Lemur Tyrosine Kinase 2 (LMTK2) Level Inversely Correlates with Phospho-Tau in Neuropathological Stages of Alzheimer’s Disease

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. CHR-IHC
2.3. FDL-IHC
2.4. Semiquantitative Analysis for CHR-IHC
2.5. FDL-IHC Image Analysis
2.6. Statistical Analysis
3. Results
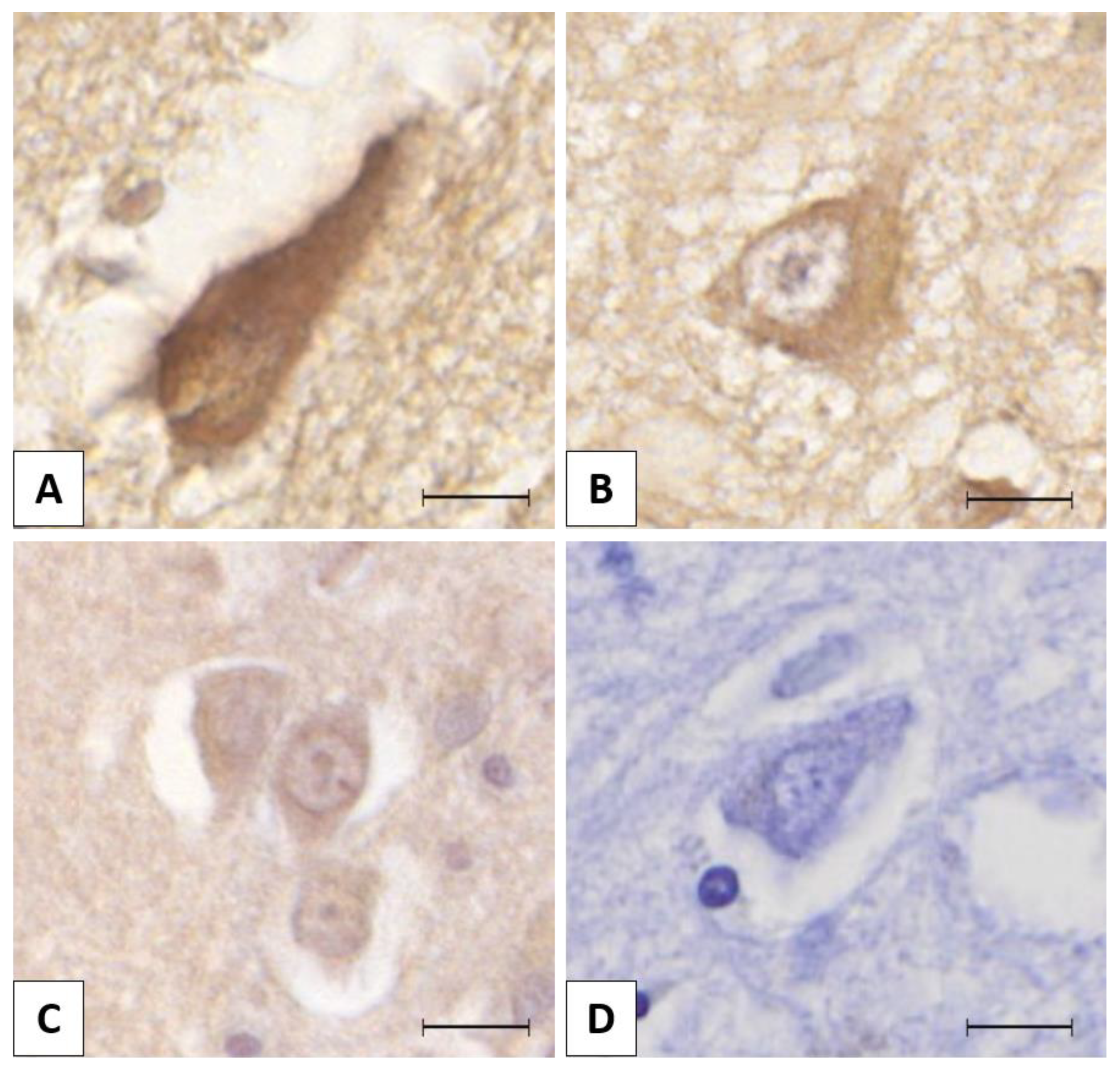
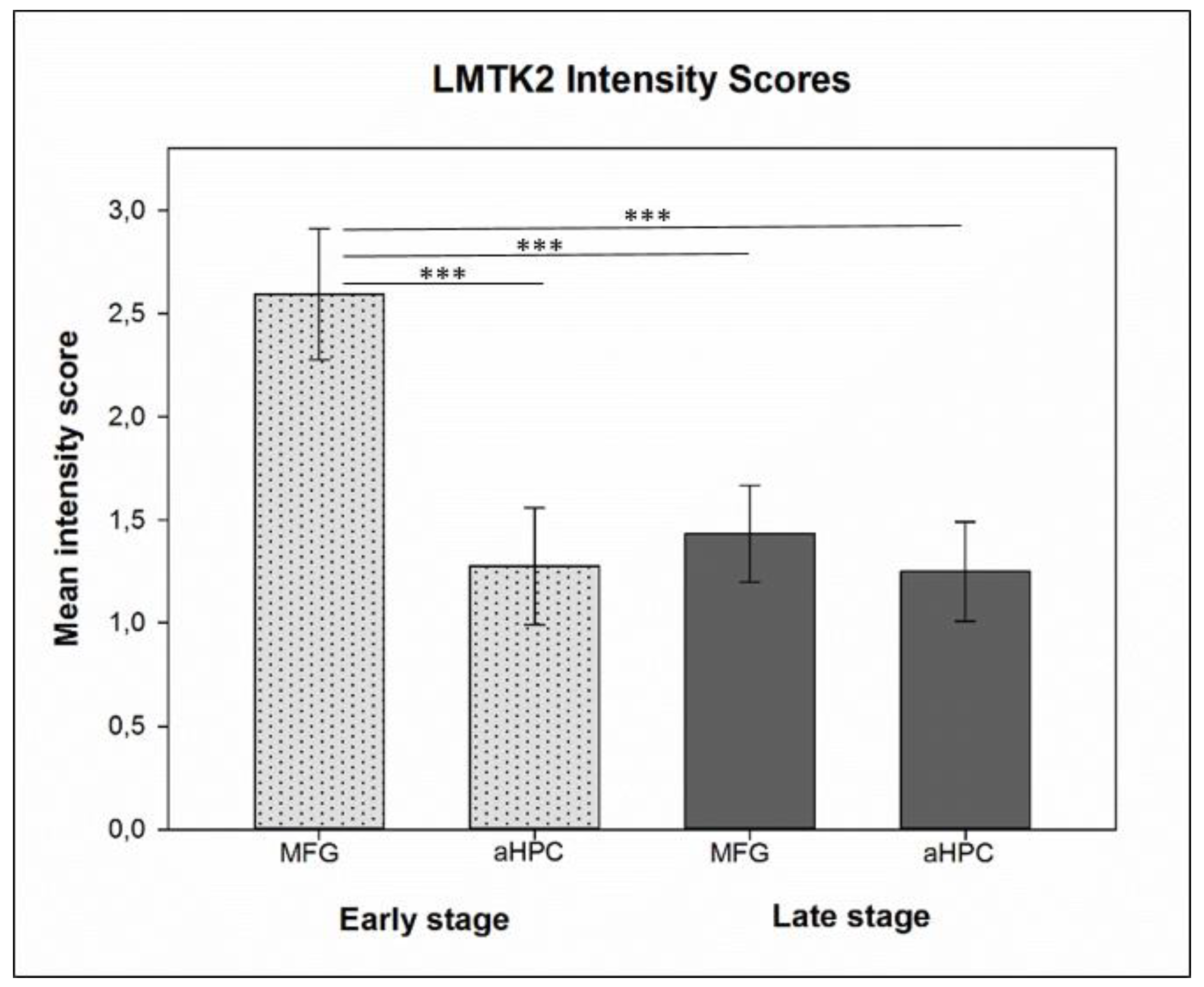
3.1. Chromogenic Immunohistochemistry (CHR-IHC)
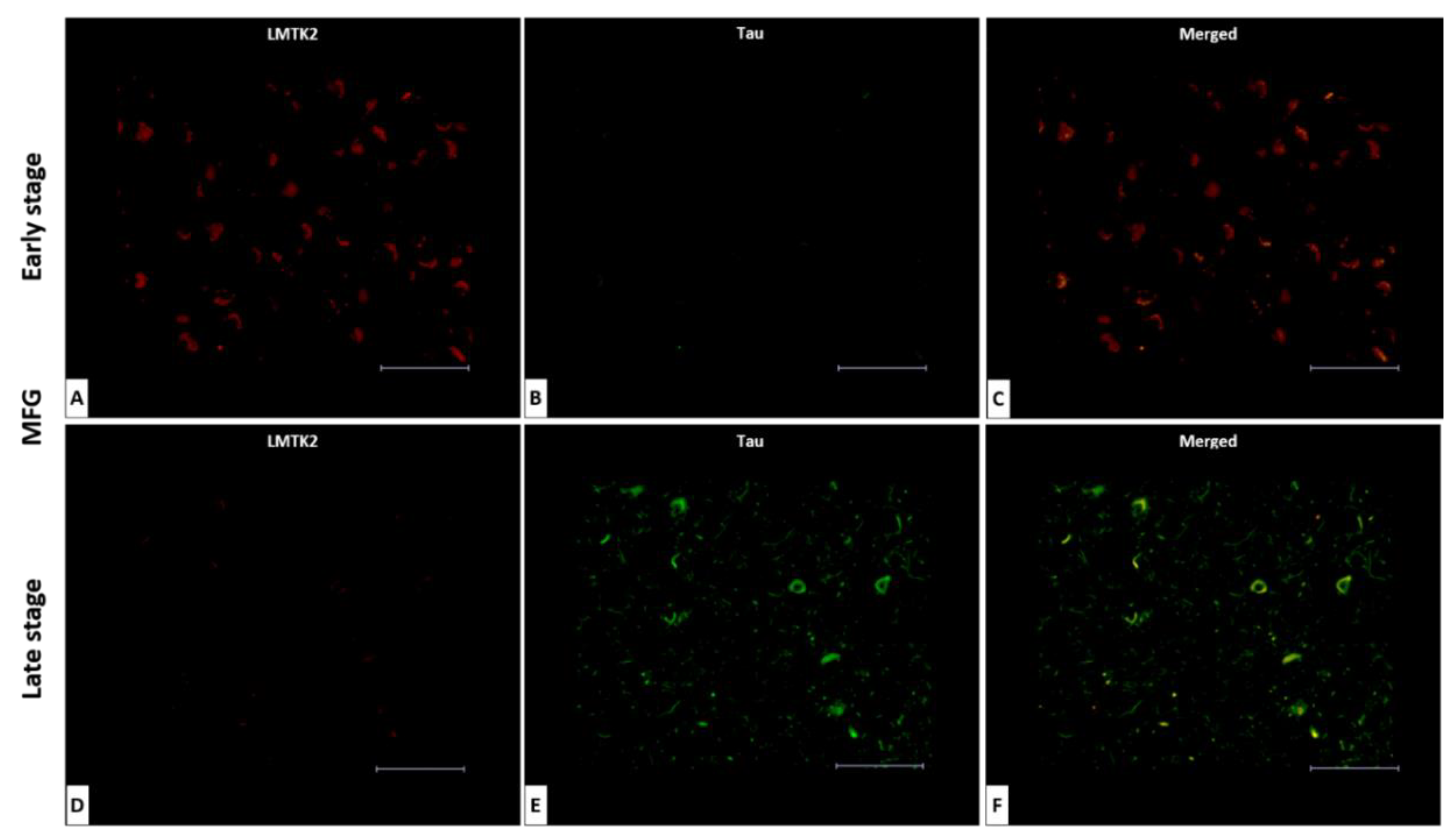
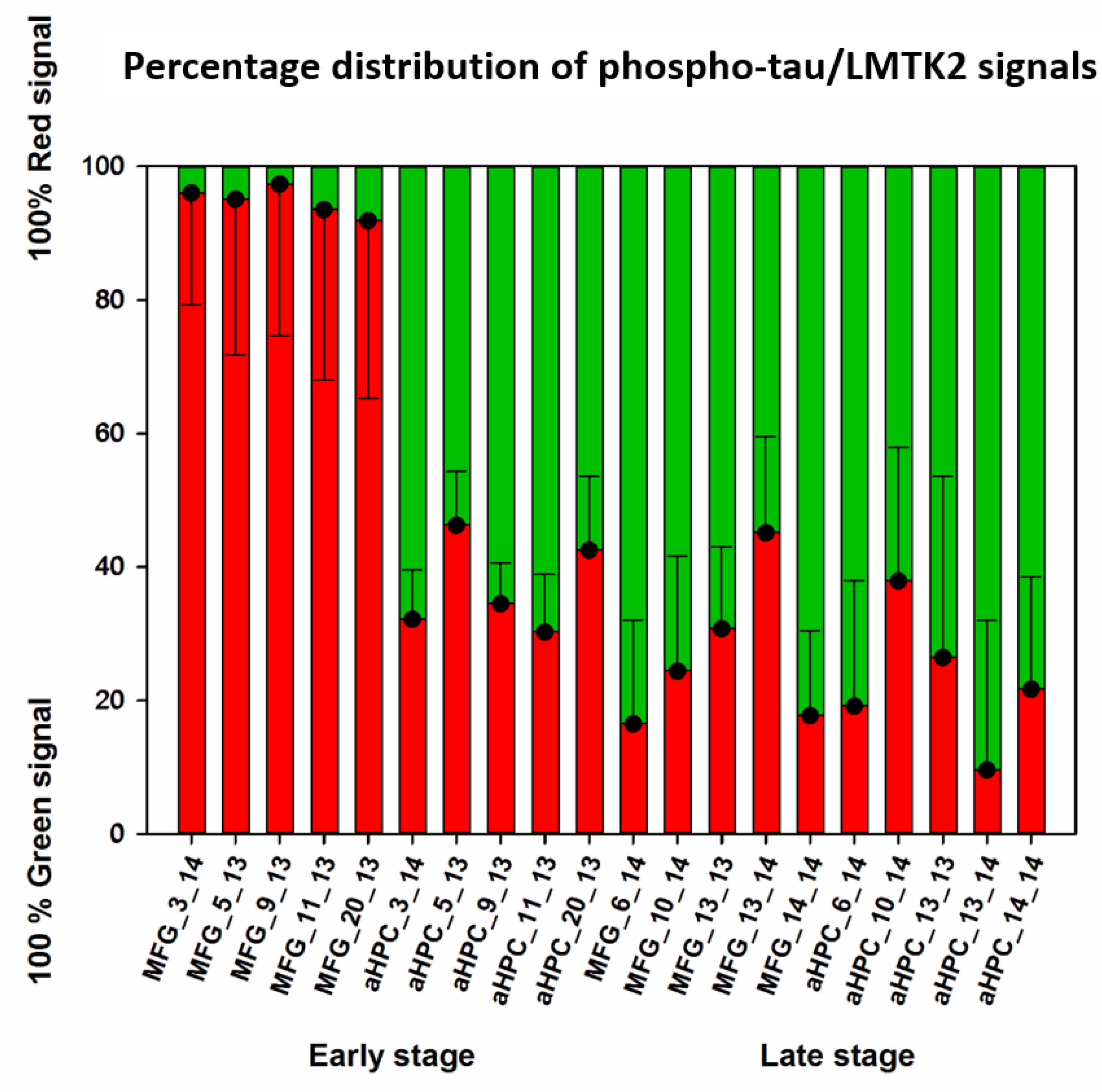
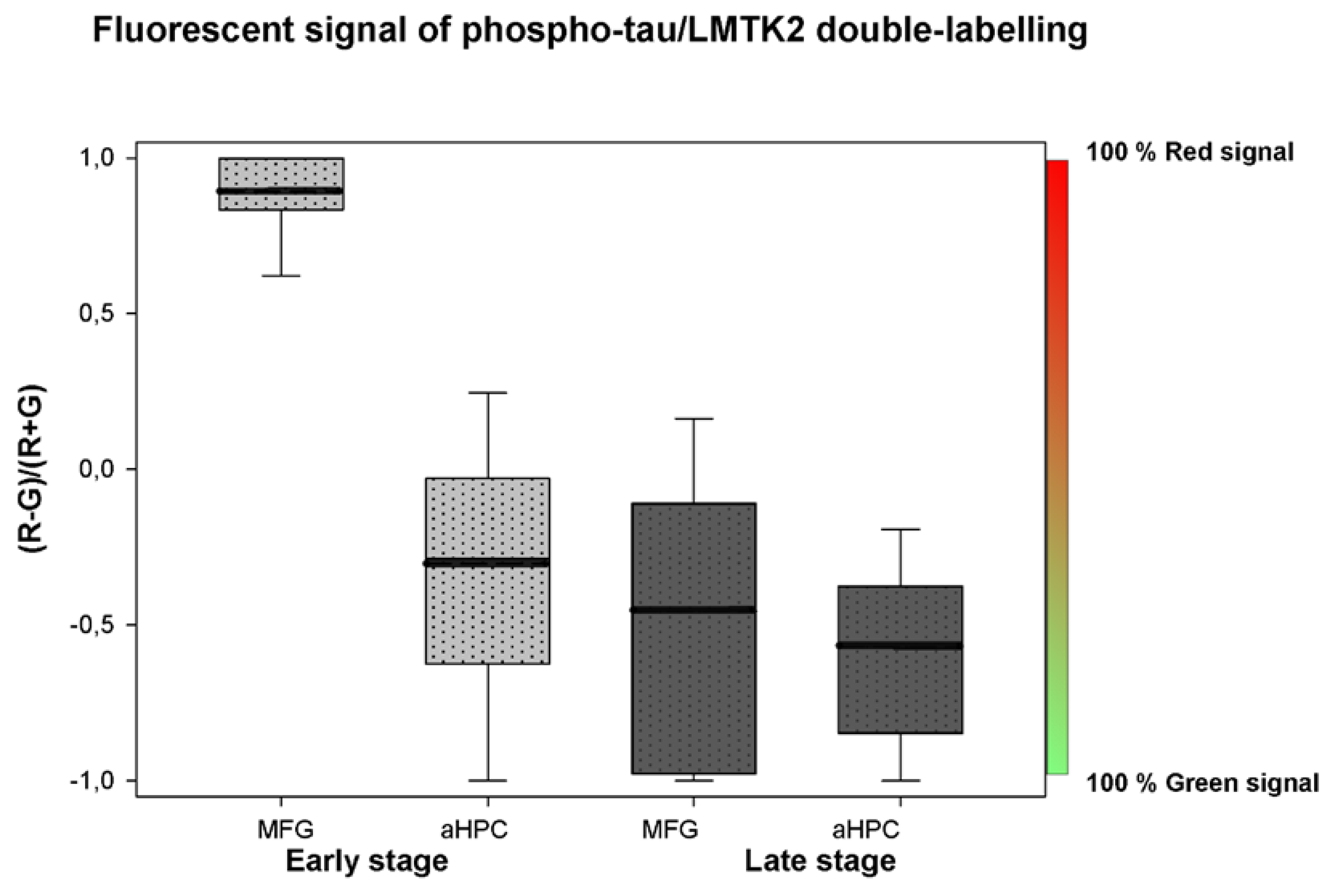
3.2. Fluorescent Double-Labelling Immunohistochemistry (FDL-IHC)
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANCOVA | Analysis of covariance |
| AD | Alzheimer’s disease |
| APOE | Apolipoprotein E |
| aHPC | Anterior hippocampus |
| CDK5 | Cyclin-dependent kinase 5 |
| CHR | Chromogenic |
| DAB | 3,3’-diaminobenzidine tetrahydrochloride |
| EDTA | Ethylenediaminetetraacetic acid |
| Fab | Fragment antigen-binding |
| Fc | Fragment crystallizable |
| FDL | Fluorescent double-labelling |
| GSK3β | Glycogen synthase kinase-3β |
| IHC | Immunohistochemistry |
| LBD | Lewy body disease |
| LMTK2 | Lemur tyrosine kinase 2 |
| MFG | Middle frontal gyrus |
| MMSE | Mini-mental state examination |
| NFT | Neurofibrillary tangles |
| PP1 | Protein phosphatase 1 |
| ROI | Region of interest |
| SNP | Single nucleotide polymorphism |
References
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [PubMed]
- Bereczki, E.; Francis, P.T.; Howlett, D.; Pereira, J.B.; Höglund, K.; Bogstedt, A.; Cedazo-Minguez, A.; Baek, J.H.; Hortobágyi, T.; Attems, J.; et al. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimer’s Dement. 2016, 12, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.S.; D’Orange, M.; Troakes, C.; Shurovi, B.N.; Engmann, O.; Noble, W.; Hortobágyi, T.; Giese, K.P. Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer’s disease. Mol. Brain 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.-H.; Hortobágyi, T.; Winblad, B.; Ballard, C.; Lehtiö, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Del Alonso, C.A.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non- paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Bencze, J.; Mórotz, G.M.; Seo, W.; Bencs, V.; Kálmán, J.; Miller, C.C.J.; Hortobagyi, T. Biological function of Lemur tyrosine kinase 2 (LMTK2): implications in neurodegeneration. Mol. Brain 2018, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mórotz, G.M.; Glennon, E.B.; Gomez-Suaga, P.; Lau, D.H.W.; Robinson, E.D.; Sedlák, É.; Vagnoni, A.; Noble, W.; Miller, C.C.J. LMTK2 binds to kinesin light chains to mediate anterograde axonal transport of cdk5/p35 and LMTK2 levels are reduced in Alzheimer’s disease brains. Acta Neuropathol. Commun. 2019, 7, 73. [Google Scholar]
- Bencze, J.; Szarka, M.; Bencs, V.; Szabó, R.N.; Smajda, M.; Aarsland, D.; Hortobágyi, T. Neuropathological characterization of Lemur tyrosine kinase 2 (LMTK2) in Alzheimer’s disease and neocortical Lewy body disease. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kawa, S.; Fujimoto, J.; Tezuka, T.; Nakazawa, T.; Yamamoto, T. Involvement of BREK, a serine/threonine kinase enriched in brain, in NGF signalling. Genes Cells 2004, 9, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brautigan, D.L. Peptide Microarray Analysis of Substrate Specificity of the Transmembrane Ser/Thr Kinase KPI-2 Reveals Reactivity with Cystic Fibrosis Transmembrane Conductance Regulator and Phosphorylase. Mol. Cell. Proteom. 2006, 5, 2124–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Brautigan, D.L. A Novel Transmembrane Ser/Thr Kinase Complexes with Protein Phosphatase-1 and Inhibitor-2. J. Boil. Chem. 2002, 277, 49605–49612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manser, C.; Guillot, F.; Vagnoni, A.; Davies, J.; Lau, K.F.; McLoughlin, D.M.; De Vos, K.J.; Miller, C.C.J. Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo. Oncogene 2012, 31, 2773–2782. [Google Scholar] [CrossRef] [Green Version]
- Conti, A.; Majorini, M.T.; Fontanella, E.; Bardelli, A.; Giacca, M.; Delia, D.; Mano, M.; Lecis, D. Lemur tyrosine kinase 2 (LMTK2) is a determinant of cell sensitivity to apoptosis by regulating the levels of the BCL2 family members. Cancer Lett. 2017, 389, 59–69. [Google Scholar] [CrossRef]
- Matarin, M.; Salih, D.A.; Yasvoina, M.; Cummings, D.M.; Guelfi, S.; Liu, W.; Solim, M.A.N.; Moens, T.G.; Paublete, R.M.; Ali, S.S.; et al. A Genome-wide Gene-Expression Analysis and Database in Transgenic Mice during Development of Amyloid or Tau Pathology. Cell Rep. 2015, 10, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Rongve, A.; Sønnesyn, H.; Skogseth, R.; Aarsland, D. A 5-year longitudinal study of patients with dementia with lewy bodies and Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, P261. [Google Scholar] [CrossRef]
- Skogseth, R.E.; Hortobágyi, T.; Soennesyn, H.; Chwiszczuk, L.; Ffytche, D.; Rongve, A.; Ballard, C.; Aarsland, D. Accuracy of Clinical Diagnosis of Dementia with Lewy Bodies versus Neuropathology. J. Alzheimer’s Dis. 2017, 59, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Lau, K.-F.; Ackerley, S.; Banner, S.J.; Shemilt, S.J.A.; Cooper, J.D.; Leigh, P.N.; Shaw, C.E.; McLoughlin, D.M.; Miller, C.C.J. Identification of a Novel, Membrane-Associated Neuronal Kinase, Cyclin-Dependent Kinase 5/p35-Regulated Kinase. J. Neurosci. 2003, 23, 4975–4983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manser, C.; Vagnoni, A.; Guillot, F.; Davies, J.; Miller, C.C.J. Cdk5/p35 phosphorylates lemur tyrosine kinase-2 to regulate protein phosphatase-1C phosphorylation and activity. J. Neurochem. 2012, 121, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Brown, H.; Pant, H.C.; Pigino, G.; DeBoer, S.; Beffert, U.; Brady, S.T. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004, 23, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.-J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of Active Glycogen Synthase Kinase 3β (GSK-3β) in Brains Staged for Alzheimer Disease Neurofibrillary Changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Grundke-Iqbal, I.; Iqbal, K.; Bogdanovic, N.; Winblad, B.; Cowburn, R.F. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 1998, 797, 267–277. [Google Scholar] [CrossRef]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; Lafrancois, J.; Wang, L.; Kondo, T.; et al. Cdk5 Is a Key Factor in Tau Aggregation and Tangle Formation In Vivo. Neuron 2003, 38, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, D.B.; Soria, J.P.; Tomasiewicz, H.G.; Wood, J.G. Phosphorylation of human tau protein by microtubule-associated kinases: GSK3β and cdk5 are key participants. J. Neurosci. Res. 2000, 62, 463–472. [Google Scholar] [CrossRef]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef]
- Plattner, F.; Angelo, M.; Giese, K.P. The Roles of Cyclin-dependent Kinase 5 and Glycogen Synthase Kinase 3 in Tau Hyperphosphorylation. J. Boil. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.D.; Cai, W.; Chen, X.; Bajaj, A.; Driver, J.; Schernhammer, E.; Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.; et al. The associations between Parkinson’s disease and cancer: The plot thickens. Transl. Neurodegener. 2015, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusakawa, G.-I. Calpain-dependent Proteolytic Cleavage of the p35 Cyclin-dependent Kinase 5 Activator to p25. J. Boil. Chem. 2000, 275, 17166–17172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; De La Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nat. 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.-H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef]
- Nath, R.; Davis, M.; Probert, A.W.; Kupina, N.C.; Ren, X.; Schielke, G.P.; Wang, K.K.W. Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem. Biophys. Res. Commun. 2000, 274, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.W.; Ando, D.M.; Taketa, D.A.; Zhou, H.; Dahlquist, F.W.; Lew, J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 2884–2889. [Google Scholar] [CrossRef] [Green Version]
- Patrick, G.N.; Zhou, P.; Kwon, Y.T.; Howley, P.M.; Tsai, L.-H. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J. Boil. Chem. 1998, 273, 24057–24064. [Google Scholar] [CrossRef] [Green Version]
- Engmann, O.; Hortobágyi, T.; Thompson, A.J.; Guadagno, J.; Troakes, C.; Soriano, S.; Al-Sarraj, S.; Kim, Y.; Giese, K.P. Cyclin-dependent kinase 5 activator p25 Is generated during memory formation and is reduced at an early stage in Alzheimer’s disease. Biol. Psychiatry 2011, 70, 159–168. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Lewis, P.A.; Hardy, J.; Martins, L.M.; Wood, N.W. Cancer and Neurodegeneration: Between the Devil and the Deep Blue Sea. PLoS Genet. 2010, 6, e1001257. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Pertsemlidis, A. Cancer and neurodegenerative disorders: pathogenic convergence through microRNA regulation. J. Mol. Cell Boil. 2011, 3, 176–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, E.; Vallano, M. Lou Role of protein kinases in neurodegenerative disease: Cyclin-dependent kinases in Alzheimer’s disease. Front. Biosci. 2005, 10, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, K.W.; Joo, Y.; Choi, Y.; Kim, M.; Lee, S.H.; Cha, S.H.; Suh, Y.H.; Kim, H.S. Swedish amyloid precursor protein mutation increases cell cycle-related proteins in vitro and in vivo. J. Neurosci. Res. 2008, 86, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Giovanni, A.; Wirtz-Brugger, F.; Keramaris, E.; Slack, R.; Park, D.S. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J. Boil. Chem. 1999, 274, 19011–19016. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.B.; Bonni, A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 2004, 72, 1–25. [Google Scholar] [CrossRef]
- Currais, A.; Hortobágyi, T.; Soriano, S. The neuronal cell cycle as a mechanism of pathogenesis in Alzheimer’s disease. Aging (Albany. NY) 2009, 1, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Malik, B.; Currais, A.; Andres, A.; Towlson, C.; Pitsi, D.; Nunes, A.; Niblock, M.; Cooper, J.; Hortobágyi, T.; Soriano, S. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the Presenilin-1 Alzheimer’s disease brain. Cell Cycle 2008, 7, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Bradbury, N.A. Lemur Tyrosine Kinase 2, a novel target in prostate cancer therapy. Oncotarget 2015, 6, 14233–14246. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Bradbury, N.A. Kinase modulation of androgen receptor signaling: implications for prostate cancer. Cancer Cell Microenviron. 2015, 2, 2. [Google Scholar]
- Harries, L.W.; Perry, J.R.; Mccullagh, P.; Crundwell, M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer 2010, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-S.; Ju, Y.S.; Lee, W.-C.; Shin, J.-Y.; Lee, J.-K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.-O.; Shin, J.-Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eeles, R.A.; The UK Genetic Prostate Cancer Study Collaborators; Kote-Jarai, Z.; Giles, G.G.; Olama, A.A.; Guy, M.; Jugurnauth, S.K.; Mulholland, S.; Leongamornlert, D.A.; Edwards, S.M.; et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 2008, 40, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Lehtinen, M.; Donovan, N.; Bonni, A. Cdc2 Phosphorylation of BAD Links the Cell Cycle to the Cell Death Machinery. Mol. Cell 2002, 9, 1005–1016. [Google Scholar] [CrossRef]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, À.; Stella, A.M.G.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by β-amyloid causes neuronal death. FASEB J. 1999, 13, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Farinelli, S.E.; Greene, L.A. Inhibitors of Cyclin-dependent Kinases Promote Survival of Post-mitotic Neuronally Differentiated PC12 Cells and Sympathetic Neurons. J. Boil. Chem. 1996, 271, 8161–8169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.S.; Levine, B.; Ferrari, G.; Greene, L.A. Cyclin Dependent Kinase Inhibitors and Dominant Negative Cyclin Dependent Kinase 4 and 6 Promote Survival of NGF-Deprived Sympathetic Neurons. J. Neurosci. 1997, 17, 8975–8983. [Google Scholar] [CrossRef]
- Freeman, R.S.; Estus, S.; Johnson, E.M. Analysis of cell cycle-related gene expression in postmitotic neurons: Selective induction of cyclin D1 during programmed cell death. Neuron 1994, 12, 343–355. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Park, D.S.; Greene, L.A.; Shelanski, M.L. Role of Cell Cycle Regulatory Proteins in Cerebellar Granule Neuron Apoptosis. J. Neurosci. 1999, 19, 8747–8756. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; A Greene, L. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 2001, 32, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Dey, I.; Bradbury, N.A. Activation of TPA-response element present in human Lemur Tyrosine Kinase 2 (lmtk2) gene increases its expression. Biochem. Biophys. Rep. 2017, 12, 140–150. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | Sex | Age (Baseline) | Final MMSE | Braak Tau Stage | APOE Gene Polymorphism |
|---|---|---|---|---|---|
| HT-05-13 | M | 70 | 25 | II | ε3–ε3 |
| HT-09-13 | M | 77 | 22 | III | ε3–ε3 |
| HT-11-13 | F | 55 | 23 | I | ε3–ε4 |
| HT-20-13 | M | 64 | 26 | I | - |
| HT-03-14 | M | 72 | 16 | III | ε3–ε3 |
| HT-13-13 | F | 80 | 13 | VI | ε3–ε4 |
| HT-14-13 | F | 84 | 13 | VI | ε3–ε3 |
| HT-06-14 | M | 77 | 6 | VI | ε3–ε4 |
| HT-10-14 | M | 68 | 7 | VI | ε4–ε4 |
| HT-14-14 | M | 55 | 0 | VI | ε3–ε3 |
| Early stage | Late stage | |||||||
|---|---|---|---|---|---|---|---|---|
| MFG | aHPC | MFG | aHPC | |||||
| Spearman Rank Order Correlation Coefficient | −1 | −1 | −1 | −1 | ||||
| p-Value | 0.0000002 | 0.0000002 | 0.0000002 | 0.0000002 | ||||
| Mean (Red; Green) | 0.946 | 0.054 | 0.349 | 0.651 | 0.273 | 0.727 | 0.215 | 0.785 |
| Standard deviation | 0.087 | 0.233 | 0.237 | 0.159 | ||||
| Standard error | 0.00175 | 0.00468 | 0.00349 | 0.00238 | ||||
| Number of pairs examined | 2464 | 2478 | 4609 | 4457 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bencze, J.; Szarka, M.; Bencs, V.; Szabó, R.N.; Módis, L.V.; Aarsland, D.; Hortobágyi, T. Lemur Tyrosine Kinase 2 (LMTK2) Level Inversely Correlates with Phospho-Tau in Neuropathological Stages of Alzheimer’s Disease. Brain Sci. 2020, 10, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10020068
Bencze J, Szarka M, Bencs V, Szabó RN, Módis LV, Aarsland D, Hortobágyi T. Lemur Tyrosine Kinase 2 (LMTK2) Level Inversely Correlates with Phospho-Tau in Neuropathological Stages of Alzheimer’s Disease. Brain Sciences. 2020; 10(2):68. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10020068
Chicago/Turabian StyleBencze, János, Máté Szarka, Viktor Bencs, Renáta Nóra Szabó, László V. Módis, Dag Aarsland, and Tibor Hortobágyi. 2020. "Lemur Tyrosine Kinase 2 (LMTK2) Level Inversely Correlates with Phospho-Tau in Neuropathological Stages of Alzheimer’s Disease" Brain Sciences 10, no. 2: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10020068