
Thalidomide Exerts Anti-Inflammatory Effects in Cutaneous Lupus by Inhibiting the IRF4/NF-ҡB and AMPK1/mTOR Pathways

 ,
,
Abstract
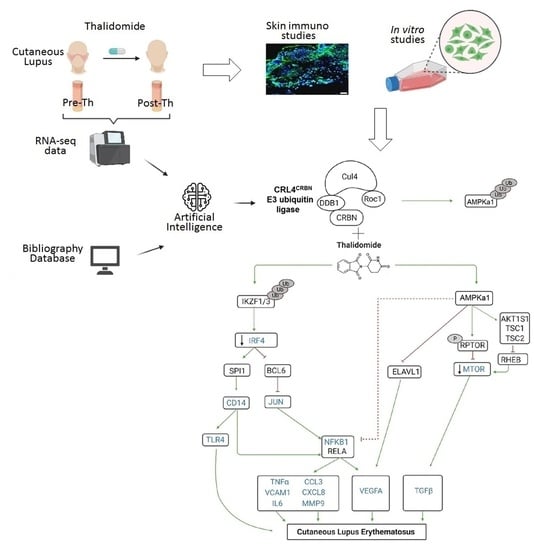
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patients
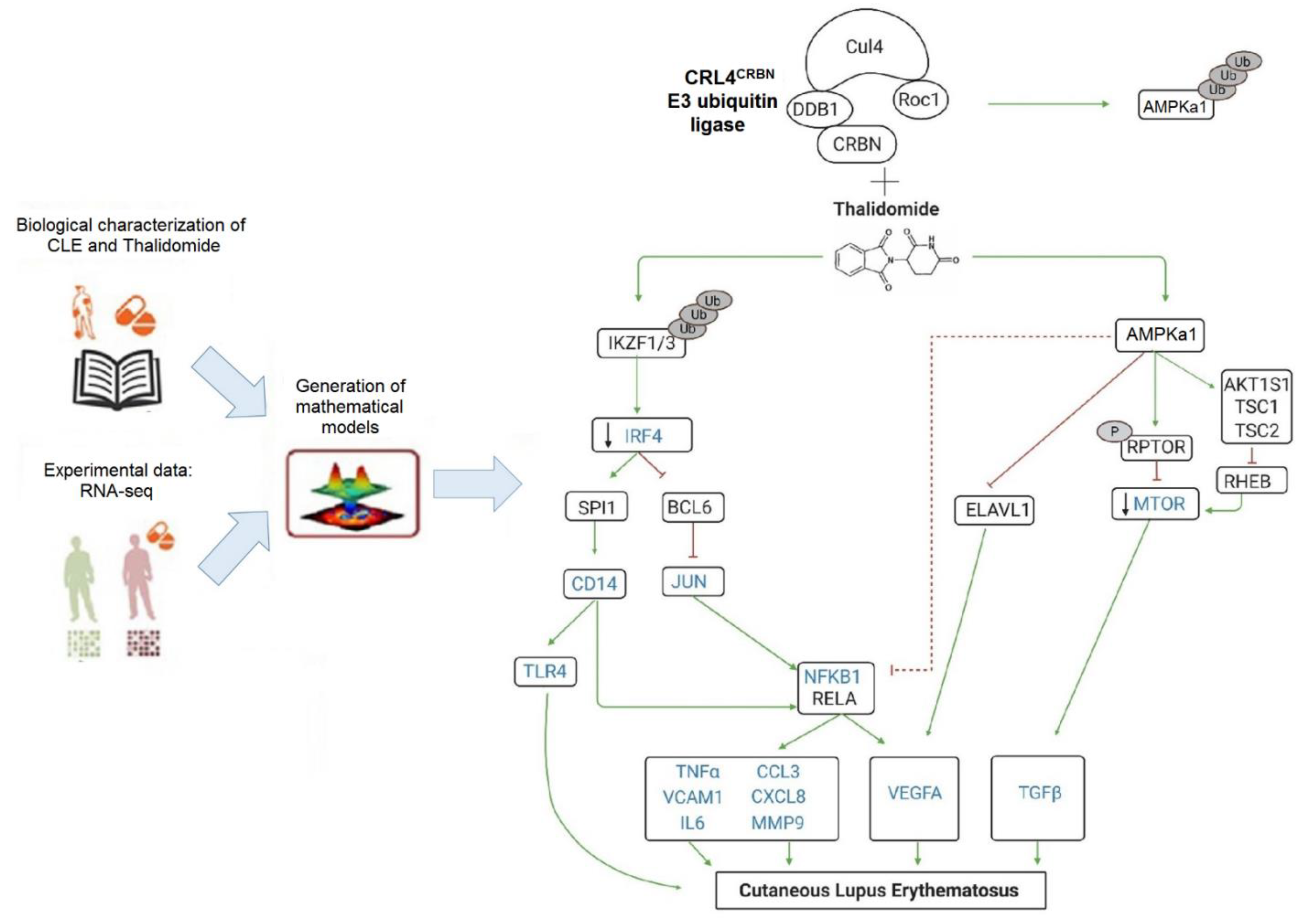
2.2. RNA-Seq and System Biology Analysis
2.3. Flow Cytometry
2.4. Immunofluorescence and Immunohistochemistry
2.5. In Vitro Ubiquitination Assay
2.6. Co-Immunoprecipitation for Cell-Based Ubiquitination Assay
2.7. Protein Extraction and Western Blot
2.8. RNA Extraction and RT-qPCR
2.9. Proliferation Assays
2.10. Cell Culture
2.11. Gene Silencing
2.12. Co-Culture Experiments
2.13. Statistical Analysis
3. Results
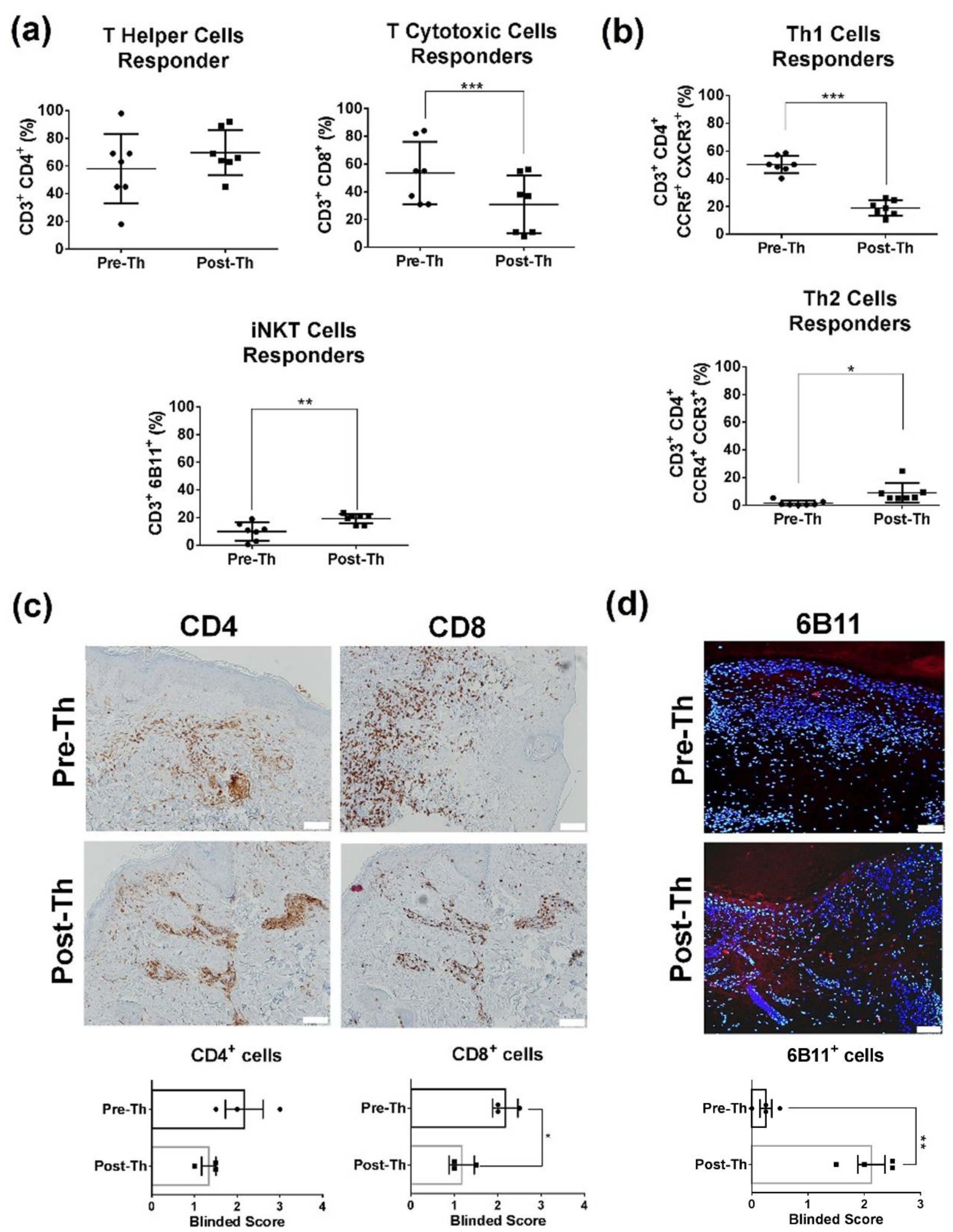
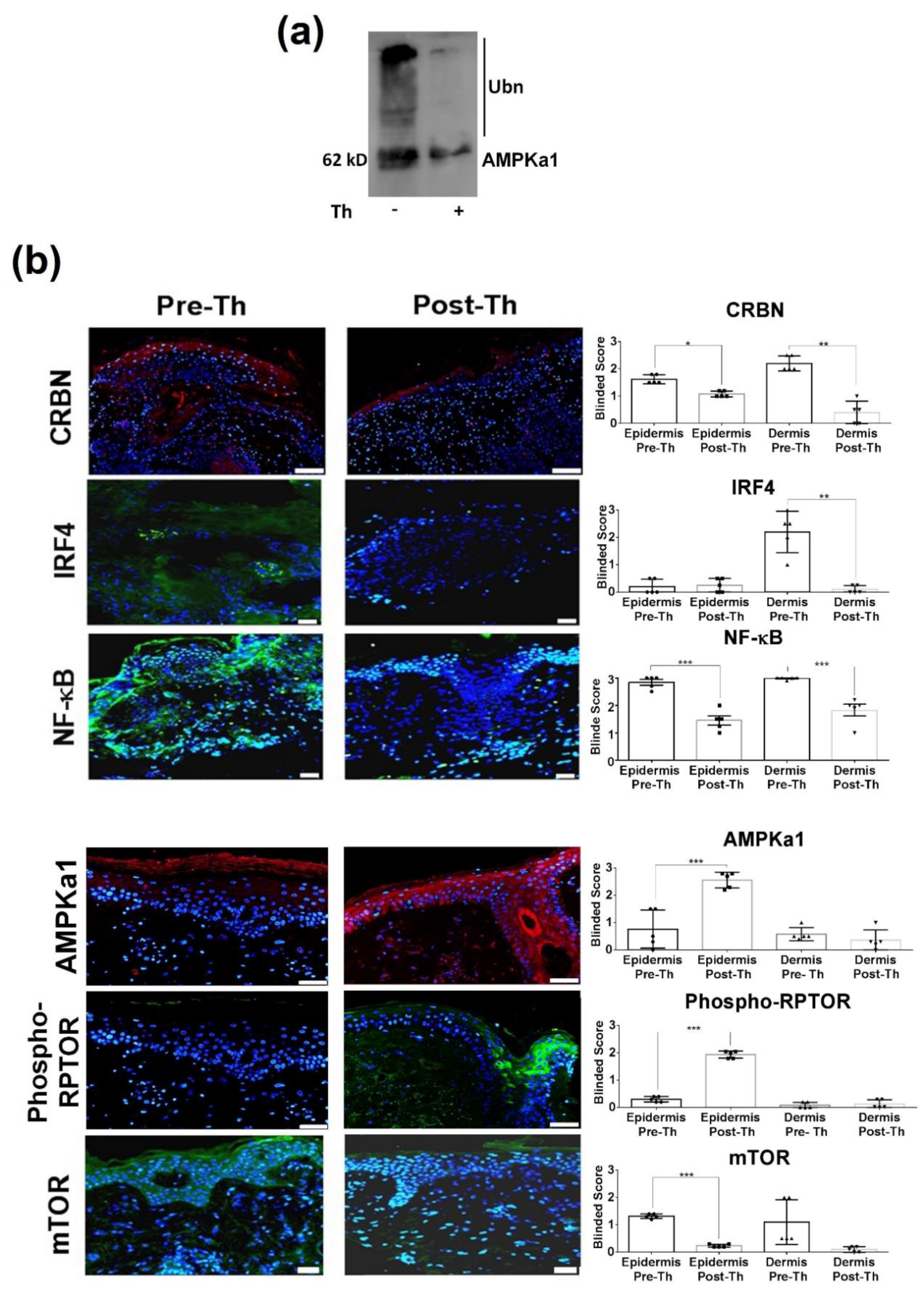
3.1. Immunoregulatory Effects in CLE Peripheral Blood and Skin
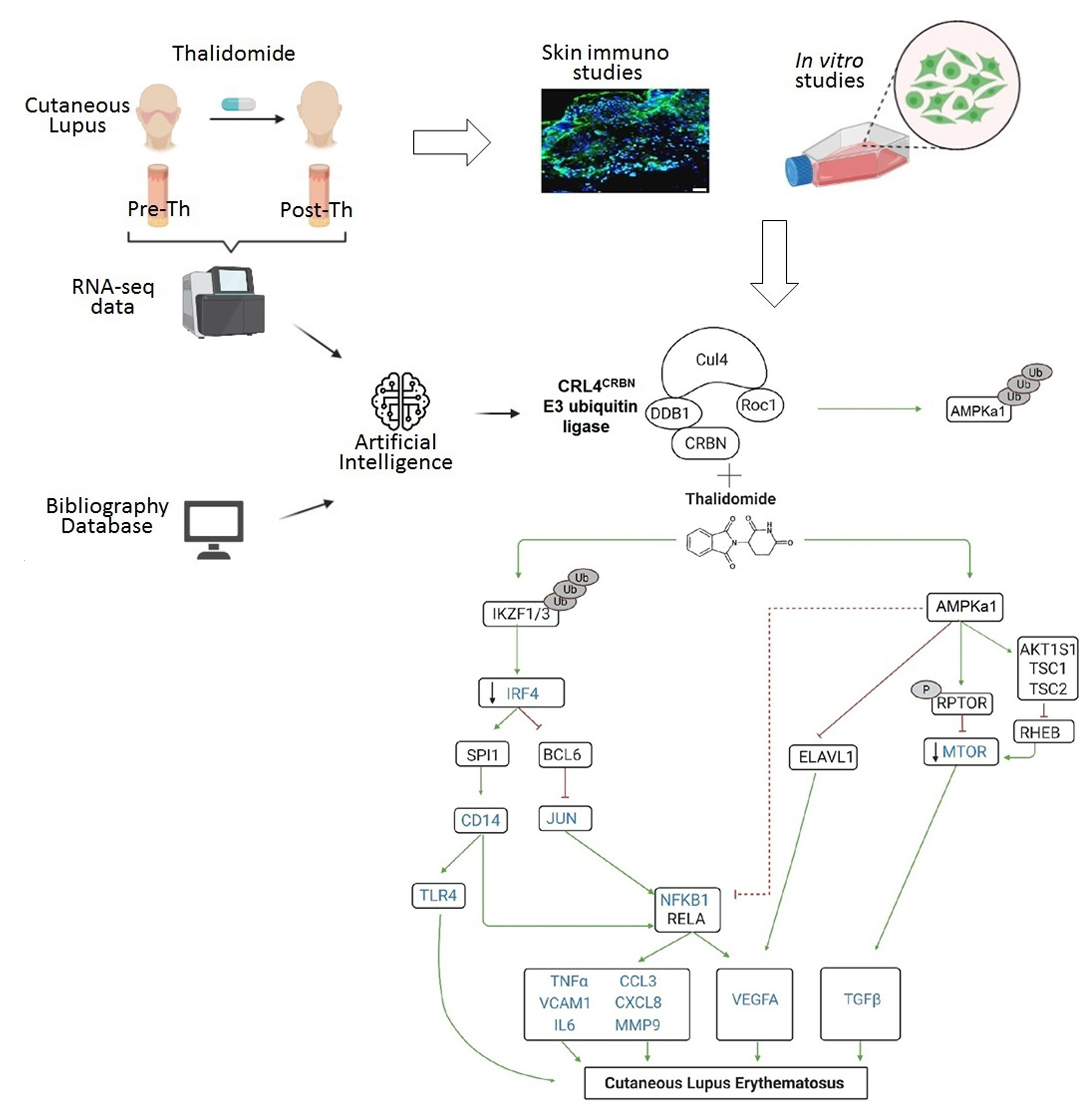
3.2. RNA-Sequencing with Therapeutic Performance Mapping System (TMPS) Analysis Revealed Thalidomide’s Mechanisms in CLE
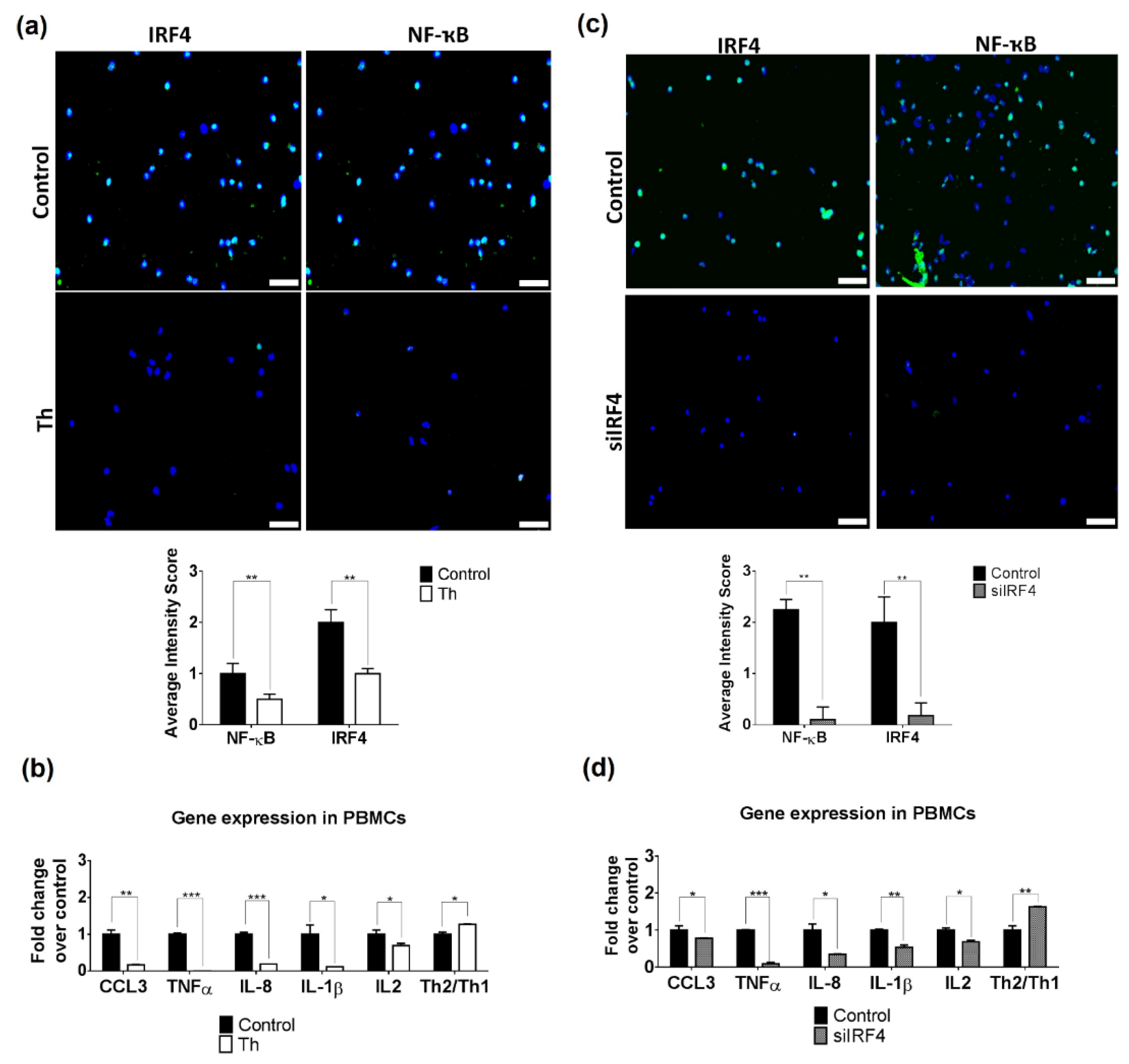
3.3. Thalidomide Modulates PBMCs via the IRF4/NF-ҡB Signalling Pathway
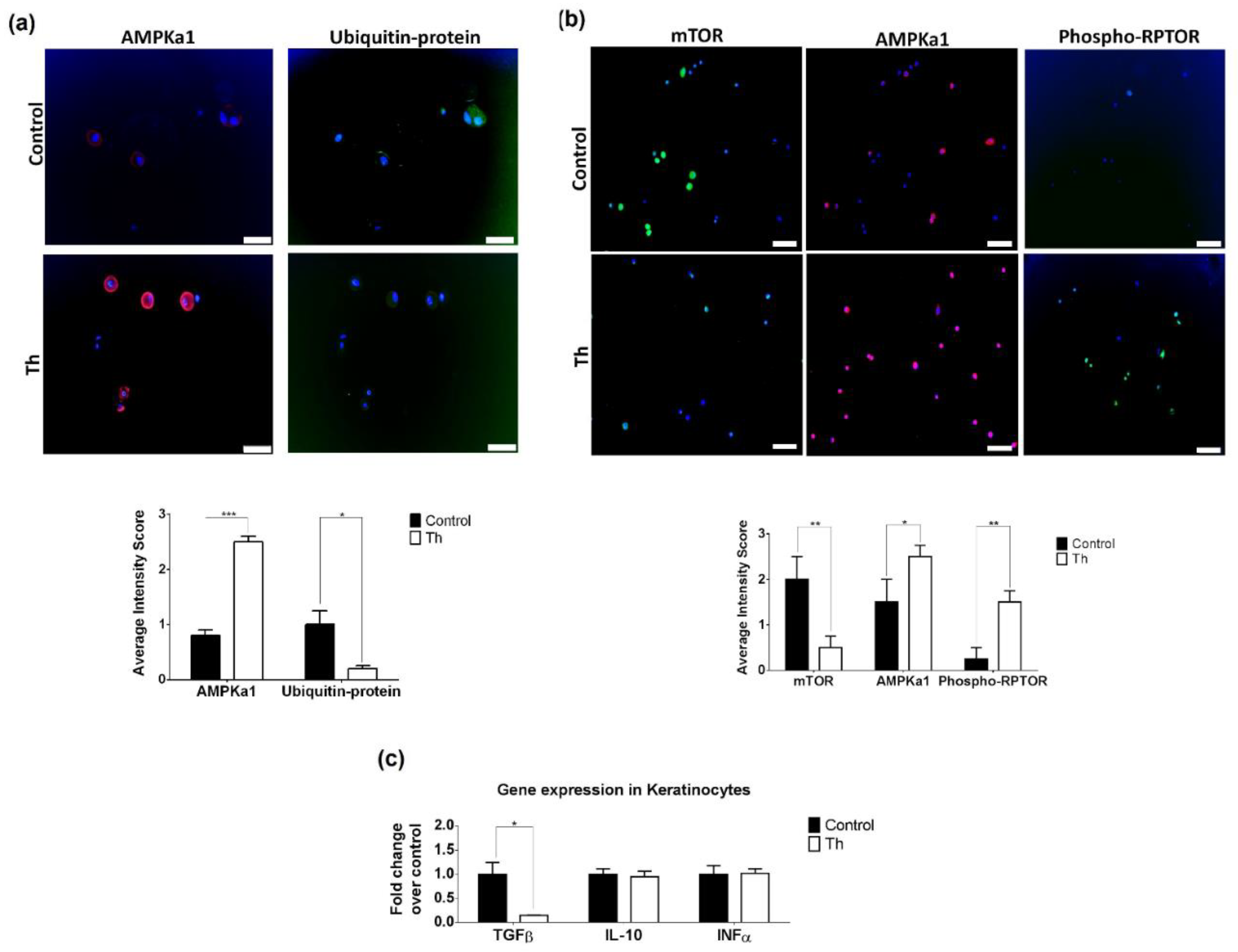
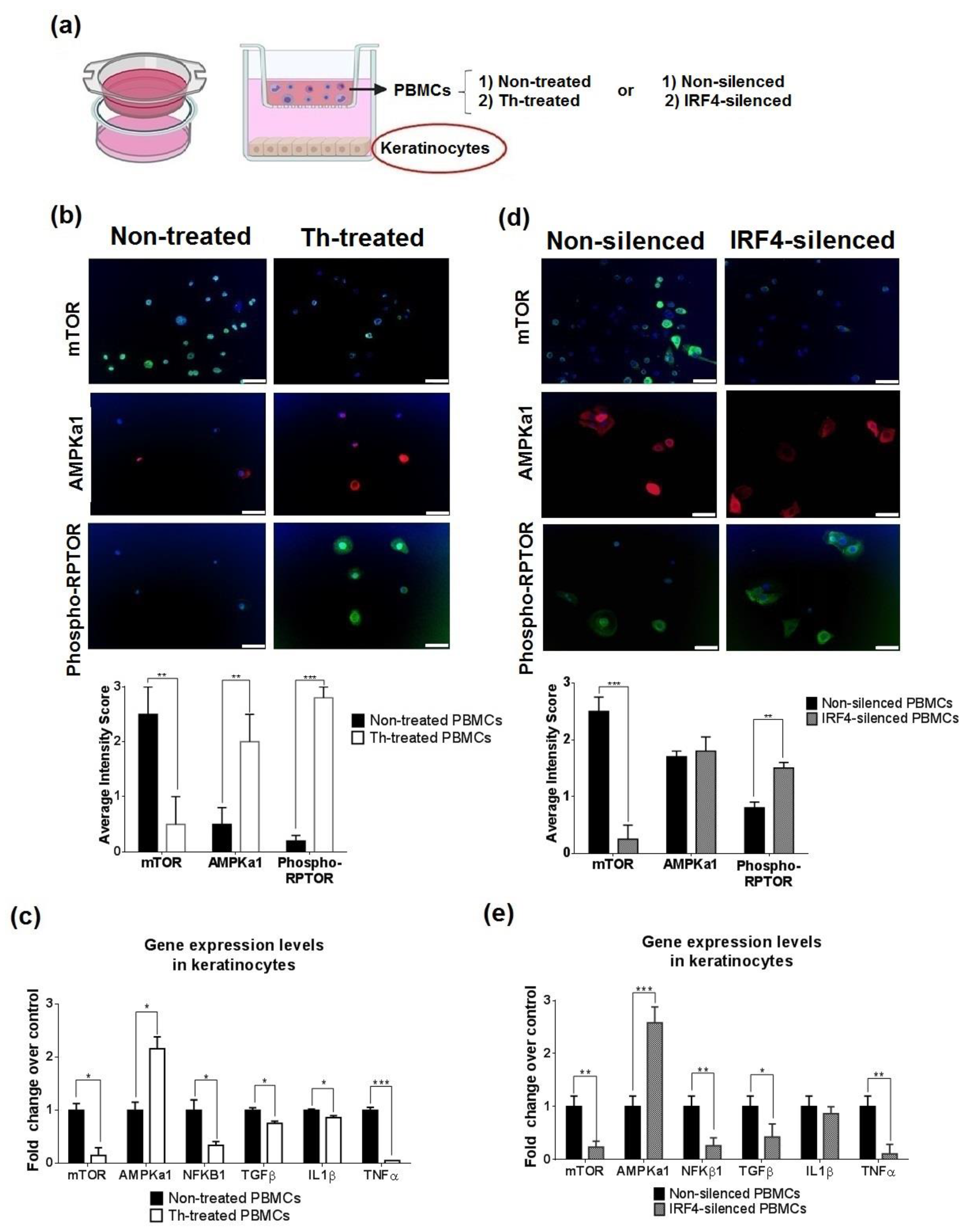
3.4. Thalidomide Modulates the AMPK1/mTOR-NF-ҡB Signalling Pathway in Keratinocytes
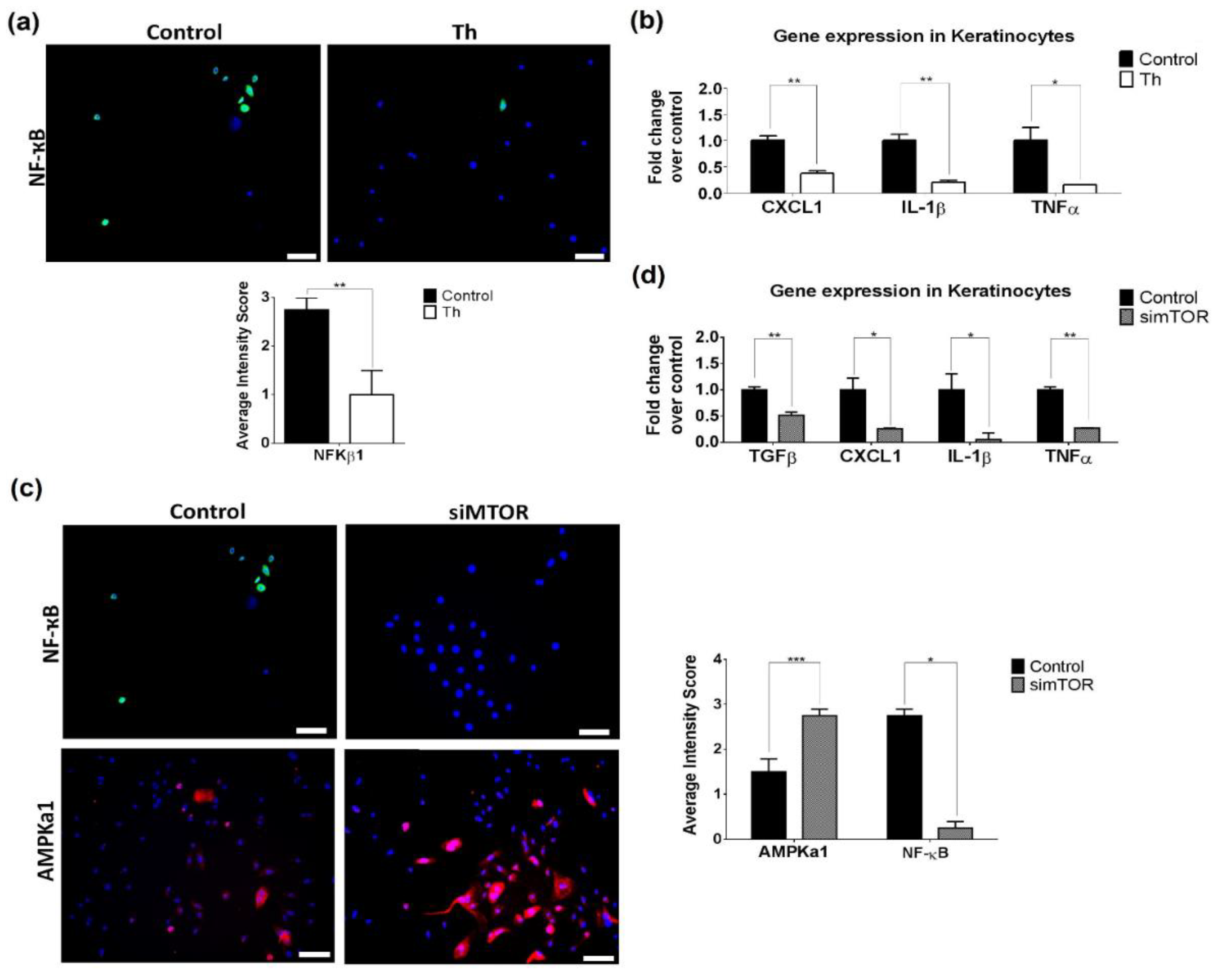
3.5. Thalidomide-Treated PBMCs Downregulate Keratinocyte mTOR Signalling Pathway
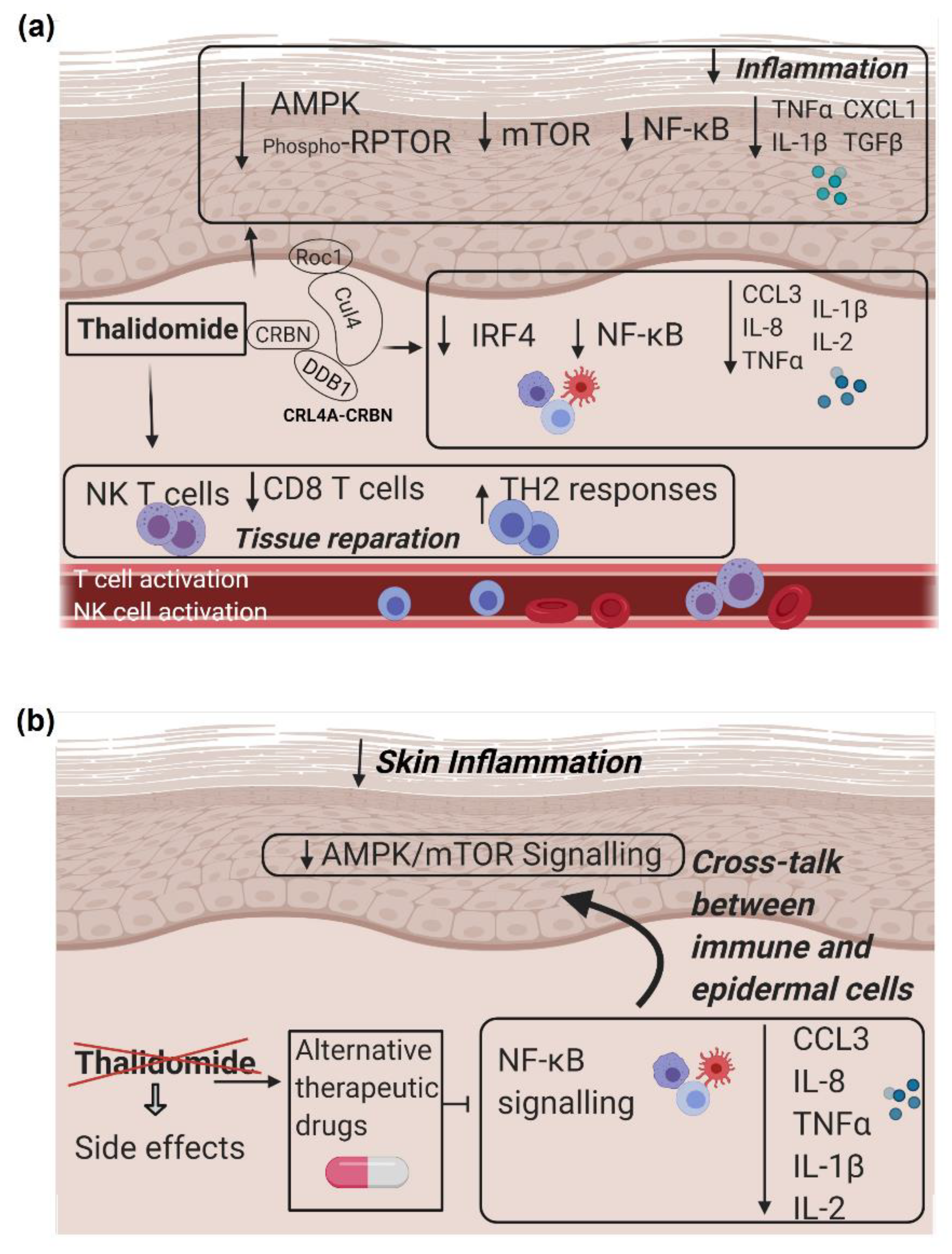
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Onkon, L.G.; Werth, V.P. Cutaneous lupus erythematosus: Diagnosis and treatment. Best Pract. Res. Clin. Rheumatol. 2013, 27, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Werth, V.P. Clinical manifestations of cutaneous lupus erythematosus. Autoimmun. Rev. 2005, 4, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Gillian, J.N.; Sontheimer, R.D. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J. Am. Acad. Derm. 1981, 4, 471–475. [Google Scholar] [CrossRef]
- Baltaci, M.; Fritsch, P. Histologic features of cutaneous lupus erythematosus. Autoimun. Rev. 2009, 8, 467–473. [Google Scholar] [CrossRef]
- Sticherling, M.; Bonsmann, G.; Kuhn, A. Diagnostic approach and treatment of cutaneous lupus erythematosus. J. Dtsch. Derm. Ges. 2008, 6, 48–59. [Google Scholar] [CrossRef]
- Cortés-Hernández, J.; Torres-Salido, M.; Castro-Marrero, J.; Vilardell-Tarres, M.; Ordi-Ros, J. Thalidoide in the treatment of refractory cutaneous lupus erythematosus: Prognostic factors of clinical outcome. Br. J. Derm. 2012, 166, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Callen, J.P. Management of “refractory” skin disease in patients with lupus erythematosus. Best Pract. Res. Clin. Rheumatol. 2005, 19, 767–784. [Google Scholar] [CrossRef]
- Hastings, R.C.; Trautman, J.R.; Enna, C.D.; Jacobson, R.R. Thalidomide in the treatment of erythema nodosum leprosum. With a note on slected laboratory abnormalities in erythema nodosum leprosum. Clin. Pharm. 1970, 11, 481–487. [Google Scholar]
- Palumbo, A.; Facon, T.; Sonneveld, P.; Bladè, J.; Offidani, M.; Gay, F.; Moreau, P.; Waage, A.; Spencer, A.; Ludwig, H.; et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood 2008, 11, 3968–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, J.B.; Gonzalez, F.F. Lupus eritematoso discoide & talidomida. Derm. Rev. Mex 1975, 19, 131–139. [Google Scholar]
- Cuadrado, M.J.; Karim, Y.; Sanna, G.; Smith, E.; Khamashta, M.A.; Hughes, G.R.V. Thalidomide for the treatment of resistant cutaneous lupus: Efficacy and safety of different therapeutic regimens. Am. J. Med. 2005, 118, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Duong, D.J.; Spigel, G.T.; Moxley, R.T., 3rd; Gaspari, A.A. American experience with low-dose thalidomide therapy for severe cutaneous lupus erythematosus. Arch. Dermatol. 1999, 135, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Bastuji-Garin, S.; Ochonisky, S.; Bouche, P.; Gherardi, R.K.; Duguet, C.; Djerradine, Z.; Poli, F.; Revuz, J.; Thalidomide Neuropathy Study Group. Incidence and risk factors for thalidomide neuropathy: A prospective study of 135 dermatologic patients. J. Investig. Derm. 2002, 119, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Schumock, G.T.; Desai, A.A.; Kwaan, H.C.; Raisch, D.W.; Newlin, R.; Stadler, W. Thalidomide-associated deep vein thrombosis and pulmonary embolism. Am. J. Med. 2002, 113, 603–606. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Handa, H. Teratogenic effects of thalidomide: Molecular mechanisms. Cell Mol. Life Sci 2011, 68, 1569–1579. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keifer, J.A.; Guttridge, D.C.; Ashburner, B.P.; Baldwin, A.S., Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J. Biol. Chem 2001, 276, 22382–22387. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor α by enhancing mRNA degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Kobayashi, N.; Yamazaki, T.; Agematsu, K. Thalidomide as an immunotherapeutic agent: The effects on neutrophil-mediated inflammation. Curr. Pharm. Des. 2005, 11, 395–401. [Google Scholar] [CrossRef]
- Kingsmore, S.F.; Lindquist, I.E.; Mudge, J.; Gessler, D.D.; Beavis, W.D. Genome-wide association studies: Progress and potential for drug discovery and development. Nat. Rev. Drug Discov. 2008, 7, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Avey, D.; Sankararaman, S.; Yim, A.K.Y.; Barve, R.; Milbrandt, J.; Mitra, R.D. Single-Cell RNA-Seq Uncovers a Robust Transcriptional Response to Morphine by Glia. Cell Rep. 2018, 24, 3619–3629. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.T.; Lee, H.W.; Lee, H.O.; Kim, S.C.; Seo, Y.J.; Chung, W.; Hye, H.E.; Do-Hyun, N.; Junhyoung, K.; Kyeung, M.J.; et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015, 16, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, S.A.; Houghtaling, B.R.; Elemento, O.; Kapoor, T.M. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol. 2012, 8, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khun, A.; Ruzicka, T. Classification of cutaneous lupus erythematosus. In Cutaneous Lupus Erythematosus; Khun, A., Lehmann, P., Ruzicka, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 53–59. [Google Scholar]
- Albrecht, J.; Taylor, L.; Berlin, J.A.; Dulay, S.; Ang, G.; Fakharzadeh, S.; Kantor, J.; Kim, E.; Militello, G.; McGinnis, K.; et al. The CLASI (Cutaneous Lupus Erythematosus Disease Area and Severity Index): An outcome instrument for cutaneous lupus erythematosus. J. Investig. Derm. 2005, 125, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000 Res. 2018, 7, 1338. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Jorba, G.; Aguirre-Plans, J.; Junet, V.; Segú-Vergés, C.; Ruiz, J.L.; Pujol, A.; Fernández-Fuentes, N.; Mas, J.M.; Oliva, B. In-silico simulated prototype-patients using TPMS technology to study a potential adverse effect of sacubitril and valsartan. PLoS ONE 2020, 15, e0228926. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Szklarzyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Hecker, N.; Ahmed, J.; von Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. Super Target goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanable, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1063. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Fionda, C.; Abruzzese, M.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé, C.; Gimenez-Barcons, M.; Ferrer, B.; Ordi-Ros, J.; Cortés-Hernández, J. Microarray study reveals a transforming growth factor-β-dependent mechanism of fibrosis in discoid lupus erythematosus. Br. J. Derm. 2016, 175, 302–313. [Google Scholar] [CrossRef]
- O’Brien, J.C.; Hosler, G.A.; Chong, B.F. Changes in T cell and B cell composition in discoid lupus erythematosus skin at different stages. J. Derm. Sci. 2017, 85, 247–249. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wu, H.; Liao, W.; Zhao, M.; Chan, V.; Li, L.; Zhena, M.; Chen, G.; Zhang, J.; Lau, C.-S.; et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J. Autoimmun. 2018, 93, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gadola, S.D.; Dulphy, N.; Salio, M.; Cerundolo, V. Valpha24-JalphaQ-independent, CD1d-restricted recognition of alpha-galactosylceramide by human CD4(+) and CD8alphabeta(+) T lymphocytes. J. Immunol. 2002, 168, 5514–5520. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.H.; Liu, N.; Klimek, V.; Hassoun, H.; Mazumder, A.; Nimer, S.D.; Jagannath, S.; Dhodapkar, M.V. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: Therapeutic implications. Blood 2006, 108, 618–621. [Google Scholar] [CrossRef] [Green Version]
- Elkhal, A.; Pichavant, M.; He, R.; Scott, J.; Meyer, E.; Goya, S.; Geha, R.S.; Umetsu, D.T. CD1d restricted natural killer T cells are not required for allergic skin inflammation. J. Allergy Clin. Immunol. 2006, 18, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Goubier, A.; Vocanson, M.; Macari, C.; Poyet, G.; Herbelin, A.; Nicolas, J.F.; Dubois, B.; Kaiserlina, D. Invariant NKT cells suppress CD8(+) T-cell-mediated allergic contact dermatitis independently of regulatory CD4(+) T cells. J. Investig. Dermatol. 2013, 133, 980–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, H.; Kawakami, K.; Ritsu, M.; Kanno, E.; Suzuki, A.; Kamimatsuno, R.; Takagi, N.; Miyasaka, T.; Ishii, K.; Imai, Y.; et al. Contribution of Invariant Natural Killer T Cells to Skin Wound Healing. Am. J. Pathol. 2015, 185, 3248–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, H.; Kawakami, K.; Kanno, E.; Suzuki, A.; Takagi, N.; Yamamoto, H.; Ishii, K.; Imai, Y.; Maruyama, R.; Tachi, M. Invariant NKT cells promote skin wound healing by preventing a prolonged neutrophilic inflammatory response. Wound Repair Regen. 2017, 25, 805–815. [Google Scholar] [CrossRef]
- Godó, M.; Sessler, T.; Hamar, P. Role of invariant natural killer T (iNKT) cells in systemic lupus erythematosus. Curr. Med. Chem. 2008, 15, 1778–1787. [Google Scholar] [CrossRef]
- Lee, H.S.; Kwon, H.S.; Park, D.E.; Woo, Y.D.; Kim, H.Y.; Kim, H.R.; Cho, S.H.; Min, K.U.; Kang, H.R.; Chang, Y.S. Thalidomide inhibits alternative activation of macrophages in vivo and in vitro: A potential mechanism of anti-asthmatic effect of thalidomide. PLoS ONE 2015, 10, e0123094. [Google Scholar] [CrossRef]
- Haslett, P.A.; Corral, L.G.; Albert, M.; Kaplan, G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J. Exp. Med. 1998, 187, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- McHugh, S.M.; Rifkin, I.R.; Deighton, J.; Wilson, A.B.; Lachmann, P.J.; Lockwood, C.M.; Ewan, P.W. The immunosuppressive drug thalidomide induces T helper cell type 2 (Th2) and concomitantly inhibits Th1 cytokine production in mitogen- and antigen-stimulated human peripheral blood mononuclear cell cultures. Clin. Exp. Immunol. 1995, 99, 160–167. [Google Scholar] [CrossRef]
- Chan, A.J.; Jang, J.C.; Nair, M.G. Tissue Remodeling and Repair during Type 2 inflammation. In The Th2 Type Immune Response in Health and Disease; Gause, W., Artis, D., Eds.; Springer: New York, NY, USA, 2016; ISBN 978-1-4939-2910-8. [Google Scholar]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Amare, G.G.; Meharie, B.G.; Belayneh, Y.M. A drug repositioning success: The repositioned therapeutic applications and mechanisms of action of thalidomide. J. Oncol. Pharm. Pract. 2021, 27, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ito, T.; Liu, S.; Ando, H.; Sakamoto, S.; Yamaguchi, Y.; Tokunaga, E.; Shibata, N.; Handa, H.; Hakoshima, T. Structural basis of thalidomide enantiomer binding to cereblon. Sci. Rep. 2018, 8, 1294. [Google Scholar] [CrossRef]
- Kowalski, T.W.; Gomes, J.D.A.; Garcia, G.B.C.; Fraga, L.R.; Paixao-Cortes, V.R.; Recamonde-Mendoza, M.; Sanseverino, M.T.V.; Shuler-Faccini, L.; Vianna, F.S.L. CRL4-Cereblon complex in Thalidomide Embryopathy: A translational investigation. Sci. Rep. 2020, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Ni, A.; Chen, H.; Wu, Y.; Li, W.; Chen, S.; Li, J. Expression of IRF-4 and IBP in skin lesions of patients with psoriasis vulgaris. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 287–290. [Google Scholar] [CrossRef]
- Agnarelli, A.; Chevassut, T.; Mancini, E.J. IRF4 in multiple myeloma-Biology, disease and therapeutic target. Leuk. Res. 2018, 72, 52–58. [Google Scholar] [CrossRef]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Ohba, Y.; Yanai, H. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 2005, 102, 15989–15994. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Carrio, J.; López, P.; Alperi-López, M.; Caminal-Montero, L.; Ballina-Garcia, F.J.; Suárez, A. IRF4 and IRGs delineate clinically relevant gene expression signatures in systemic lupus erythematosus and rheumatoid arthritis. Front. Immunol. 2018, 9, 3085. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.R.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.; Breider, M.; et al. Rate of CRL4(CRBN) substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Lee, K.M.; Jo, S.; Kim, H.; Lee, J.; Park, C.S. Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta. 2011, 1813, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Yuan, J.; Li, Y.F.; Li, R.S. Thalidomide decreases high glucose-induced extracellular matrix protein synthesis in mesangial cells via the AMPK pathway. Exp. Med. 2019, 17, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, Y.; Wang, B.; Li, R. Renal-protective effect of thalidomide in streptozotocin-induced diabetic rats through anti-inflammatory pathway. Drug Des. Dev. Ther. 2018, 12, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.; Li, X.; Deng, Y.; Chang, H.W.; Kim, D.Y. AMPK is down-regulated by the CRL4A-CRBN axis through the polyubiquitination of AMPKa isoforms. FASEB J. 2019, 33, 6539–6550. [Google Scholar] [CrossRef] [PubMed]
- Cibrian, D.; Fuente, H.; Sánchez-Madrid, F. Metabolic pathways that control skin homeostasis and inflammation. Trends Mol. Med. 2020, 26, 975–986. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Dong, L.; Yang, Z.W.; Zhang, J.; Zhang, S.L.; Niu, M.J.; Xia, J.W.; Gong, Y.; Zhu, N.; et al. Crosstalk between the Akt/mTORC1 and NF-kb signaling pathways promotes hypoxia-induced pulmonary hypertension by increasing DPP4 expression in PASMCs. Acta Pharm. Sin. 2019, 40, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Domingo, S.; Ferrer, B.; Moliné, T.; Ordi-Ros, J.; Cortés-Hernández, J. MicroRNA Expression Profiling Identifies miR-31 and miR-485-3p as Regulators in the Pathogenesis of Discoid Cutaneous Lupus. J. Investig. Derm. 2019, 139, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.; Ulvmar, M.; Toftgard, R. The two-faced NF-kappaB in the skin. Int. Rev. Immunol. 2008, 27, 205–223. [Google Scholar] [CrossRef]
- Verma, I.M. Nuclear factor (NF)-kappaB proteins: Therapeutic targets. Ann. Rheum Dis. 2004, 63 (Suppl. S2), ii57–ii61. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.; Degitz, K.; Quirling, M.; Jilg, N.; Page, S.; Brand, K. Involvement of NF-κB signalling in skin physiology and disease. Cell. Signal 2003, 15, 1–7. [Google Scholar] [CrossRef]
- Zhang, C.; Xiao, C.; Dang, E.; Cao, J.; Zhu, Z.; Fu, M.; Yao, X.; Liu, Y.; Jin, B.; Wang, G.; et al. CD100-Plexin-B2 promotes the inflammation in psoriasis by activating NF-kB ant the inflammasome in keratinocytes. J. Investig. Dermatol. 2018, 138, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Homey, B.; Dieu-Nosjean, M.C.; Wiesenborn, A.; Massacrier, C.; Pin, J.J.; Oldham, E.; Catron, D.; Buchanan, M.E.; Müller, A.; Malefyt, R.W.; et al. Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J. Immunol. 2000, 164, 6621–6632. [Google Scholar] [CrossRef] [Green Version]
- Laggner, U.; Meglio, P.D.; Perera, G.K.; Hundhausen, C.; Lacy, K.E.; Ali, N.; Smith, C.H.; Hayday, A.C.; Nickoloff, B.J.; Nestle, F.O. Identificiation of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J. Immunol. 2011, 187, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Muto, S.; Jung, K.; Itai, A.; Matsuda, H. Topical application with a new NF-kappaB inhibitor improves atopic dermatitis in NC/NgaTnd mice. J. Investig. Derm. 2007, 127, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kB pathway for the therapy of diseases: Mechanism an clinical study. Signal. Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, S.; Solé, C.; Moliné, T.; Ferrer, B.; Cortés-Hernández, J. Thalidomide Exerts Anti-Inflammatory Effects in Cutaneous Lupus by Inhibiting the IRF4/NF-ҡB and AMPK1/mTOR Pathways. Biomedicines 2021, 9, 1857. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121857
Domingo S, Solé C, Moliné T, Ferrer B, Cortés-Hernández J. Thalidomide Exerts Anti-Inflammatory Effects in Cutaneous Lupus by Inhibiting the IRF4/NF-ҡB and AMPK1/mTOR Pathways. Biomedicines. 2021; 9(12):1857. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121857
Chicago/Turabian StyleDomingo, Sandra, Cristina Solé, Teresa Moliné, Berta Ferrer, and Josefina Cortés-Hernández. 2021. "Thalidomide Exerts Anti-Inflammatory Effects in Cutaneous Lupus by Inhibiting the IRF4/NF-ҡB and AMPK1/mTOR Pathways" Biomedicines 9, no. 12: 1857. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121857