
The Impact of Steatosis on Chronic Hepatitis C Progression and Response to Antiviral Treatments

 ,
,  and
and
Abstract
:1. Introduction
2. The Impact of HCV-Associated Steatosis on Necroinflammation, Fibrosis, and HCC
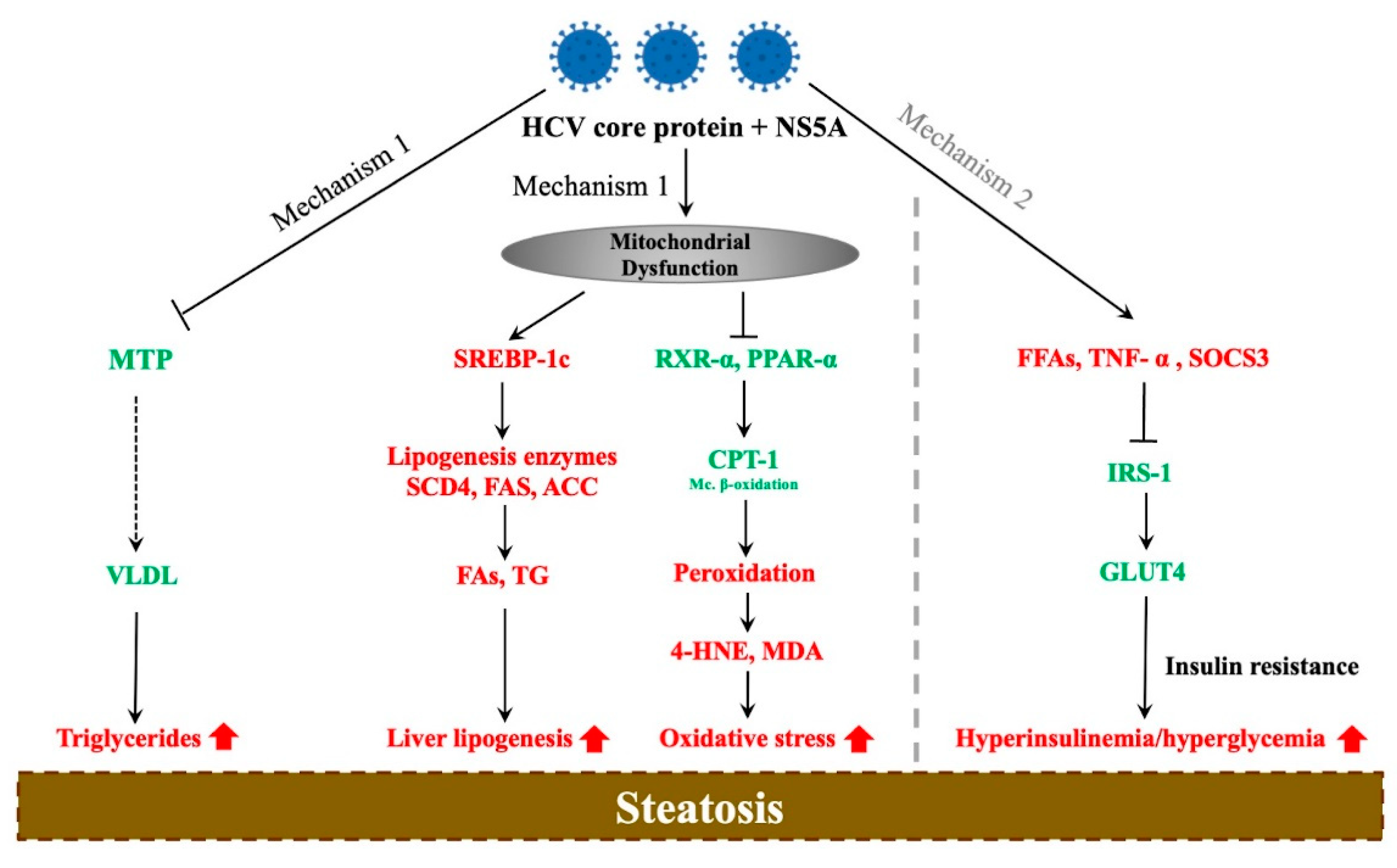
3. Molecular Mechanisms of HCV-Associated Steatosis
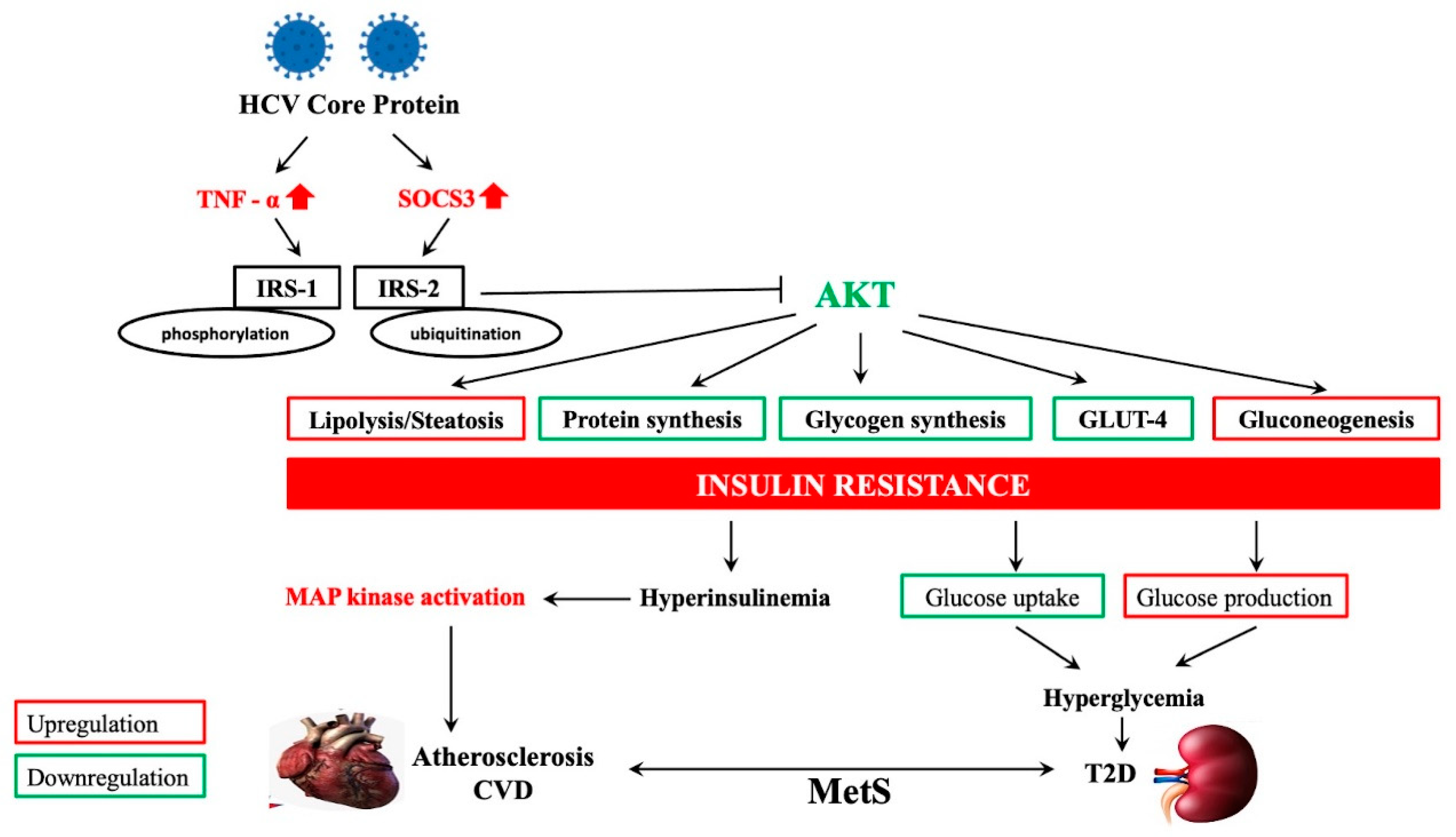
4. HCV-Related Steatosis and Extrahepatic Manifestations
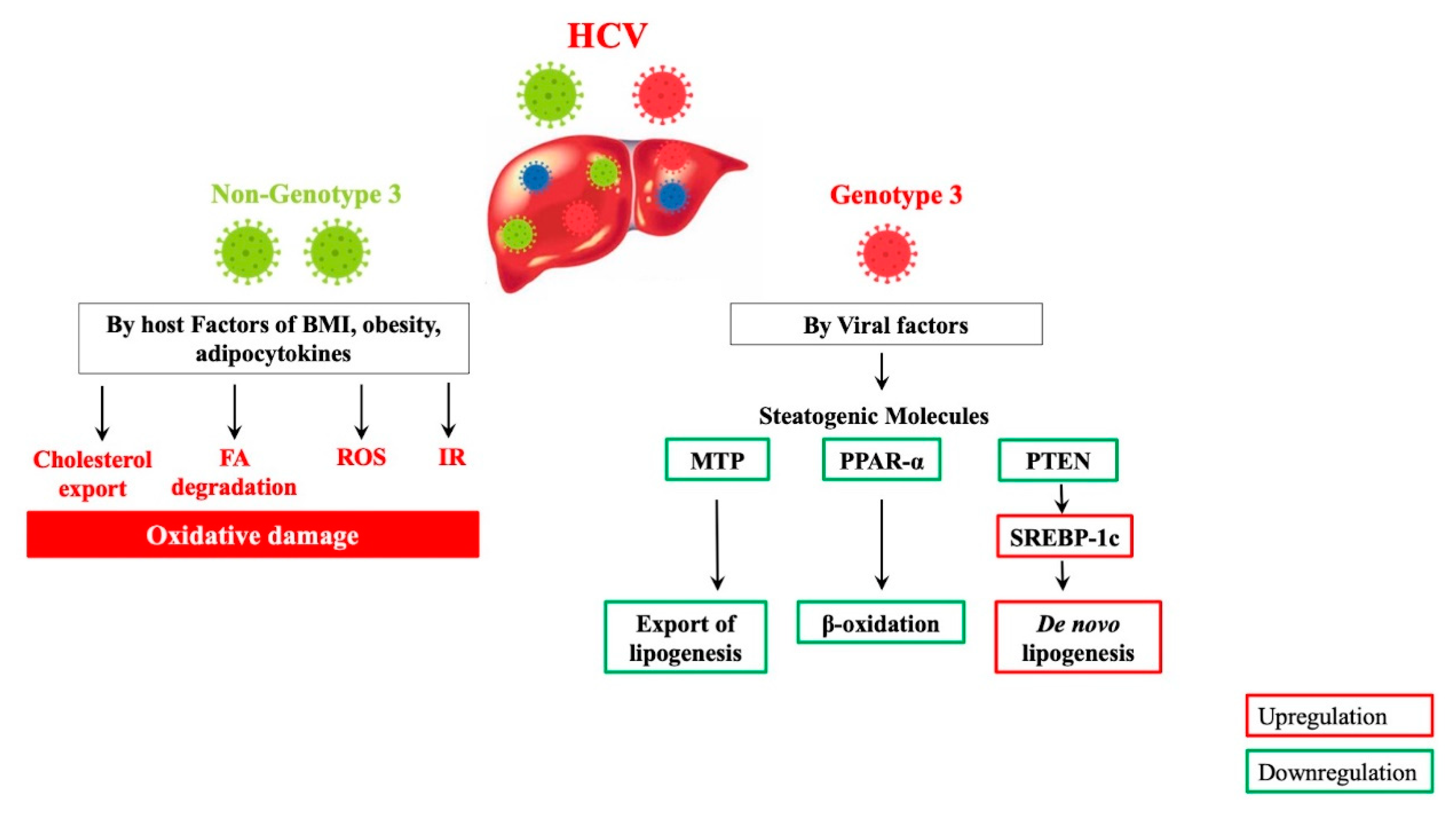
5. Molecular Mechanisms of HCV Genotype-Specific Steatogenesis
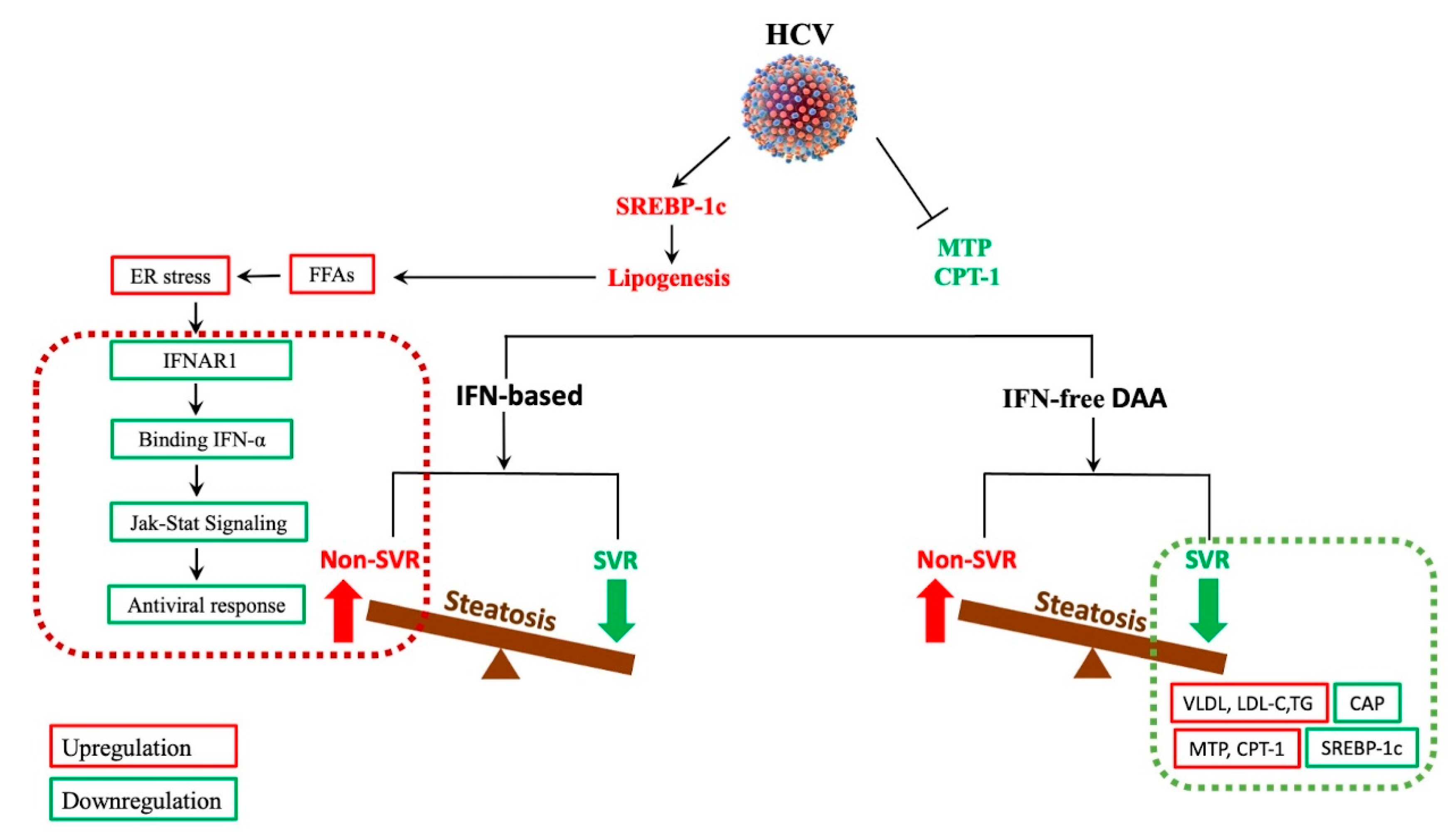
6. The Impact of Antiviral Therapy on HCV-Related Steatosis, Extrahepatic Manifestations, and HCC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D. From nonalcoholic fatty liver disease to metabolic dysfunction-associated fatty liver disease: Is it time for a change of terminology? Hepatoma Res. 2020, 6, 64. [Google Scholar] [CrossRef]
- Marjot, T.; Moolla, A.; Cobbold, J.F.; Hodson, L.; Tomlinson, J.W. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr. Rev. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, M.; Goldkamp, W.J.; Neuschwander-Tetri, B.A. NASH: The Emerging Most Common Form of Chronic Liver Disease. Mo. Med. 2018, 115, 225–229. [Google Scholar]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features.: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef]
- Salomao, M.; Yu, W.M.; Brown, R.S., Jr.; Emond, J.C.; Lefkowitch, J.H. Steatohepatitic hepatocellular carcinoma (SH-HCC): A distinctive histological variant of HCC in hepatitis C virus-related cirrhosis with associated NAFLD/NASH. Am. J. Surg. Pathol. 2010, 34, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Chan, W.K.; Chitturi, S.; Chawla, Y.; Dan, Y.Y.; Duseja, A.; Fan, J.; Goh, K.L.; Hamaguchi, M.; Hashimoto, E.; et al. Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-Part 1: Definition, risk factors and assessment. J. Gastroenterol. Hepatol. 2018, 33, 70–85. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210.e192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.C. PNPLA3-A Potential Therapeutic Target for Personalized Treatment of Chronic Liver Disease. Front. Med. 2019, 6, 304. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef]
- Modaresi Esfeh, J.; Ansari-Gilani, K. Steatosis and hepatitis C. Gastroenterol. Rep. 2016, 4, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.F.; Huang, C.F.; Yeh, M.L.; Dai, C.Y.; Yu, M.L.; Chuang, W.L. Updates in the management and treatment of HCV genotype 3, what are the remaining challenges? Expert Rev. Anti Infect. Ther. 2018, 16, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Farrell, G.C.; Fung, C.; George, J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology 2002, 36, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Bondini, S.; Younossi, Z.M. Non-alcoholic fatty liver disease and hepatitis C infection. Minerva Gastroenterol. Dietol. 2006, 52, 135–143. [Google Scholar] [PubMed]
- Ohya, K.; Akuta, N.; Suzuki, F.; Fujiyama, S.; Kawamura, Y.; Kominami, Y.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; et al. Predictors of treatment efficacy and liver stiffness changes following therapy with Sofosbuvir plus Ribavirin in patients infected with HCV genotype 2. J. Med. Virol. 2018, 90, 919–925. [Google Scholar] [CrossRef]
- Saldarriaga, O.A.; Dye, B.; Pham, J.; Wanninger, T.G.; Millian, D.; Kueht, M.; Freiberg, B.; Utay, N.; Stevenson, H.L. Comparison of liver biopsies before and after direct-acting antiviral therapy for hepatitis C and correlation with clinical outcome. Sci. Rep. 2021, 11, 14506. [Google Scholar] [CrossRef]
- Ogasawara, N.; Kobayashi, M.; Akuta, N.; Kominami, Y.; Fujiyama, S.; Kawamura, Y.; Sezaki, H.; Hosaka, T.; Suzuki, F.; Saitoh, S.; et al. Serial changes in liver stiffness and controlled attenuation parameter following direct-acting antiviral therapy against hepatitis C virus genotype 1b. J. Med. Virol. 2018, 90, 313–319. [Google Scholar] [CrossRef]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, L.E.; Gambardella, M.; Andreana, A.; Tripodi, M.-F.; Utili, R.; Ruggiero, G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001, 33, 1358–1364. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; McHutchison, J.; Manns, M.; Goodman, Z.; Zeuzem, S.; Younossi, Z.; Albrecht, J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003, 38, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Afsari, A.; Lee, E.; Shokrani, B.; Boortalary, T.; Sherif, Z.A.; Nouraie, M.; Laiyemo, A.O.; Alkhalloufi, K.; Brim, H.; Ashktorab, H. Clinical and Pathological Risk Factors Associated with Liver Fibrosis and Steatosis in African-Americans with Chronic Hepatitis C. Dig. Dis. Sci. 2017, 62, 2159–2165. [Google Scholar] [CrossRef]
- Ramesh, S.; Sanyal, A.J. Hepatitis C and nonalcoholic fatty liver disease. Semin. Liver Dis. 2004, 24, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Hézode, C.; Roudot-Thoraval, F.; Bastie, A.; Zafrani, E.S.; Pawlotsky, J.M.; Dhumeaux, D. Worsening of steatosis is an independent factor of fibrosis progression in untreated patients with chronic hepatitis C and paired liver biopsies. Gut 2003, 52, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Cross, T.J.; Quaglia, A.; Hughes, S.; Joshi, D.; Harrison, P.M. The impact of hepatic steatosis on the natural history of chronic hepatitis C infection. J. Viral Hepat. 2009, 16, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Perumalswami, P.; Kleiner, D.E.; Lutchman, G.; Heller, T.; Borg, B.; Park, Y.; Liang, T.J.; Hoofnagle, J.H.; Ghany, M.G. Steatosis and progression of fibrosis in untreated patients with chronic hepatitis C infection. Hepatology 2006, 43, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Boyer, N.; Guimont, M.C.; Cazals-Hatem, D.; Tubach, F.; Nahon, K.; Daïkha, H.; Vidaud, D.; Martinot, M.; Vidaud, M.; et al. Liver fibrosis is not associated with steatosis but with necroinflammation in French patients with chronic hepatitis C. Gut 2003, 52, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Rubbia-Brandt, L.; Fabris, P.; Paganin, S.; Leandro, G.; Male, P.J.; Giostra, E.; Carlotto, A.; Bozzola, L.; Smedile, A.; Negro, F. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut 2004, 53, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Negro, F. Steatosis and insulin resistance in response to treatment of chronic hepatitis C. J. Viral Hepat. 2012, 19 (Suppl. 1), 42–47. [Google Scholar] [CrossRef]
- Rubbia-Brandt, L.; Quadri, R.; Abid, K.; Giostra, E.; Malé, P.J.; Mentha, G.; Spahr, L.; Zarski, J.P.; Borisch, B.; Hadengue, A.; et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J. Hepatol. 2000, 33, 106–115. [Google Scholar] [CrossRef]
- Marcellin, P.; Asselah, T.; Boyer, N. Fibrosis and disease progression in hepatitis C. Hepatology 2002, 36, S47–S56. [Google Scholar] [PubMed]
- Romero-Gómez, M.; Del Mar Viloria, M.; Andrade, R.J.; Salmerón, J.; Diago, M.; Fernández-Rodríguez, C.M.; Corpas, R.; Cruz, M.; Grande, L.; Vázquez, L.; et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 2005, 128, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Maeno, T.; Okumura, A.; Ishikawa, T.; Kato, K.; Sakakibara, F.; Sato, K.; Ayada, M.; Hotta, N.; Tagaya, T.; Fukuzawa, Y.; et al. Mechanisms of increased insulin resistance in non-cirrhotic patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2003, 18, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Teoh, N.C.; Farrell, G.C. Management of chronic hepatitis C virus infection: A new era of disease control. Intern. Med. J. 2004, 34, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Kukla, M.; Piotrowski, D.; Waluga, M.; Hartleb, M. Insulin resistance and its consequences in chronic hepatitis C. Clin. Exp. Hepatol. 2015, 1, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, M.; Hosokawa, T.; Matsunaga, K.; Hirayama, I.; Tanaka, T.; Sato, M.; Yasui, Y.; Tamaki, N.; Ueda, K.; Tsuchiya, K.; et al. Hepatic steatosis in chronic hepatitis C is a significant risk factor for developing hepatocellular carcinoma independent of age, sex, obesity, fibrosis stage and response to interferon therapy. Hepatol. Res. 2010, 40, 870–877. [Google Scholar] [CrossRef]
- Tanaka, A.; Uegaki, S.; Kurihara, H.; Aida, K.; Mikami, M.; Nagashima, I.; Shiga, J.; Takikawa, H. Hepatic steatosis as a possible risk factor for the development of hepatocellular carcinoma after eradication of hepatitis C virus with antiviral therapy in patients with chronic hepatitis C. World J. Gastroenterol. 2007, 13, 5180–5187. [Google Scholar] [CrossRef] [Green Version]
- Ohata, K.; Hamasaki, K.; Toriyama, K.; Matsumoto, K.; Saeki, A.; Yanagi, K.; Abiru, S.; Nakagawa, Y.; Shigeno, M.; Miyazoe, S.; et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 2003, 97, 3036–3043. [Google Scholar] [CrossRef]
- Takuma, Y.; Nouso, K.; Makino, Y.; Saito, S.; Takayama, H.; Takahara, M.; Takahashi, H.; Murakami, I.; Takeuchi, H. Hepatic steatosis correlates with the postoperative recurrence of hepatitis C virus-associated hepatocellular carcinoma. Liver Int. 2007, 27, 620–626. [Google Scholar] [CrossRef]
- Pekow, J.R.; Bhan, A.K.; Zheng, H.; Chung, R.T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007, 109, 2490–2496. [Google Scholar] [CrossRef]
- Ji, D.; Chen, G.-F.; Niu, X.-X.; Zhang, M.; Wang, C.; Shao, Q.; Wu, V.; Wang, Y.; Cheng, G.; Hurwitz, S.J.; et al. Non-alcoholic fatty liver disease is a risk factor for occurrence of hepatocellular carcinoma after sustained virologic response in chronic hepatitis C patients: A prospective four-years follow-up study. Metabolism Open 2021, 10, 100090. [Google Scholar] [CrossRef]
- Nkontchou, G.; Ziol, M.; Aout, M.; Lhabadie, M.; Baazia, Y.; Mahmoudi, A.; Roulot, D.; Ganne-Carrie, N.; Grando-Lemaire, V.; Trinchet, J.C.; et al. HCV genotype 3 is associated with a higher hepatocellular carcinoma incidence in patients with ongoing viral C cirrhosis. J. Viral Hepat. 2011, 18, e516–e522. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [Green Version]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. Jama 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in chronic hepatitis C: Why does it really matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Jahan, S.; Khaliq, S.; Ijaz, B.; Ahmad, W.; Samreen, B.; Hassan, S. Interaction of the hepatitis C virus (HCV) core with cellular genes in the development of HCV-induced steatosis. Arch. Virol. 2010, 155, 1735–1753. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Pazienza, V.; Pugnale, P.; Bruttin, F.; Rubbia-Brandt, L.; Juge-Aubry, C.E.; Meier, C.A.; Hadengue, A.; Negro, F. Peroxisome proliferator-activated receptor-α and -γ mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment. Pharmacol. Ther. 2006, 23, 107–114. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Suzuki, T.; Shimoike, T.; Suzuki, R.; Moriya, K.; Shintani, Y.; Fujie, H.; Matsuura, Y.; Koike, K.; Miyamura, T. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 2002, 35, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dharancy, S.; Malapel, M.; Desreumaux, P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor α and carnitine palmitoyl acyl-CoA transferase 1A. World J. Gastroenterol. 2005, 11, 7591. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, A.J. Pathophysiology of Nonalcoholic Steatohepatitis. J. Clin. Gastroenterol. 2006, 40, S17–S29. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Mizuta, T. Interaction between hepatitis C virus and metabolic factors. World J. Gastroenterol. 2014, 20, 2888–2901. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, X.M.; Li, A.L.; Yang, G.; Hu, H.N. Hepatitis C Virus Increases Free Fatty Acids Absorption and Promotes its Replication Via Down-Regulating GADD45α Expression. Med. Sci. Monit. 2016, 22, 2347–2356. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, R.; Fouda, S.; Sainu, A.; Pappachan, J.M. Metabolic complications of hepatitis C virus infection. World J. Gastroenterol. 2021, 27, 1267–1282. [Google Scholar] [CrossRef]
- Fabrizi, F.; Donato, F.M.; Messa, P. Hepatitis C and Its Metabolic Complications in Kidney Disease. Ann. Hepatol. 2017, 16, 851–861. [Google Scholar] [CrossRef]
- Younossi, Z.; Park, H.; Henry, L.; Adeyemi, A.; Stepanova, M. Extrahepatic Manifestations of Hepatitis C: A Meta-analysis of Prevalence, Quality of Life, and Economic Burden. Gastroenterology 2016, 150, 1599–1608. [Google Scholar] [CrossRef]
- Nevola, R.; Acierno, C.; Pafundi, P.C.; Adinolfi, L.E. Chronic hepatitis C infection induces cardiovascular disease and type 2 diabetes: Mechanisms and management. Minerva Med. 2021, 112, 188–200. [Google Scholar] [CrossRef]
- Shyu, Y.C.; Huang, T.S.; Chien, C.H.; Yeh, C.T.; Lin, C.L.; Chien, R.N. Diabetes poses a higher risk of hepatocellular carcinoma and mortality in patients with chronic hepatitis B: A population-based cohort study. J. Viral Hepat. 2019, 26, 718–726. [Google Scholar] [CrossRef]
- Yen, Y.H.; Chang, K.C.; Tsai, M.C.; Tseng, P.L.; Lin, M.T.; Wu, C.K.; Lin, J.T.; Hu, T.H.; Wang, J.H.; Chen, C.H. Elevated body mass index is a risk factor associated with possible liver cirrhosis across different etiologies of chronic liver disease. J. Formos. Med. Assoc. 2018, 117, 268–275. [Google Scholar] [CrossRef]
- Li, X.; Gao, Y.; Xu, H.; Hou, J.; Gao, P. Diabetes mellitus is a significant risk factor for the development of liver cirrhosis in chronic hepatitis C patients. Sci. Rep. 2017, 7, 9087. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Y.; Younossi, Z.M. Obesity-associated nonalcoholic fatty liver disease. Clin. Liver Dis. 2014, 18, 19–31. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Kee, K.M.; Wang, J.H.; Hsu, N.T.; Hsiao, C.C.; Chen, Y.; Lu, S.N. Association between chronic viral hepatitis and metabolic syndrome in southern Taiwan: A large population-based study. Aliment. Pharmacol. Ther. 2018, 48, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, P.; Dwivedi, A.K.; Dodoo, C.A.; Shokar, N.K.; Salinas, J.; Lakshmanaswamy, R. The association between metabolic syndrome and Hepatitis C virus infection in the United States. Cancer Causes Control 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Guaraldi, G.; Nascimbeni, F.; Romagnoli, D.; Zona, S.; Targher, G. Fatty liver is associated with an increased risk of diabetes and cardiovascular disease—Evidence from three different disease models: NAFLD, HCV and HIV. World J. Gastroenterol. 2016, 22, 9674–9693. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Li, C.; Sattar, N. Metabolic syndrome and incident diabetes: Current state of the evidence. Diabetes Care 2008, 31, 1898–1904. [Google Scholar] [CrossRef] [Green Version]
- Benetos, A.; Thomas, F.; Pannier, B.; Bean, K.; Jégo, B.; Guize, L. All-cause and cardiovascular mortality using the different definitions of metabolic syndrome. Am. J. Cardiol. 2008, 102, 188–191. [Google Scholar] [CrossRef]
- Dragsbæk, K.; Neergaard, J.S.; Laursen, J.M.; Hansen, H.B.; Christiansen, C.; Beck-Nielsen, H.; Karsdal, M.A.; Brix, S.; Henriksen, K. Metabolic syndrome and subsequent risk of type 2 diabetes and cardiovascular disease in elderly women: Challenging the current definition. Medicine 2016, 95, e4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, T.; Hamaguchi, M.; Kojima, T.; Hashimoto, Y.; Ohbora, A.; Kato, T.; Nakamura, N.; Fukui, M. The impact of non-alcoholic fatty liver disease on incident type 2 diabetes mellitus in non-overweight individuals. Liver Int. 2016, 36, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.V.; Allison, M.A.; Lima, J.A.; Bluemke, D.A.; Abbasi, S.A.; Ouyang, P.; Jerosch-Herold, M.; Ding, J.; Budoff, M.J.; Murthy, V.L. Liver fat, statin use, and incident diabetes: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2015, 242, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Ballestri, S.; Targher, G.; Loria, P. Diagnosis and management of cardiovascular risk in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 629–650. [Google Scholar] [CrossRef]
- Mantovani, A.; Ballestri, S.; Lonardo, A.; Targher, G. Cardiovascular Disease and Myocardial Abnormalities in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1246–1267. [Google Scholar] [CrossRef]
- Lonardo, A.; Sookoian, S.; Pirola, C.J.; Targher, G. Non-alcoholic fatty liver disease and risk of cardiovascular disease. Metabolism 2016, 65, 1136–1150. [Google Scholar] [CrossRef]
- Ambachew, S.; Eshetie, S.; Geremew, D.; Endalamaw, A.; Melku, M. Prevalence of Type 2 Diabetes Mellitus among Hepatitis C Virus-Infected Patients: A Systematic Review and Meta-Analysis. Dubai Diabetes Endocrinol. J. 2018, 24, 29–37. [Google Scholar] [CrossRef]
- Petta, S.; Maida, M.; Macaluso, F.S.; Barbara, M.; Licata, A.; Craxì, A.; Cammà, C. Hepatitis C Virus Infection Is Associated With Increased Cardiovascular Mortality: A Meta-Analysis of Observational Studies. Gastroenterology 2016, 150, 145―155.e144; quiz e115―e146. [Google Scholar] [CrossRef] [Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; Jayyousi, A.; DeFronzo, R.A.; Asaad, N.; Al-Suwaidi, J. Insulin Resistance the Link between T2DM and CVD: Basic Mechanisms and Clinical Implications. Curr. Vasc. Pharmacol. 2019, 17, 153–163. [Google Scholar] [CrossRef]
- Maruyama, S.; Koda, M.; Oyake, N.; Sato, H.; Fujii, Y.; Horie, Y.; Murawaki, Y. Myocardial injury in patients with chronic hepatitis C infection. J. Hepatol. 2013, 58, 11–15. [Google Scholar] [CrossRef]
- Lonardo, A.; Adinolfi, L.E.; Restivo, L.; Ballestri, S.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Loria, P. Pathogenesis and significance of hepatitis C virus steatosis: An update on survival strategy of a successful pathogen. World J. Gastroenterol. 2014, 20, 7089–7103. [Google Scholar] [CrossRef]
- Oem, J.K.; Jackel-Cram, C.; Li, Y.P.; Zhou, Y.; Zhong, J.; Shimano, H.; Babiuk, L.A.; Liu, Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J. Gen. Virol. 2008, 89, 1225–1230. [Google Scholar] [CrossRef]
- Clément, S.; Peyrou, M.; Sanchez-Pareja, A.; Bourgoin, L.; Ramadori, P.; Suter, D.; Vinciguerra, M.; Guilloux, K.; Pascarella, S.; Rubbia-Brandt, L.; et al. Down-regulation of phosphatase and tensin homolog by hepatitis C virus core 3a in hepatocytes triggers the formation of large lipid droplets. Hepatology 2011, 54, 38–49. [Google Scholar] [CrossRef]
- d’Avigdor, W.M.H.; Budzinska, M.A.; Lee, M.; Lam, R.; Kench, J.; Stapelberg, M.; McLennan, S.V.; Farrell, G.; George, J.; McCaughan, G.W.; et al. Virus Genotype-Dependent Transcriptional Alterations in Lipid Metabolism and Inflammation Pathways in the Hepatitis C Virus-infected Liver. Sci. Rep. 2019, 9, 10596. [Google Scholar] [CrossRef] [Green Version]
- Hourioux, C.; Patient, R.; Morin, A.; Blanchard, E.; Moreau, A.; Trassard, S.; Giraudeau, B.; Roingeard, P. The genotype 3-specific hepatitis C virus core protein residue phenylalanine 164 increases steatosis in an in vitro cellular model. Gut 2007, 56, 1302–1308. [Google Scholar] [CrossRef]
- Fartoux, L.; Poujol-Robert, A.; Guéchot, J.; Wendum, D.; Poupon, R.; Serfaty, L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut 2005, 54, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, L.E.; Ingrosso, D.; Cesaro, G.; Cimmino, A.; D’Antò, M.; Capasso, R.; Zappia, V.; Ruggiero, G. Hyperhomocysteinemia and the MTHFR C677T polymorphism promote steatosis and fibrosis in chronic hepatitis C patients. Hepatology 2005, 41, 995–1003. [Google Scholar] [CrossRef]
- Vidali, M.; Tripodi, M.F.; Ivaldi, A.; Zampino, R.; Occhino, G.; Restivo, L.; Sutti, S.; Marrone, A.; Ruggiero, G.; Albano, E.; et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J. Hepatol. 2008, 48, 399–406. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [Green Version]
- Marciano, S.; Borzi, S.M.; Dirchwolf, M.; Ridruejo, E.; Mendizabal, M.; Bessone, F.; Sirotinsky, M.E.; Giunta, D.H.; Trinks, J.; Olivera, P.A.; et al. Pre-treatment prediction of response to peginterferon plus ribavirin in chronic hepatitis C genotype 3. World J. Hepatol. 2015, 7, 703–709. [Google Scholar] [CrossRef]
- Shah, S.R.; Patel, K.; Marcellin, P.; Foster, G.R.; Manns, M.; Kottilil, S.; Healey, L.; Pulkstenis, E.; Subramanian, G.M.; McHutchison, J.G.; et al. Steatosis is an independent predictor of relapse following rapid virologic response in patients with HCV genotype 3. Clin. Gastroenterol. Hepatol. 2011, 9, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Restivo, L.; Zampino, R.; Guerrera, B.; Ruggiero, L.; Adinolfi, L.E. Steatosis is the predictor of relapse in HCV genotype 3- but not 2-infected patients treated with 12 weeks of pegylated interferon-α-2a plus ribavirin and RVR. J. Viral Hepat. 2012, 19, 346–352. [Google Scholar] [CrossRef]
- Toyoda, H.; Kumada, T.; Kiriyama, S.; Tanikawa, M.; Hisanaga, Y.; Kanamori, A.; Tada, T.; Kitabatake, S.; Murakami, Y. Association between hepatic steatosis and hepatic expression of genes involved in innate immunity in patients with chronic hepatitis C. Cytokine 2013, 63, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, F.; Aboulnasr, F.M.; Chandra, P.K.; Hazari, S.; Poat, B.; Baker, D.P.; Balart, L.A.; Dash, S. Free fatty acids induce ER stress and block antiviral activity of interferon alpha against hepatitis C virus in cell culture. Virol. J. 2012, 9, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiramatsu, N.; Oze, T.; Takehara, T. Suppression of hepatocellular carcinoma development in hepatitis C patients given interferon-based antiviral therapy. Hepatol. Res. 2015, 45, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Suda, G.; Furusyo, N.; Toyoda, H.; Kawakami, Y.; Ikeda, H.; Suzuki, M.; Arataki, K.; Mori, N.; Tsuji, K.; Katamura, Y.; et al. Daclatasvir and asunaprevir in hemodialysis patients with hepatitis C virus infection: A nationwide retrospective study in Japan. J. Gastroenterol. 2018, 53, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, E.A.; Adams, J.L.; Kramer, J.; Sahota, A.K.; Silverberg, M.J.; Shenkman, E.; Nelson, D.R. Assessing the Safety of Direct-Acting Antiviral Agents for Hepatitis C. JAMA Netw. Open 2019, 2, e194765. [Google Scholar] [CrossRef] [Green Version]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef] [Green Version]
- Czaja, A.J.; Carpenter, H.A.; Santrach, P.J.; Moore, S.B. Host- and disease-specific factors affecting steatosis in chronic hepatitis C. J. Hepatol. 1998, 29, 198–206. [Google Scholar] [CrossRef]
- Tsukuda, Y.; Suda, G.; Tsunematsu, S.; Ito, J.; Sato, F.; Terashita, K.; Nakai, M.; Sho, T.; Maehara, O.; Shimazaki, T.; et al. Anti-adipogenic and antiviral effects of l-carnitine on hepatitis C virus infection. J. Med. Virol. 2017, 89, 857–866. [Google Scholar] [CrossRef]
- Kawagishi, N.; Suda, G.; Nakamura, A.; Kimura, M.; Maehara, O.; Suzuki, K.; Nakamura, A.; Ohara, M.; Izumi, T.; Umemura, M.; et al. Liver steatosis and dyslipidemia after HCV eradication by direct acting antiviral agents are synergistic risks of atherosclerosis. PLoS ONE 2018, 13, e0209615. [Google Scholar] [CrossRef] [Green Version]
- Sidorkiewicz, M. Hepatitis C Virus Uses Host Lipids to Its Own Advantage. Metabolites 2021, 11. [Google Scholar] [CrossRef]
- Meissner, E.G.; Lee, Y.J.; Osinusi, A.; Sims, Z.; Qin, J.; Sturdevant, D.; McHutchison, J.; Subramanian, M.; Sampson, M.; Naggie, S.; et al. Effect of sofosbuvir and ribavirin treatment on peripheral and hepatic lipid metabolism in chronic hepatitis C virus, genotype 1-infected patients. Hepatology 2015, 61, 790–801. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yatsuhashi, H.; Abiru, S.; Yamasaki, K.; Komori, A.; Nagaoka, S.; Saeki, A.; Uchida, S.; Bekki, S.; Kugiyama, Y.; et al. Rapid Increase in Serum Low-Density Lipoprotein Cholesterol Concentration during Hepatitis C Interferon-Free Treatment. PLoS ONE 2016, 11, e0163644. [Google Scholar] [CrossRef]
- Hwang, H.W.; Yu, J.H.; Jin, Y.-J.; Suh, Y.J.; Lee, J.-W. Correlation between the small dense LDL level and nonalcoholic fatty liver disease: Possibility of a new biomarker. Medicine 2020, 99, e21162. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Gaubatz, J.W.; Sun, W.; Dodge, R.C.; Crosby, J.R.; Jiang, J.; Couper, D.; Virani, S.S.; Kathiresan, S.; Boerwinkle, E.; et al. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: The Atherosclerosis Risk In Communities (ARIC) study. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Kim, Y.S.; Kim, S.G.; Lee, Y.N.; Jeong, S.W.; Jang, J.Y.; Lee, S.H.; Kim, H.S.; Kim, B.S. The impact of pegylated interferon and ribavirin combination treatment on lipid metabolism and insulin resistance in chronic hepatitis C patients. Clin. Mol. Hepatol. 2014, 20, 38–46. [Google Scholar] [CrossRef]
- Patton, H.M.; Patel, K.; Behling, C.; Bylund, D.; Blatt, L.M.; Vallée, M.; Heaton, S.; Conrad, A.; Pockros, P.J.; McHutchison, J.G. The impact of steatosis on disease progression and early and sustained treatment response in chronic hepatitis C patients. J. Hepatol. 2004, 40, 484–490. [Google Scholar] [CrossRef]
- Zeuzem, S.; Hultcrantz, R.; Bourliere, M.; Goeser, T.; Marcellin, P.; Sanchez-Tapias, J.; Sarrazin, C.; Harvey, J.; Brass, C.; Albrecht, J. Peginterferon alfa-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J. Hepatol. 2004, 40, 993–999. [Google Scholar] [CrossRef]
- Kobayashi, N.; Iijima, H.; Tada, T.; Kumada, T.; Yoshida, M.; Aoki, T.; Nishimura, T.; Nakano, C.; Takata, R.; Yoh, K.; et al. Changes in liver stiffness and steatosis among patients with hepatitis C virus infection who received direct-acting antiviral therapy and achieved sustained virological response. Eur. J. Gastroenterol. Hepatol. 2018, 30, 546–551. [Google Scholar] [CrossRef]
- Petta, S.; Adinolfi, L.E.; Fracanzani, A.L.; Rini, F.; Caldarella, R.; Calvaruso, V.; Cammà, C.; Ciaccio, M.; Di Marco, V.; Grimaudo, S.; et al. Hepatitis C virus eradication by direct-acting antiviral agents improves carotid atherosclerosis in patients with severe liver fibrosis. J. Hepatol. 2018, 69, 18–24. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Narciso, V.; Nevola, R.; Rinaldi, L.; Calvaruso, V.; Staiano, L.; Di Marco, V.; et al. Impact of hepatitis C virus clearance by direct-acting antiviral treatment on the incidence of major cardiovascular events: A prospective multicentre study. Atherosclerosis 2020, 296, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, M.N.D.; Ambrosino, P.; Buonomo, A.R.; Pinchera, B.; Calcaterra, I.; Crispo, M.; Scotto, R.; Borgia, F.; Mattia, C.; Gentile, I. Direct-acting antivirals improve endothelial function in patients with chronic hepatitis: A prospective cohort study. Intern. Emerg. Med. 2020, 15, 263–271. [Google Scholar] [CrossRef]
- Cacoub, P.; Saadoun, D. Extrahepatic Manifestations of Chronic HCV Infection. N. Engl. J. Med. 2021, 384, 1038–1052. [Google Scholar] [CrossRef]
- Butt, A.A.; Yan, P.; Shuaib, A.; Abou-Samra, A.B.; Shaikh, O.S.; Freiberg, M.S. Direct-Acting Antiviral Therapy for HCV Infection Is Associated With a Reduced Risk of Cardiovascular Disease Events. Gastroenterology 2019, 156, 987–996.e988. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Nevola, R.; Coppola, C.; Narciso, V.; Rinaldi, L.; Calvaruso, V.; Pafundi, P.C.; Lombardi, R.; et al. Reduced incidence of type 2 diabetes in patients with chronic hepatitis C virus infection cleared by direct-acting antiviral therapy: A prospective study. Diabetes Obes. Metab. 2020, 22, 2408–2416. [Google Scholar] [CrossRef]
- Farrag, H.M.; Monir, M.S.; Abdel-Dayem, W.S.; Ali, H.A.; Ibrahim, A.M. Global longitudinal strain as a predictor of short-term effect of oral antiviral regimens on myocardium in Egyptian patients with chronic viral hepatitis C. Egypt Heart J. 2021, 73, 6. [Google Scholar] [CrossRef]
- Chen, J.Y.; Cheng, P.N.; Chiu, Y.C.; Chiu, H.C.; Tsai, W.C.; Tsai, L.M. Persistent augmentation of central arterial stiffness following viral clearance by direct-acting antivirals in chronic hepatitis C. J. Viral Hepat. 2021, 28, 159–167. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Nevola, R.; Guerrera, B.; D’Alterio, G.; Marrone, A.; Giordano, M.; Rinaldi, L. Hepatitis C virus clearance by direct-acting antiviral treatments and impact on insulin resistance in chronic hepatitis C patients. J. Gastroenterol. Hepatol. 2018, 33, 1379–1382. [Google Scholar] [CrossRef]
- Gualerzi, A.; Bellan, M.; Smirne, C.; Tran Minh, M.; Rigamonti, C.; Burlone, M.E.; Bonometti, R.; Bianco, S.; Re, A.; Favretto, S.; et al. Improvement of insulin sensitivity in diabetic and non diabetic patients with chronic hepatitis C treated with direct antiviral agents. PLoS ONE 2018, 13, e0209216. [Google Scholar] [CrossRef]
- Reig, M.; Mariño, Z.; Perelló, C.; Iñarrairaegui, M.; Ribeiro, A.; Lens, S.; Díaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, L.; Rinaldi, L.; Di Francia, R.; Coppola, N.; Guerrera, B.; Imparato, M.; Monari, C.; Nevola, R.; Rosato, V.; Fontanella, L.; et al. Hepatocellular Carcinoma in Hcv Cirrhosis after Viral Clearance with Direct Acting Antiviral Therapy: Preliminary Evidence and Possible Meanings. Wcrj 2016, 3, e748. [Google Scholar]
- Rinaldi, L.; Perrella, A.; Guarino, M.; De Luca, M.; Piai, G.; Coppola, N.; Pafundi, P.C.; Ciardiello, F.; Fasano, M.; Martinelli, E.; et al. Incidence and risk factors of early HCC occurrence in HCV patients treated with direct acting antivirals: A prospective multicentre study. J. Transl. Med. 2019, 17, 292. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ye, J.; Song, M.; Zhou, M.; Hu, Y. Ribavirin augments doxorubicin’s efficacy in human hepatocellular carcinoma through inhibiting doxorubicin-induced eIF4E activation. J. Biochem. Mol. Toxicol. 2018, 32, e22007. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nakatsuka, K.; Shimizu, M.; Tamura, H.; Shinya, E.; Atsukawa, M.; Harimoto, H.; Takahashi, H.; Sakamoto, C. Ribavirin modulates the conversion of human CD4(+) CD25(-) T cell to CD4(+) CD25(+) FOXP3(+) T cell via suppressing interleukin-10-producing regulatory T cell. Immunology 2012, 137, 259–270. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Number of Patients | Antiviral Therapy | Antiviral Response | HCC Occurrence/Recurrence (%) | HCC-Related Risk Factors | |
|---|---|---|---|---|---|---|
| SVR | Non-SVR | |||||
| Kurosaki et al., 2010 [47] | 1279 | IFN | 393 | 886 | 68/1279~5% | High-grade steatosis, advanced fibrosis, non-SVR, older age, male sex, high BMI |
| Tanaka et al., 2007 [48] | 266 | IFN | 266 | 0 | 6/266~2.6% | Steatosis, older age, fibrosis |
| Ohata et al., 2003 [49] | 161 | IFN (71/161) | 20 | 51 | 71/71~100% | Steatosis, aging, cirrhosis, no IFN treatment |
| Takuma et al., 2007 [50] | 88 | Curative resection | ND | ND | 55/88~63% | Steatosis, fibrosis stage, surgical procedure outcome, number and size of tumor |
| Pekow et al., 2007 [51] | 94 | Liver transplantation | ND | ND | 32/94~34% | Steatosis, older age, AFP |
| Ji, D., et al., 2021 [52] | 1735 | IFN-free DAAs and PR | 1336 | 399 | 54/1336~4.4% | NAFLD, older age, higher AFP level, higher liver stiffness measurement, diabetes mellitus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siphepho, P.Y.; Liu, Y.-T.; Shabangu, C.S.; Huang, J.-F.; Huang, C.-F.; Yeh, M.-L.; Yu, M.-L.; Wang, S.-C. The Impact of Steatosis on Chronic Hepatitis C Progression and Response to Antiviral Treatments. Biomedicines 2021, 9, 1491. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101491
Siphepho PY, Liu Y-T, Shabangu CS, Huang J-F, Huang C-F, Yeh M-L, Yu M-L, Wang S-C. The Impact of Steatosis on Chronic Hepatitis C Progression and Response to Antiviral Treatments. Biomedicines. 2021; 9(10):1491. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101491
Chicago/Turabian StyleSiphepho, Phumelele Yvonne, Yi-Ting Liu, Ciniso Sylvester Shabangu, Jee-Fu Huang, Chung-Feng Huang, Ming-Lun Yeh, Ming-Lung Yu, and Shu-Chi Wang. 2021. "The Impact of Steatosis on Chronic Hepatitis C Progression and Response to Antiviral Treatments" Biomedicines 9, no. 10: 1491. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101491