Pharmacological Inhibition of O-GlcNAc Transferase Promotes mTOR-Dependent Autophagy in Rat Cortical Neurons



Abstract
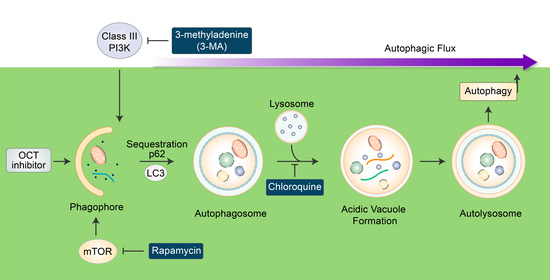
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cortical Neuron Cultures
2.3. Immunocytochemical Analysis
2.4. Immunoblot Analysis
2.5. Autophagic Flux Counting
2.6. Dot Blot Analysis
2.7. Statistical Analysis
3. Results
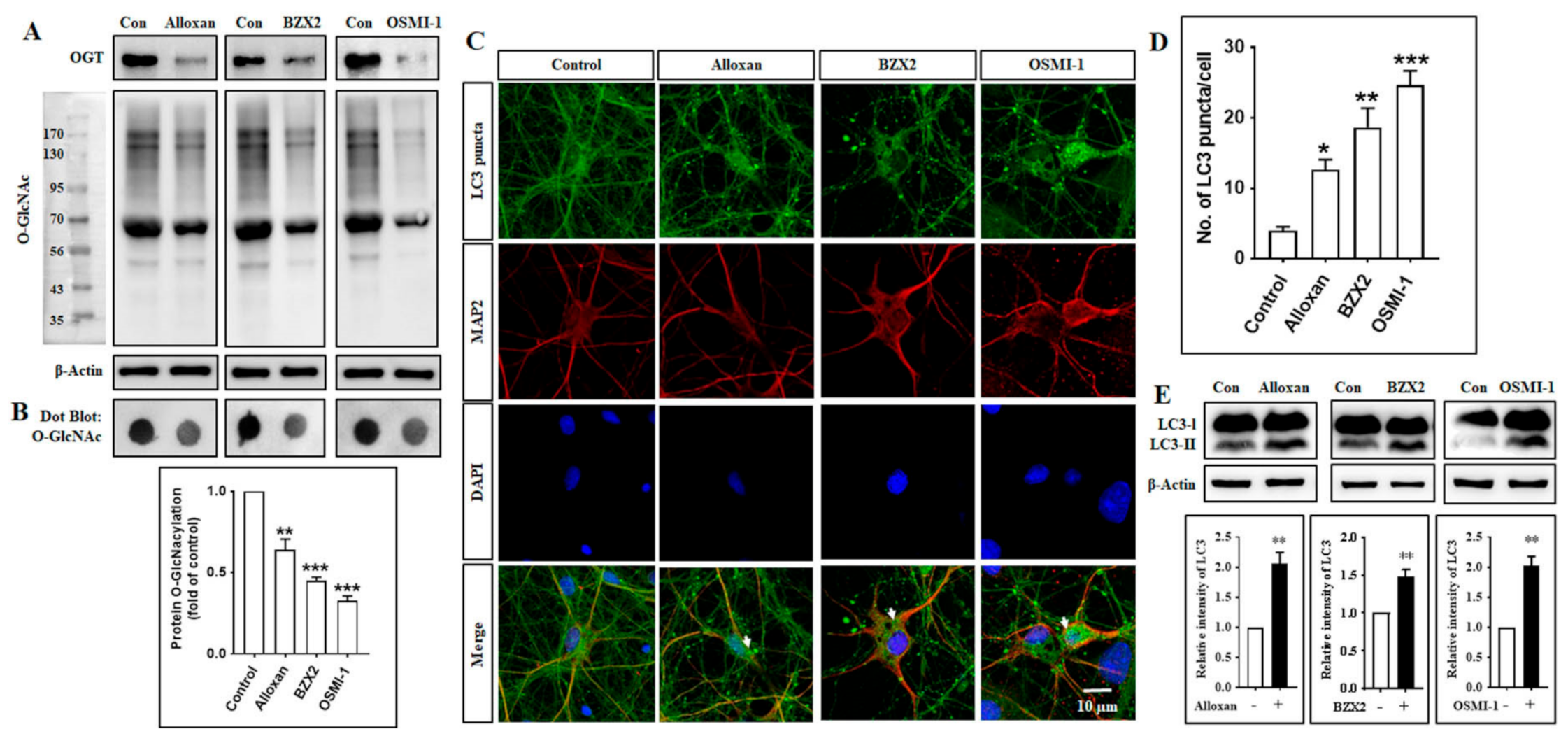
3.1. Inhibition of O-GlcNAc Transferase (OGT) Stimulates Autophagy in Rat Cortical Neurons and Neuronal Cell Lines
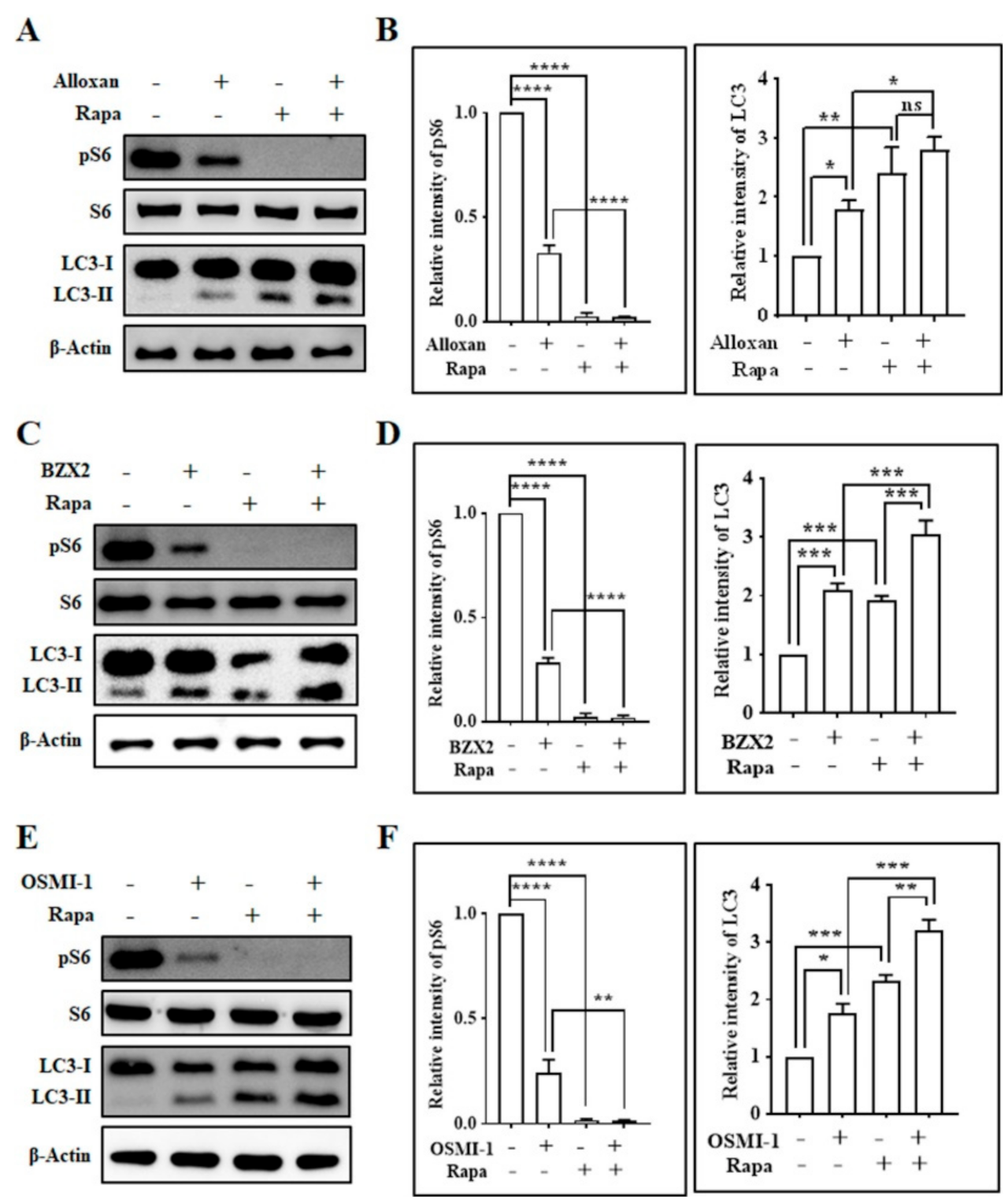
3.2. OGT Inhibitors Induce Autophagy in Cortical Neurons in an mTOR-Dependent Manner
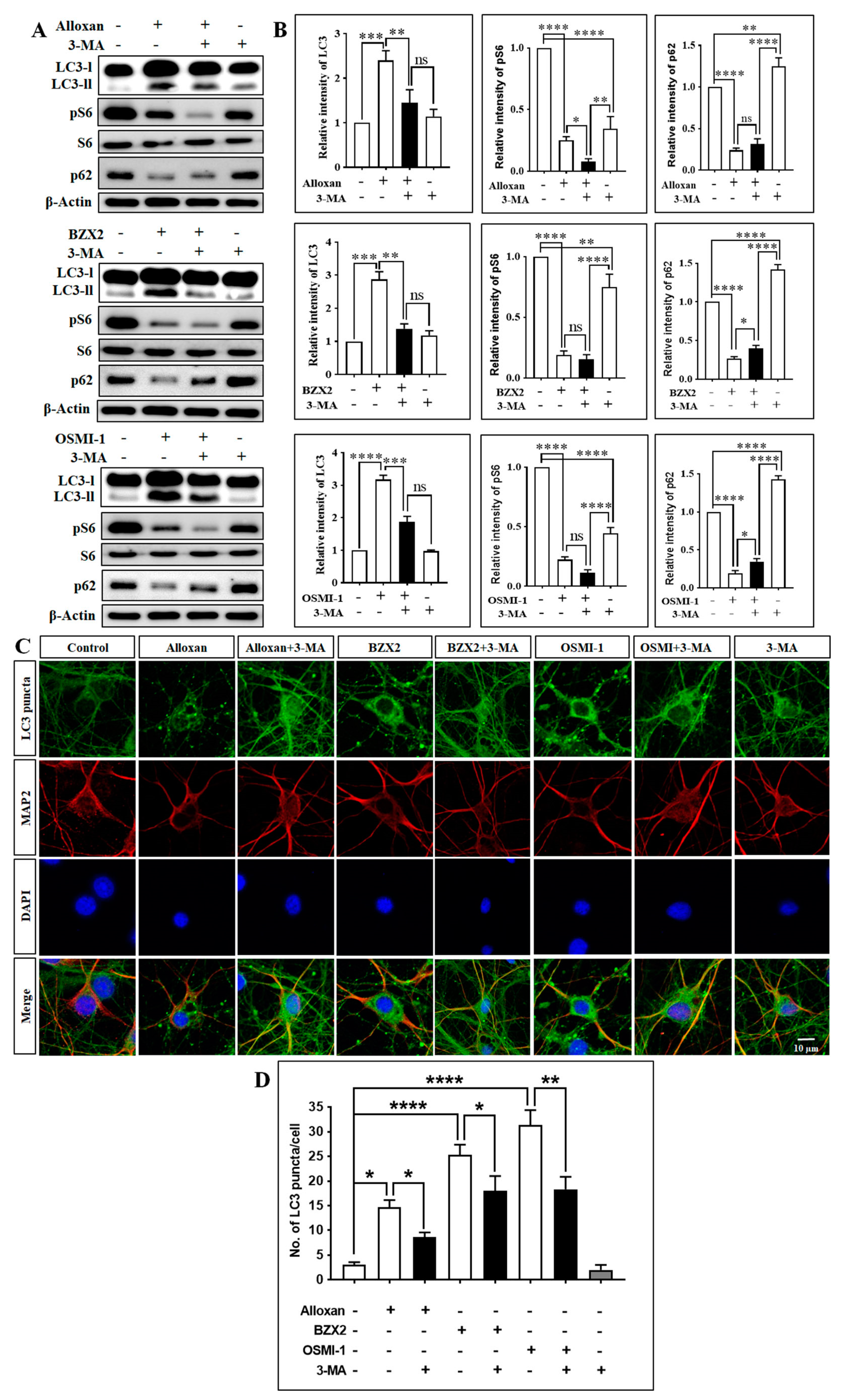
3.3. Autophagy Inhibitor Blocks OGT Inhibitor-Mediated Autophagy in Cortical Neurons
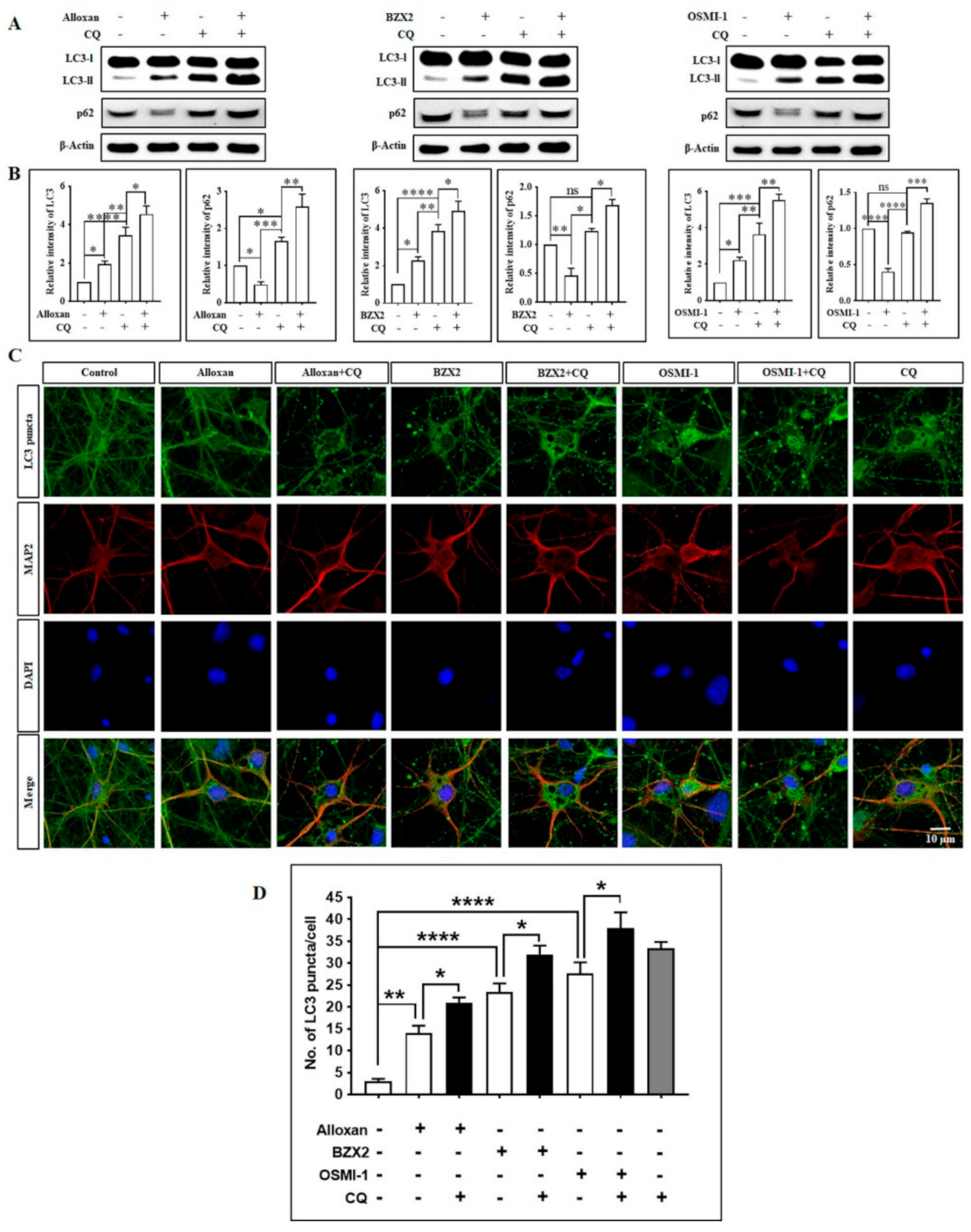
3.4. OGT Inhibitors Induce Autophagic Flux and Degrade the Autophagy Substrate p62/SQSTM1 in Cortical Neurons
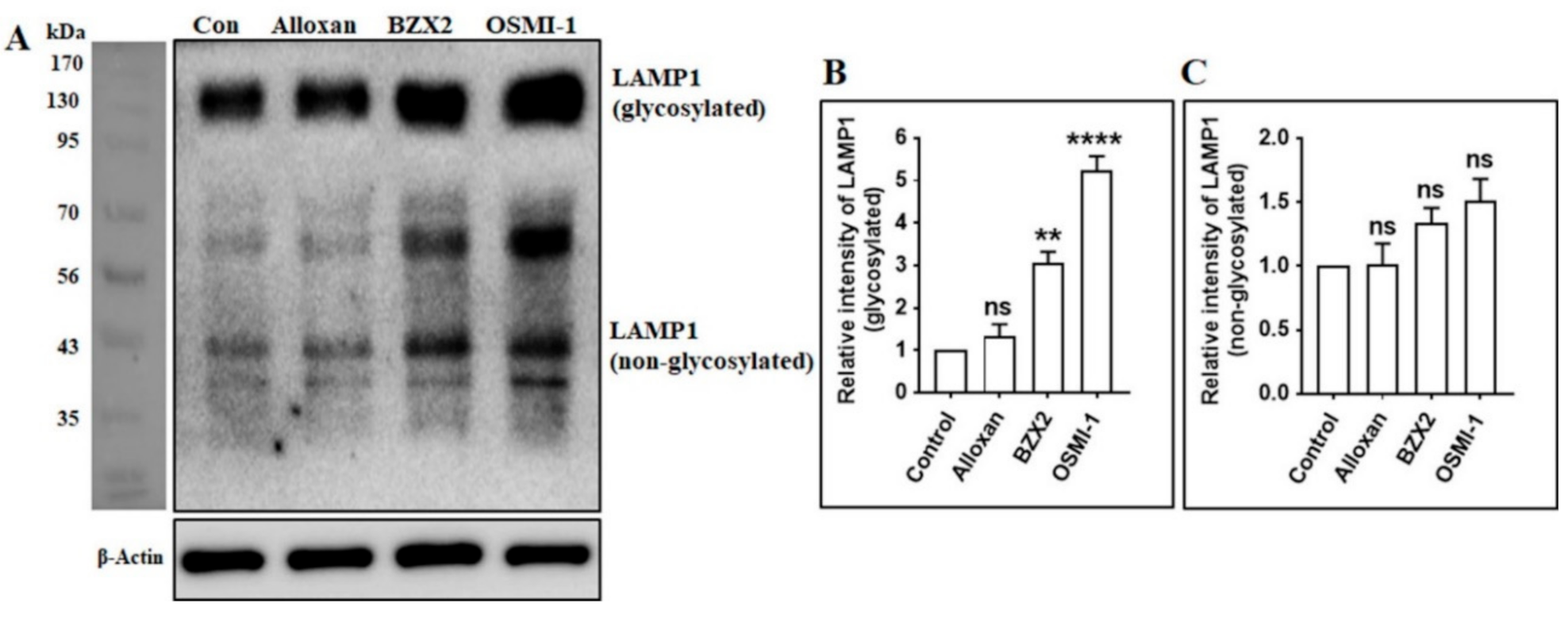
3.5. OGT Inhibitors Increase the Expression of Lysosome-Associated Membrane Protein 1 (LAMP1) in Cortical Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, A.C.; Jensen, E.H.; Rexach, J.E.; Vinters, H.V.; Hsieh-Wilson, L.C. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 15120–15125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.; Rhim, H. Functional significance of O-GlcNAc modification in regulating neuronal properties. Pharmacol. Res. 2018, 129, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Hwang, H.; Rahman, A.; Chung, C.; Rhim, H. Elevated O-GlcNAcylation induces an antidepressant-like phenotype and decreased inhibitory transmission in medial prefrontal cortex. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rexach, J.E.; Clark, P.M.; Mason, D.E.; Neve, R.L.; Peters, E.C.; Hsieh-Wilson, L.C. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat. Chem. Biol. 2012, 8, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Jones, S.P. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 2011, 40, 895–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Liang, Q.; Li, L.; Hu, Z.; Wu, F.; Zhang, P.; Ma, Y.; Zhao, B.; Kovács, A.L.; Zhang, Z.; et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 2014, 16, 1215–1226. [Google Scholar] [CrossRef]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross Talk Between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.-X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, E.G.; Albarran, E.; White, C.W.; Bieri, G.; Sanchez-Diaz, C.; Pratt, K.; Snethlage, C.E.; Ding, J.B.; Villeda, S.A. Neuronal O-GlcNAcylation improves cognitive function in the aged mouse brain. Curr. Biol. 2019, 29, 3359–3369.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Pak, J.; Jang, I.; Cho, J.W. Inhibition of mTOR affects protein stability of OGT. Biochem. Biophys. Res. Commun. 2014, 453, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hwang, H.; Cho, Y.; Rhim, H. Modulation of O-GlcNAcylation regulates autophagy in cortical astrocytes. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hwang, H.; Nah, S.-Y.; Rhim, H. Gintonin stimulates autophagic flux in primary cortical astrocytes. J. Ginseng Res. 2018, 44, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Bishayee, K.; Sadra, A.; Huh, S.-O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Bishayee, K.; Habib, K.; Sadra, A.; Huh, S.-O. 18α-Glycyrrhetinic acid lethality for neuroblastoma cells via de-regulating the Beclin-1/Bcl-2 complex and inducing apoptosis. Biochem. Pharmacol. 2016, 117, 97–112. [Google Scholar] [CrossRef]
- Rahman, A.; Rhim, A.H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.; Rahman, A.; Kabir, T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Boyer-Guittaut, M.; Dodson, M.; Chatham, J.C.; Darleyusmar, V.M.; Zhang, J. Regulation of autophagy by protein post-translational modification. Lab. Investig. 2015, 95, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimorim, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Dang, Y.; Dai, F.; Guo, Z.; Wu, J.; She, X.; Pei, Y.; Chen, Y.; Ling, W.; Wu, C.; et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J. Biol. Chem. 2003, 278, 29278–29287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, P.K.; Parihar, R.; Dwivedi, V.; Lakhotia, S.; Ganesh, S. Decreased O-Linked GlcNAcylation protects from cytotoxicity mediated by huntingtin Exon1 protein fragment*. J. Biol. Chem. 2014, 289, 13543–13553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapakse, A.G.; Ming, X.-F.; Carvas, J.M.; Yang, Z. The hexosamine biosynthesis inhibitor azaserine prevents endothelial inflammation and dysfunction under hyperglycemic condition through antioxidant effects. Am. J. Physiol. Circ. Physiol. 2009, 296, H815–H822. [Google Scholar] [CrossRef] [Green Version]
- Tallent, M.K.; Varghis, N.; Skorobogatko, Y.; Hernandez-Cuebas, L.; Whelan, K.; Vocadlo, D.J.; Vosseller, K. In vivo modulation of O-GlcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. J. Biol. Chem. 2008, 284, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.L.; Qin, H. O-GlcNAcylation regulates primary ciliary length by promoting microtubule disassembly. iScience 2019, 12, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Haque, M.; Nam, G.; Ryoo, N.; Rhim, H.; Kim, Y.K. Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors. Int. J. Mol. Sci. 2015, 16, 20212–20224. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.-W.; Thomas, C.J.; Walker, S. A Small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 2015, 10, 1392–1397. [Google Scholar] [CrossRef]
- Ryoo, N.; Rahman, A.; Hwang, H.; Ko, S.K.; Nah, S.-Y.; Kim, H.-C.; Rhim, H. Ginsenoside Rk1 is a novel inhibitor of NMDA receptors in cultured rat hippocampal neurons. J. Ginseng Res. 2019, 44, 490–495. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Biever, A.; Valjent, E.; Puighermanal, E. Ribosomal protein S6 phosphorylation in the nervous system: From regulation to function. Front. Mol. Neurosci. 2015, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.-X.; Lu, J.-H.; et al. Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Rahman, R.; Zaman, T.; Uddin, S.; Islam, R.; Abdel-Daim, M.M.; Rhim, H. Emerging potential of naturally pccurring autophagy modulators against neurodegeneration. Curr. Pharm. Des. 2020, 26, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Yang, X.; Zhang, X.; Liu, J. The autophagic inhibitor 3-methyladenine potently stimulates PKA-dependent lipolysis in adipocytes. Br. J. Pharmacol. 2012, 168, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Lerner, C.A.; Nacarelli, T.; Crowe, E.; Torres, C.; Sell, C. p62/SQSTM1 at the interface of aging, autophagy, and disease. AGE 2014, 36, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Shintani, T. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Neisch, A.L.; Ayala, C.I.; Kim, J.; Beltrame, A.; Braden, C.R.; Gardner, M.K.; Hays, T.S.; Neufeld, T.P. Coordination of autophagosome–lysosome fusion and transport by a Klp98A–Rab14 complex inDrosophila. J. Cell Sci. 2016, 129, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Baba, K.; Kuwada, S.; Nakao, A.; Li, X.; Okuda, N.; Nishida, A.; Mitsuda, S.; Fukuoka, N.; Kakeya, H.; Kataoka, T. Different localization of lysosomal-associated membrane protein 1 (LAMP1) in mammalian cultured cell lines. Histochem. Cell Biol. 2020, 153, 199–213. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; Zhang, L. Discovery of cell-permeable O-GlcNAc transferase inhibitors via tethering in situ click chemistry. J. Med. Chem. 2016, 60, 263–272. [Google Scholar] [CrossRef]
- Kang, E.-S.; Han, D.; Park, J.; Kwak, T.K.; Oh, M.A.; Lee, S.A.; Choi, S.; Park, Z.Y.; Kim, Y.; Lee, J.W. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic β cells. Exp. Cell Res. 2008, 314, 2238–2248. [Google Scholar] [CrossRef] [Green Version]
- Gloster, T.M.; Zandberg, W.F.; Heinonen, J.E.; Shen, D.L.; Deng, L.; Vocadlo, D.J. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 2011, 7, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Lazarus, M.B.; Pasquina, L.; Sliz, P.; Walker, S. A neutral diphosphate mimic crosslinks the active site of human O-GlcNAc transferase. Nat. Chem. Biol. 2011, 8, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meenatchi, P.; Purushothaman, A.; Maneemegalai, S. Antioxidant, antiglycation and insulinotrophic properties of Coccinia grandis (L.) in vitro: Possible role in prevention of diabetic complications. J. Tradit. Complement. Med. 2017, 7, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.N.; Alborn, W.E.; Knierman, M.D.; Konrad, R.J. Alloxan is an inhibitor of O-GlcNAc-selective N-acetyl-β-d-glucosaminidase. Biochem. Biophys. Res. Commun. 2006, 350, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shen, D.L.; Lafont, D.; Vercoutter-Edouart, A.-S.; Mortuaire, M.; Shi, Y.; Maniti, O.; Girard-Egrot, A.; Lefebvre, T.; Pinto, B.M.; et al. Design of glycosyltransferase inhibitors targeting human O-GlcNAc transferase (OGT). MedChemComm 2014, 5, 1172–1178. [Google Scholar] [CrossRef]
- Dorfmueller, H.C.; Borodkin, V.S.; Blair, D.E.; Pathak, S.; Navratilova, I.; Van Aalten, D.M.F. Substrate and product analogues as human O-GlcNAc transferase inhibitors. Amino Acids 2011, 40, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.S.; Kannan, N.D. A system-level approach to investigate alloxan-induced toxicity in microtubule-binding protein to lead type 2 diabetes mellitus. Mol. Divers. 2020, 1. [Google Scholar] [CrossRef]
- Lenzen, S. The mechanisms of alloxan and streptozotocin-induced diabetes. Diabetology 2008, 51, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Yi, W. O-GlcNAcylation, a sweet link to the pathology of diseases. J. Zhejiang Univ. Sci. B 2019, 20, 437–448. [Google Scholar] [CrossRef]
- Hwang, H.; Rhim, H. Acutely elevated O-GlcNAcylation suppresses hippocampal activity by modulating both intrinsic and synaptic excitability factors. Sci. Rep. 2019, 9, 7287. [Google Scholar] [CrossRef]
- Wang, P.; Hanover, J.A. Nutrient-driven O-GlcNAc cycling influences autophagic flux and neurodegenerative proteotoxicity. Autophagy 2013, 9, 604–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Lazarus, B.D.; Forsythe, M.E.; Love, D.C.; Krause, M.; Hanover, J.A. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 17669–17674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Lee, Y.; Pak, J.W.; Kim, H.; Choi, H.; Kim, J.-W.; Roth, J.; Cho, J.W. O-GlcNAc modification is essential for the regulation of autophagy in Drosophila melanogaster. Cell. Mol. Life Sci. 2015, 72, 3173–3183. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Ouyang, X.; Benavides, G.; Redmann, M.; Cofield, S.S.; Shacka, J.J.; Chatham, J.C.; Darley-Usmar, V.; Zhang, J. O-GlcNAc regulation of autophagy and α-synuclein homeostasis; implications for Parkinson’s disease. Mol. Brain 2017, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Fabrizi, C.; Pompili, E.; De Vito, S.; Somma, F.; Catizone, A.; Ricci, G.; Lenzi, P.; Fornai, F.; Fumagalli, L. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. NeuroToxicology 2016, 52, 12–22. [Google Scholar] [CrossRef]
- Abounit, K.; Scarabelli, T.M.; McCauley, R.B. Autophagy in mammalian cells. World J. Biol. Chem. 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Ma, X.; Li, H.; He, Y.; Hao, J. The emerging link between O-GlcNAcylation and neurological disorders. Cell. Mol. Life Sci. 2017, 74, 3667–3686. [Google Scholar] [CrossRef]
- Kim, C.; Nam, D.W.; Park, S.Y.; Song, H.; Hong, H.S.; Boo, J.H.; Jung, E.S.; Kim, Y.; Baek, J.Y.; Kim, K.S.; et al. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol. Aging 2013, 34, 275–285. [Google Scholar] [CrossRef]
- Yuzwa, A.S.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamano, T.; Hayashi, K.; Shirafuji, N.; Nakamoto, Y. The implications of autophagy in Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al Mamun, A.; Rahman, A.; Behl, T.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging proof of protein misfolding and interactions in multifactorial Alzheimer’s disease. Curr. Top. Med. Chem. 2020, 20, 2380–2390. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez-Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.; Al Mamun, A.; Jakaria; Shanmugam, T.; Ahmad, J.; Rahman, A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total. Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Cho, Y.; Hwang, H.; Rhim, H. Pharmacological Inhibition of O-GlcNAc Transferase Promotes mTOR-Dependent Autophagy in Rat Cortical Neurons. Brain Sci. 2020, 10, 958. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10120958
Rahman MA, Cho Y, Hwang H, Rhim H. Pharmacological Inhibition of O-GlcNAc Transferase Promotes mTOR-Dependent Autophagy in Rat Cortical Neurons. Brain Sciences. 2020; 10(12):958. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10120958
Chicago/Turabian StyleRahman, Md. Ataur, Yoonjeong Cho, Hongik Hwang, and Hyewhon Rhim. 2020. "Pharmacological Inhibition of O-GlcNAc Transferase Promotes mTOR-Dependent Autophagy in Rat Cortical Neurons" Brain Sciences 10, no. 12: 958. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10120958