol. 2000, 121, 135–146. [Google Scholar] [CrossRef]
- Mahamed, S.; Mitchell, G.S. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp. Physiol. 2007, 92, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.S.; Baker, T.L.; Nanda, S.A.; Fuller, D.D.; Zabka, A.G.; Hodgeman, B.A.; Bavis, R.W.; Mack, K.J.; Olson, E.B., Jr. Invited review: Intermittent hypoxia and respiratory plasticity. J. Appl. Physiol. 2001, 90, 2466–2475. [Google Scholar] [CrossRef]
- Baker, T.L.; Mitchell, G.S. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J. Physiol. 2000, 529, 215–219. [Google Scholar] [CrossRef]
- Baker-Herman, T.L.; Mitchell, G.S. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J. Neurosci. 2002, 22, 6239–6246. [Google Scholar] [CrossRef] [Green Version]
- Fuller, D.D.; Zabka, A.G.; Baker, T.L.; Mitchell, G.S. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J. Appl. Physiol. 2001, 90, 2001–2006. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, P.M.; Mitchell, G.S. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J. Physiol. 2009, 587, 5469–5481. [Google Scholar] [CrossRef]
- MacFarlane, P.M.; Vinit, S.; Mitchell, G.S. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: Differential requirement for spinal NADPH oxidase activity. Neuroscience 2011, 178, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Baker-Herman, T.L.; Fuller, D.D.; Bavis, R.W.; Zabka, A.G.; Golder, F.J.; Doperalski, N.J.; Johnson, R.A.; Watters, J.J.; Mitchell, G.S. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat. Neurosci. 2004, 7, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.S.; Nichols, N.L.; Macfarlane, P.M.; Mitchell, G.S. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J. Appl. Physiol. 2012, 113, 1184–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramham, C.R.; Messaoudi, E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog. Neurobiol. 2005, 76, 99–125. [Google Scholar] [CrossRef]
- Carter, A.R.; Chen, C.; Schwartz, P.M.; Segal, R.A. Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J. Neurosci. 2002, 22, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Dale, E.A.; Fields, D.P.; Devinney, M.J.; Mitchell, G.S. Phrenic motor neuron TrkB expression is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. Exp. Neurol. 2017, 287, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devinney, M.J.; Fields, D.P.; Huxtable, A.G.; Peterson, T.J.; Dale, E.A.; Mitchell, G.S. Phrenic long-term facilitation requires PKCθ activity within phrenic motor neurons. J. Neurosci. 2015, 35, 8107–8117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, N.L.; Gowing, G.; Satriotomo, I.; Nashold, L.J.; Dale, E.A.; Suzuki, M.; Avalos, P.; Mulcrone, P.L.; McHugh, J.; Svendsen, C.N.; et al. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am. J. Resp. Crit. Care Med. 2013, 187, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Golder, F.J.; Ranganathan, L.; Satriotomo, I.; Hoffman, M.; Lovett-Barr, M.R.; Watters, J.J.; Baker-Herman, T.L.; Mitchell, G.S. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J. Neurosci. 2008, 28, 2033–2042. [Google Scholar] [CrossRef]
- Hoffman, M.S.; Mitchell, G.S. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J. Physiol. 2011, 589, 1397–1407. [Google Scholar] [CrossRef]
- Fields, D.P.; Springborn, S.R.; Mitchell, G.S. Spinal 5-HT7 receptors induce phrenic motor facilitation via EPAC-mTORC1 signaling. J. Neurophysiol. 2015, 114, 2015–2022. [Google Scholar] [CrossRef] [Green Version]
- Nichols, N.L.; Dale, E.A.; Mitchell, G.S. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J. Appl. Physiol. 2012, 112, 1678–1688. [Google Scholar] [CrossRef] [Green Version]
- Conde, S.V.; Monteiro, E.C. Hypoxia induces adenosine release from the rat carotid body. J. Neurochem. 2004, 89, 1148–1156. [Google Scholar] [CrossRef]
- Dale, N.; Frenguelli, B.G. Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 2009, 7, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Gourine, A.V.; Llaudet, E.; Dale, N.; Spyer, K.M. Release of ATP in the ventral medulla during hypoxia in rats: Role in hypoxic ventilatory response. J. Neurosci. 2005, 25, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Devinney, M.J.; Nichols, N.L.; Mitchell, G.S. Sustained hypoxia elicits competing spinal mechanisms of phrenic motor facilitation. J. Neurosci. 2016, 36, 7877–7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, D.P.; Mitchell, G.S. Divergent cAMP signaling differentially regulates serotonin-induced spinal motor plasticity. Neuropharmacology 2017, 113, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Perim, R.R.; Fields, D.P.; Mitchell, G.S. Cross-talk inhibition between 5-HT(2B) and 5-HT(7) receptors in phrenic motor facilitation via NADPH oxidase and PKA. Am. J. Physiol. Reg. Integrat. Comp. Physiol. 2018, 314, R709–R715. [Google Scholar] [CrossRef]
- Hayes, H.B.; Jayaraman, A.; Herrmann, M.; Mitchell, G.S.; Rymer, W.Z.; Trumbower, R.D. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: A randomized trial. Neurology 2014, 82, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Trumbower, R.D.; Jayaraman, A.; Mitchell, G.S.; Rymer, W.Z. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil. Neural Repair 2012, 26, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Lovett-Barr, M.R.; Satriotomo, I.; Muir, G.D.; Wilkerson, J.E.; Hoffman, M.S.; Vinit, S.; Mitchell, G.S. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J. Neurosci. 2012, 32, 3591–3600. [Google Scholar] [CrossRef]
- Hoffman, M.S.; Golder, F.J.; Mahamed, S.; Mitchell, G.S. Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J. Physiol. 2010, 588, 255–266. [Google Scholar] [CrossRef]
- Satriotomo, I.; Dale, E.A.; Dahlberg, J.M.; Mitchell, G.S. Repetitive acute intermittent hypoxia increases expression of proteins associated with plasticity in the phrenic motor nucleus. Exp. Neurol. 2012, 237, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devivo, M.J. Epidemiology of traumatic spinal cord injury: Trends and future implications. Spinal Cord 2012, 50, 365–372. [Google Scholar] [CrossRef]
- DeVivo, M.J.; Chen, Y. Trends in new injuries, prevalent cases, and aging with spinal cord injury. Arch. Phys. Med. Rehabil. 2011, 92, 332–338. [Google Scholar] [CrossRef]
- Goshgarian, H.G. The crossed phrenic phenomenon: A model for plasticity in the respiratory pathways following spinal cord injury. J. Appl. Physiol. 2003, 94, 795–810. [Google Scholar] [CrossRef]
- Raineteau, O.; Schwab, M.E. Plasticity of motor systems after incomplete spinal cord injury. Nat. Rev. Neurosci. 2001, 2, 263–273. [Google Scholar] [CrossRef]
- Navarrete-Opazo, A.; Alcayaga, J.; Sepúlveda, O.; Rojas, E.; Astudillo, C. Repetitive intermittent hypoxia and locomotor training enhances walking function in incomplete spinal cord injury subjects: A randomized, triple-blind, placebo-controlled clinical trial. J. Neurotrauma 2017, 34, 1803–1812. [Google Scholar] [CrossRef]
- Navarrete-Opazo, A.; Vinit, S.; Dougherty, B.J.; Mitchell, G.S. Daily acute intermittent hypoxia elicits functional recovery of diaphragm and inspiratory intercostal muscle activity after acute cervical spinal injury. Exp. Neurol. 2015, 266, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser-Loose, E.J.; Hassan, A.; Mitchell, G.S.; Muir, G.D. Delayed intervention with intermittent hypoxia and task training improves forelimb function in a rat model of cervical spinal injury. J. Neurotrauma 2015, 32, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, L.; Urbin, M.A.; Mitchell, G.S.; Perez, M.A. Acute intermittent hypoxia enhances corticospinal synaptic plasticity in humans. Elife 2018, 7, e34304. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Opazo, A.A.; Vinit, S.; Mitchell, G.S. Adenosine 2A receptor inhibition enhances intermittent hypoxia-induced diaphragm but not intercostal long-term facilitation. J. Neurotrauma 2014, 31, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Opazo, A.; Dougherty, B.J.; Mitchell, G.S. Enhanced recovery of breathing capacity from combined adenosine 2A receptor inhibition and daily acute intermittent hypoxia after chronic cervical spinal injury. Exp. Neurol. 2017, 287, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, B.J.; Kopp, E.S.; Watters, J.J. Nongenomic actions of 17-β estradiol restore respiratory neuroplasticity in young ovariectomized female rats. J. Neurosci. 2017, 37, 6648–6660. [Google Scholar] [CrossRef] [Green Version]
- Nichols, N.L.; Satriotomo, I.; Harrigan, D.J.; Mitchell, G.S. Acute intermittent hypoxia induced phrenic long-term facilitation despite increased SOD1 expression in a rat model of ALS. Exp. Neurol. 2015, 273, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Nichols, N.L.; Satriotomo, I.; Allen, L.L.; Grebe, A.M.; Mitchell, G.S. Mechanisms of enhanced phrenic long-term facilitation in SOD1(G93A) Rats. J. Neurosci. 2017, 37, 5834–5845. [Google Scholar] [CrossRef] [Green Version]
- Satriotomo, I.; Nichols, N.L.; Dale, E.A.; Emery, A.T.; Dahlberg, J.M.; Mitchell, G.S. Repetitive acute intermittent hypoxia increases growth/neurotrophic factor expression in non-respiratory motor neurons. Neuroscience 2016, 322, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Acute Intermittent Hypoxia and Breathing in Neuromuscular Disease (AIH in ALS). Available online: https://clinicaltrials.gov/ct2/show/NCT03645031?term=acute+intermittent+hypoxia&cond=ALS&draw=2&rank=1 (accessed on 3 August 2021).
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Ribeiro, F.F.; Xapelli, S.; Miranda-Lourenço, C.; Tanqueiro, S.R.; Fonseca-Gomes, J.; Diógenes, M.J.; Ribeiro, J.A.; Sebastião, A.M. Purine nucleosides in neuroregeneration and neuroprotection. Neuropharmacology 2016, 104, 226–242. [Google Scholar] [CrossRef]


| Human | Sample/Model | Tissue/Sample Examined | Finding | Reference |
|---|---|---|---|---|
| A2AR | Postmortem samples from human patients with ALS | Spinal cord | Upregulation of A2AR | [64] |
| A2AR | Patients with ALS | Lymphocytes | Upregulation of A2AR | [68] |
| Adenosine | Patients with ALS | CSF | Increase of adenosine level | [61] |
| ADK | Patients with ALS | Reactive astrocyte/Spinal cord | Upregulation of ADK | [69] |
| A1R | Postmortem samples from human patients with ALS | Spinal cord | No significant change of A1R | [64] |
| Mouse | ||||
| A2AR | SOD1G93A mice | Spinal cord | Upregulation of A2AR (early symptomatic stage) | [64] |
| A2AR | SOD1G93A mice | Spinal cord | Decrease of A2AR (end stage) | [65] |
| A2AR | SOD1G93A mice | Hippocampus | Increased adenosine A2AR levels in hippocampus (pre-symptomatic and symptomatic stage). Impairment of LTP and NMDA receptor function. | [66] |
| A1R | SOD1G93A mice | Spinal cord | No significant change of A1R (symptomatic onset period: P100–110) | [64] |
| Experimental Model | Cell/Brain Area | Compound | Findings | Reference | |
|---|---|---|---|---|---|
| In vitro | |||||
| A2AR antagonist | Embryonic SD rat spinal cord cultures | Motor neurons | Istradefylline (1 μM) | Istradefylline protected against kainate-induced motor neuron death | [70] |
| A2AR antagonist, A2AR +/− | SOD1G93A+ astrocyte induced cell death | Embryonic stem cell-derived motor neuron (ESMN) | Istradefylline (1, 10 μM) A2AR +/− | Pharmacological inhibition (istradefylline) and partial genetic ablation of A2AR (A2AR +/−) significantly protected ESMN from SODG93A+ astrocyte-induced cell death | [71] |
| A2AR agonist, antagonist | Motor neuron cell line | NSC34 cells | Agonist: JMF1907 (30 μM) Antagonist: SCH58261 (10 μM) | JMF1907 enhanced the activity of adenylyl cyclase (AC) and suppressed the aberrant AMPK activity induced by AICAR, the AMPK-triggered mislocalization of TDP-43. These effects of JMF1907 were blocked using an A2AR-selective antagonist (SCH58261) | [72] |
| ADA | C9orf72 or sporadic ALS patients derived induced astrocyte | Astrocyte | RNA and protein levels of ADA were reduced in C9orf72 and sporadic ALS patient cell models. C9orf72 and sporadic ALS induced astrocytes were more susceptible to adenosine-mediated toxicity | [62] | |
| D2R agonist, A2AR agonist | Motor neuron | Cell line: NSC34 cells | A2AR agonist: T1–11 (30 μM) D2R agonist: quinpirole (1 μM) | Activation of D2R (quinpirole) negatively regulated A2AR-evoked cAMP signaling, without significantly affecting the binding affinity of T1–11 toward A2AR in NSC34 cells Activation of D2R suppressed A2AR-mediated protection of TDP-43 mislocalization in NSC34 cells | [73] |
| In vivo | |||||
| A2AR antagonist | SOD1G93A mice | Spinal cord | Istradefylline (3 mg/kg, ip) starting at P90–95 by daily ip injection (before symptomatic onset period). Disease onset: 121 ± 1.7 day | Istradefylline significantly delayed disease progression | [64] |
| A2AR antagonist | SOD1G93A mice | Hippocampus | Istradefylline (3 mg/kg/day) via drinking water (7.5 μg/mL) starting from 11 weeks to 16–18 weeks (symptomatic) old (early symptomatic disease stage) | Istradefylline rescued LTP impairment and A2AR levels | [66] |
| A2AR agonist | SOD1G93A mice | Spinal cord | CGS21680 (5 mg/kg/day, ip): Starting at 8 weeks of age (before the clinical manifestation of the disease) | CGS21680 treatment slowed the onset of motor neuron degeneration (12 weeks) and muscle weakness | [74] |
| A2AR agonist | SOD1G93A mice (Electrophysiological recordings) | Neuromuscular junction | CGS21680 (5 nM) pre-symptomatic mice (4–6 weeks) symptomatic mice (12–14 weeks) | In pre-symptomatic mice (4–6 weeks) the excitatory A2AR-mediated effects on neuromuscular transmission are exacerbated In symptomatic mice (12–14 weeks) the excitatory A2AR-mediated effects on neuromuscular transmission were absent | [75] |
| A2AR agonist | TDP-43 transgenic mice | Spinal cord | JMF1907 (111 mg/ mouse/day, sc) using ALZET osmotic minipump. Starting from 6 weeks to 23 weeks old (from presymptomatic) | JMF1907 markedly reduced the activation of AMPK JMF1907 also improved motor function based on rotarod performance and forelimb grip strength | [72] |
| A2AR agonist, antagonist, A1R antagonist | SOD1G93A mice | Motor performance, survival | A2AR agonist: CGS21680 (2.5 mg/kg, ip): five times per week. A2AR antagonist: istradefylline (3 mg/kg/day) via drinking water (0.25 mg/mL). A1R antagonist: DPCPX (0.75 mg/kg, ip): five times per week. Starting from 70 days of age (presymptomatic stage) | Neither the stimulation nor the blockade of adenosine A2AR modified the progressive loss of motor skills or survival of SODG93A mice. Blockade of adenosine A1R from the presymptomatic stage significantly attenuated motor disease progression and induced a non-significant increase of median survival in ALS mice. | [76] |
| A1/A2AR antagonist | SOD1G93A mice | Spinal cord | Caffeine (0.3 mg/mL) via drinking water. Starting from 70 days of age (before the onset of symptoms) | Caffeine intake significantly shortened the survival of SODG93A mice | [65] |
| D2R agonist, A2AR agonist | A315T TDP-43 transgenic mice | Spinal cord, motor performance (grip strength) | A2AR agonist: T1–11 (0.25 mg/mL) via drinking water. D2R agonist: Quinpirole (6 mg/kg, ip/day). Starting from 7 to 10 weeks old | Activation of D2R inhibited the A2AR -mediated beneficial effects (rescuing effect of T1–11 on TDP-43 mislocalization and impaired grip strength) | [73] |
| A1R agonist, antagonist, A2AR agonist, antagonist | SOD1G93A mice | Neuromuscular junction | A1R agonist: CPA (50 nM) A1R antagonist: DPCPX (50 nM) A2AR agonist: CGS21680 (5 nM) A2AR antagonist: SCH58261 (50 nM) pre-symptomatic mice (4–6 weeks) symptomatic mice (12–14 weeks) | In pre-symptomatic mice (4–6 weeks), there is a loss of A1R-A2AR functional crosstalk. In symptomatic mice (12–14 weeks), there is increased A1R tonic activation | [77] |
| In vivo (phrenic motor neurons) | |||||
| A2AR antagonist | Intrapleural CtB-Saporin injected rats (neurotoxic model of respiratory motor neuron death) | phrenic motor neuron | Istradefylline twice daily, for a total dose of 1 mg/kg/day | Increased A2AR expression following CtB-Saporin injections. Istradefylline reduced phrenic motor neuron death and preserved diaphragm EMG activity | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, A.; Cross, B.; Uchida, S.; Kerrick Walker, J.; Ristuccia, R. How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis? Biomedicines 2021, 9, 1027. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9081027
Mori A, Cross B, Uchida S, Kerrick Walker J, Ristuccia R. How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis? Biomedicines. 2021; 9(8):1027. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9081027
Chicago/Turabian StyleMori, Akihisa, Brittany Cross, Shinichi Uchida, Jill Kerrick Walker, and Robert Ristuccia. 2021. "How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis?" Biomedicines 9, no. 8: 1027. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9081027