
Proteomics Profiling with SWATH-MS Quantitative Analysis of Changes in the Human Brain with HIV Infection Reveals a Differential Impact on the Frontal and Temporal Lobes

 ,
,
Abstract
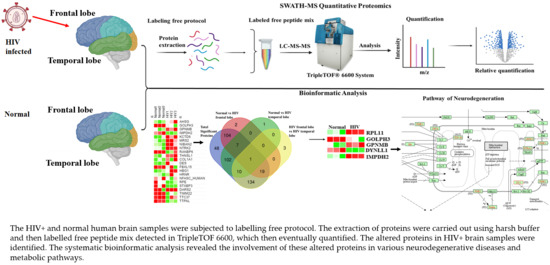
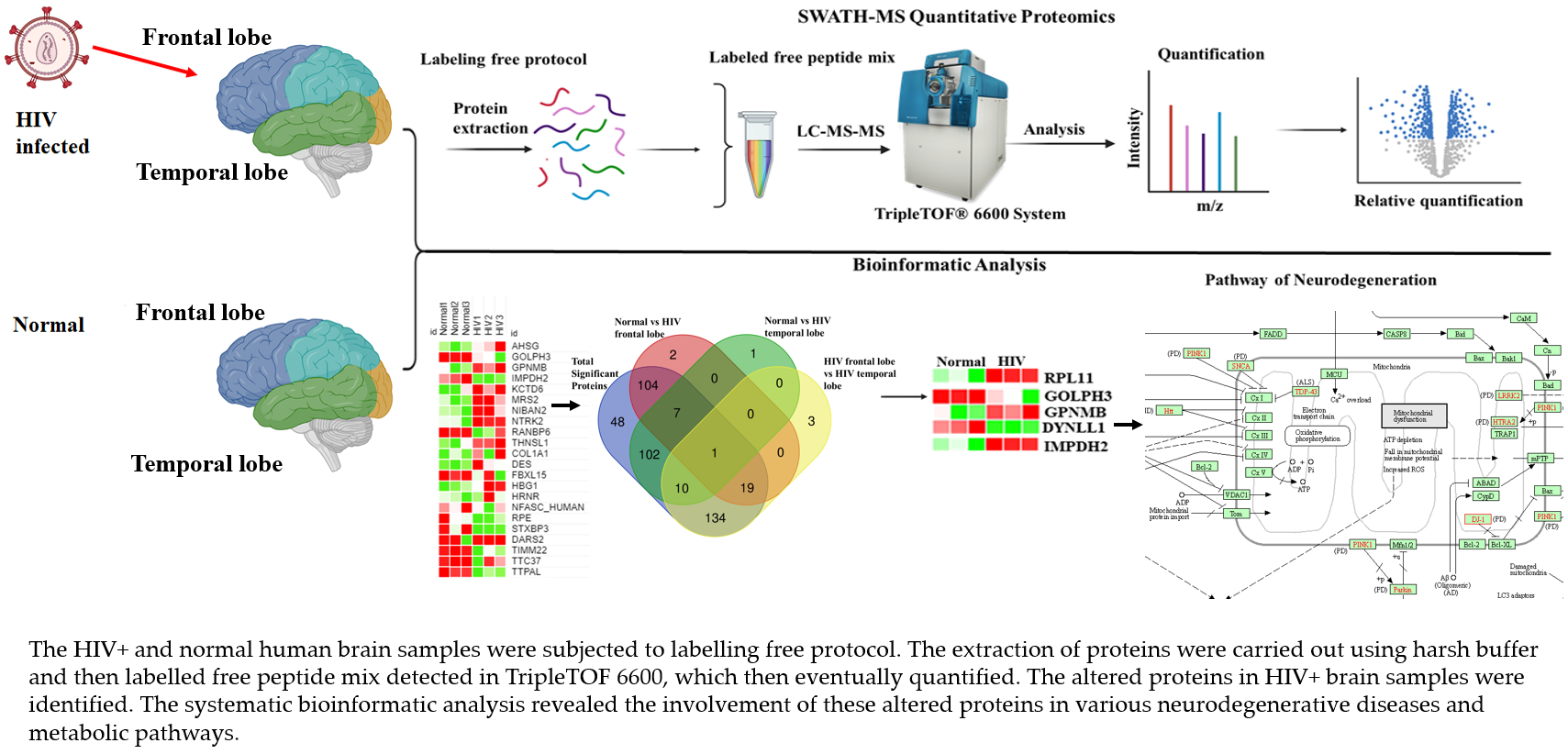
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Brain Samples
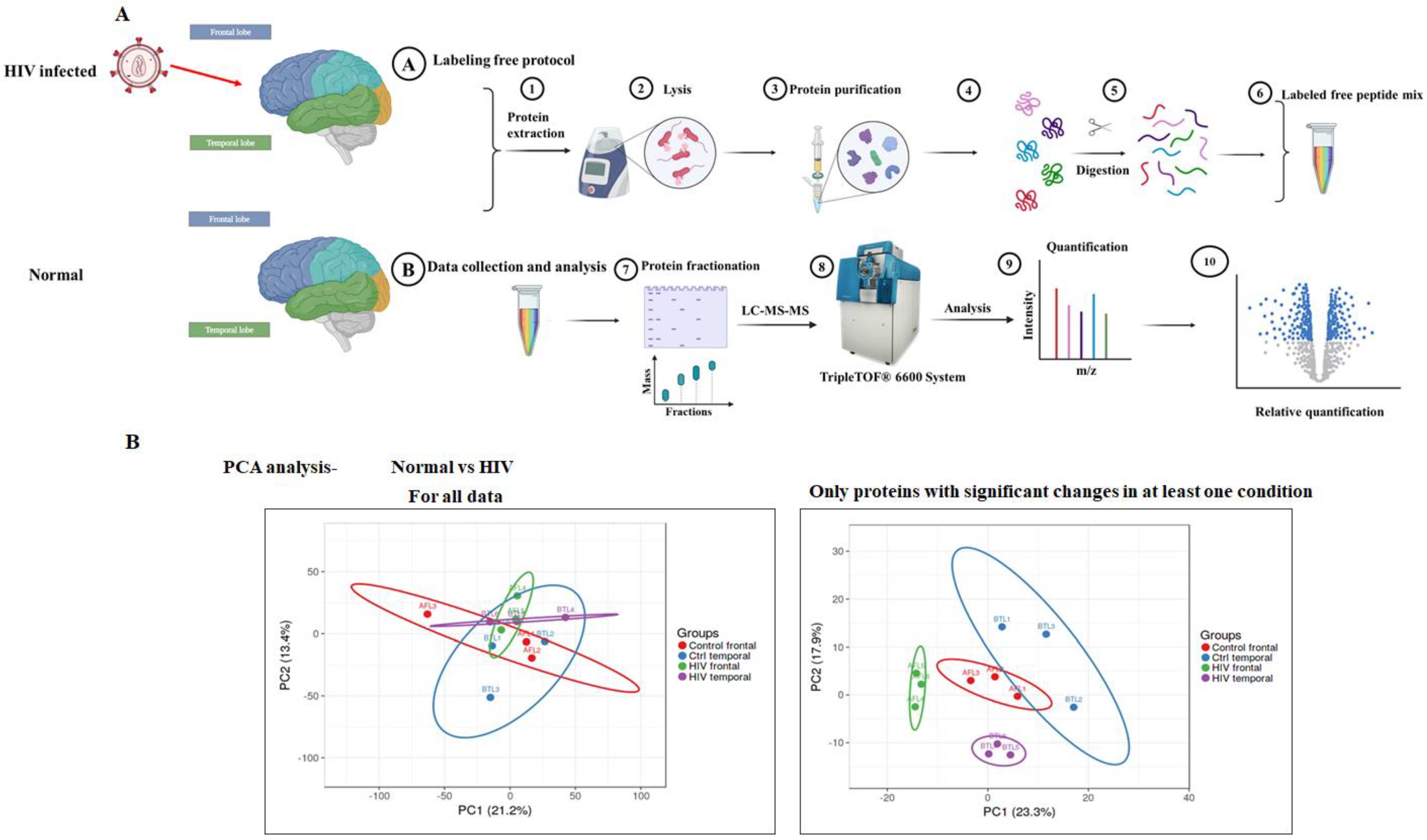
2.2. Sample Preparation
2.3. LC-MS/MS Analysis
2.4. Protein Identification and Quantification
2.5. Analysis of DE Proteins Using a Bioinformatics Approach
2.6. Data Analysis of DE Proteins
2.7. Statistical Analysis
3. Results
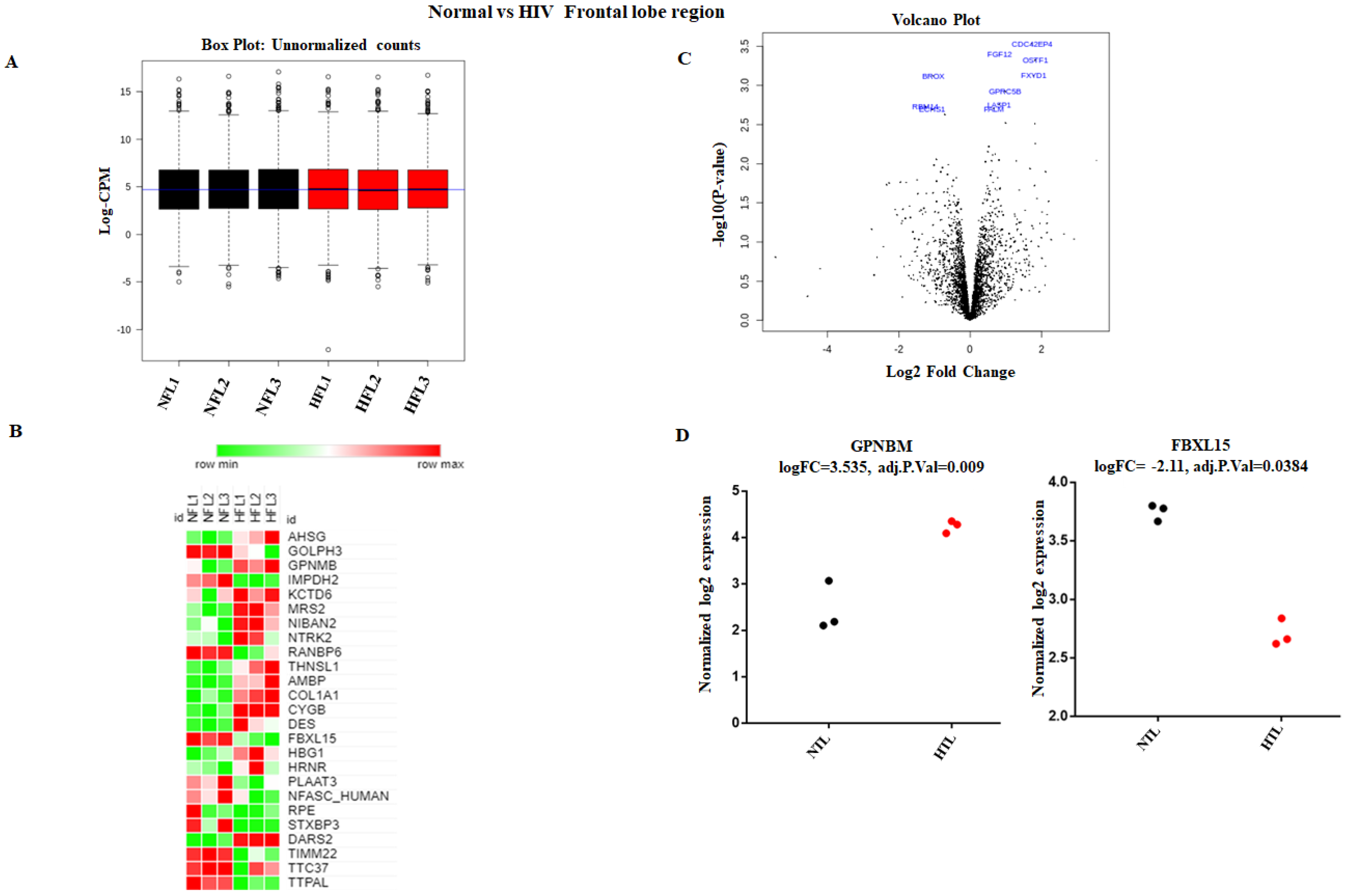
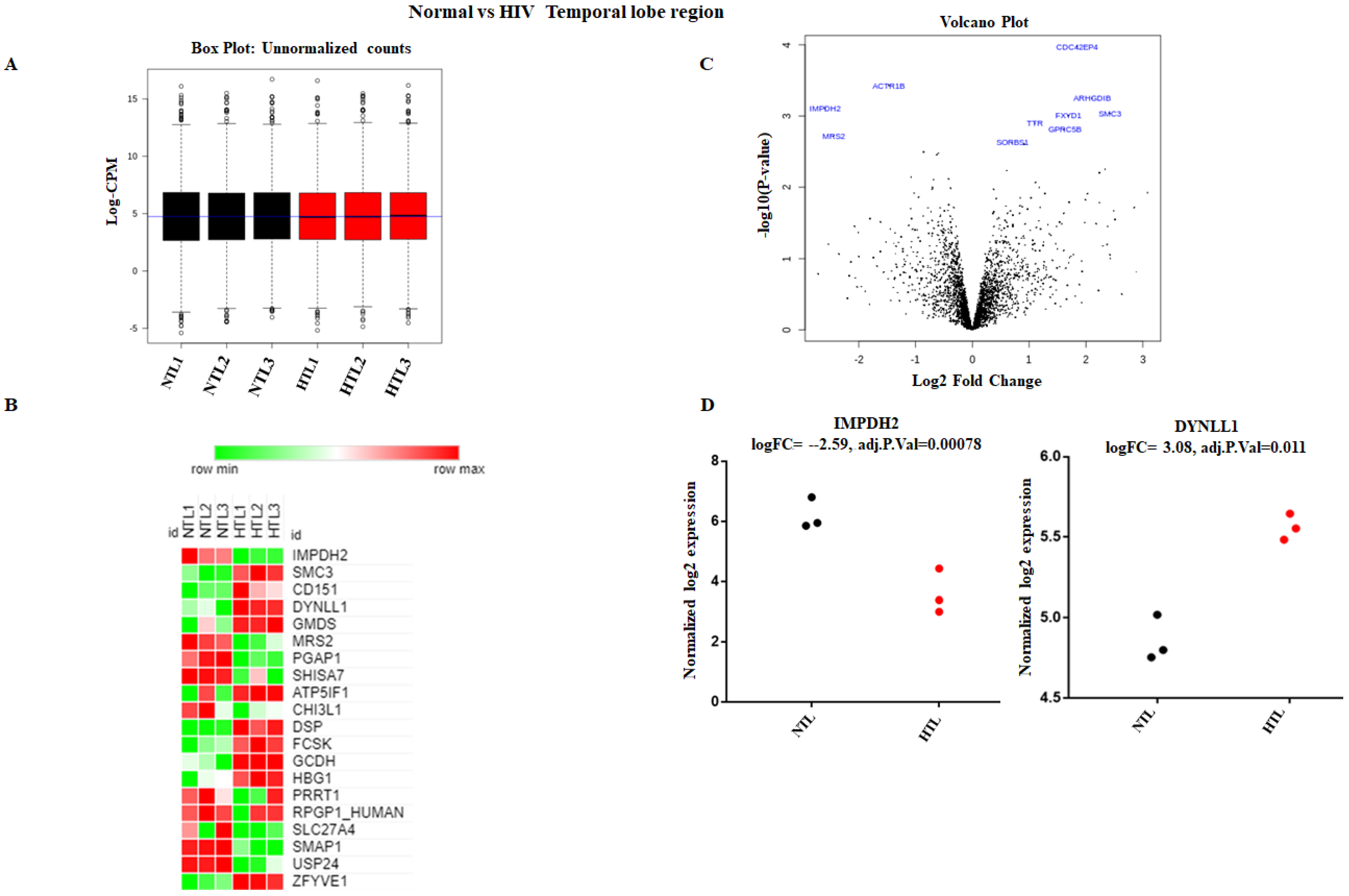
3.1. Strategy for Proteome Analysis of Normal Human Brain and HIV+ Brain Samples
3.2. Accurate Mapping and Quantification of Protein Changes in the Frontal and Temporal Lobes of the Brain
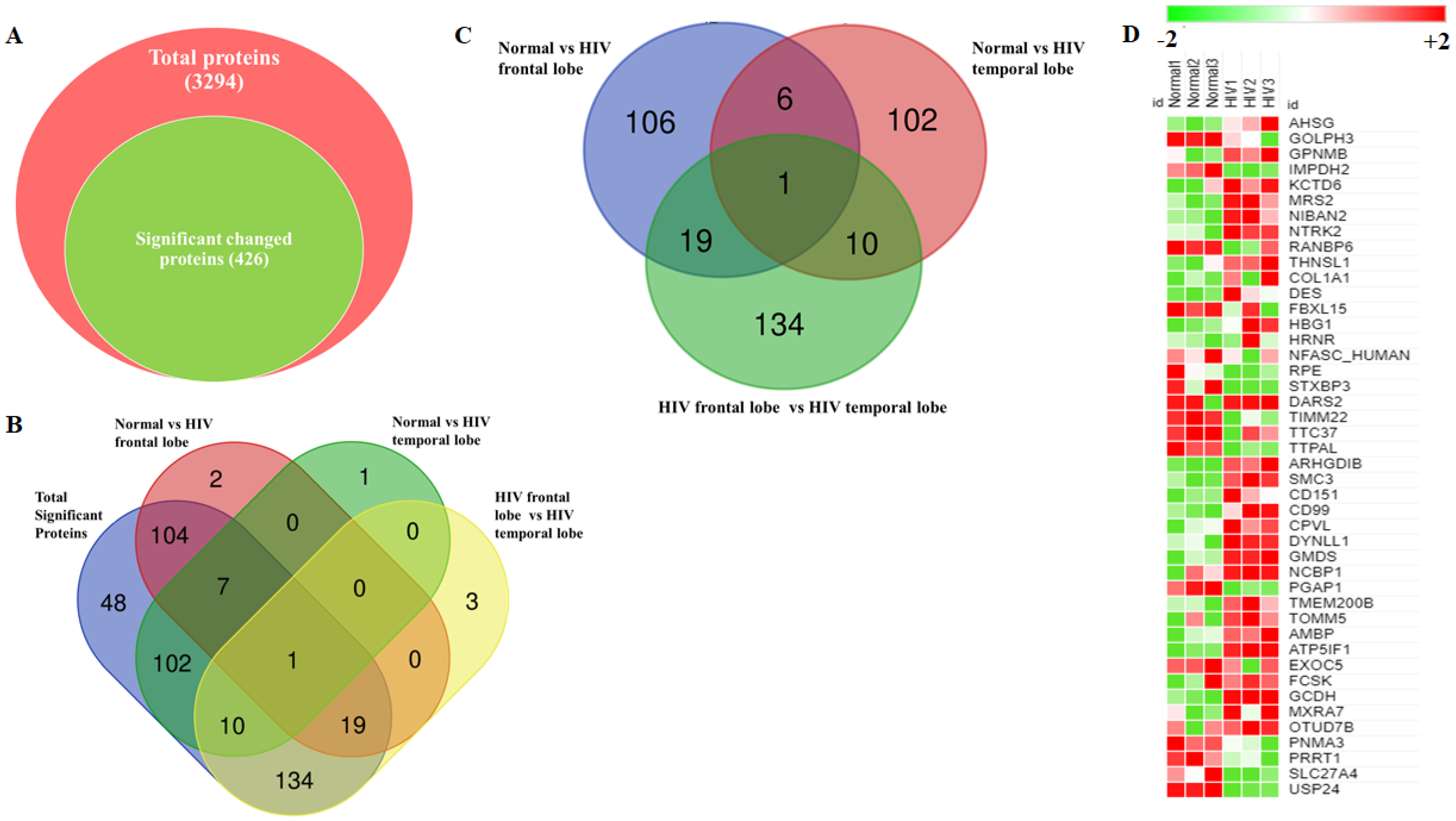
3.3. Identification and Quantitation of Global Proteomes in the Frontal and Temporal Lobes of HIV+ Brains
3.4. Identification of DE Proteins in HIV+ Brains
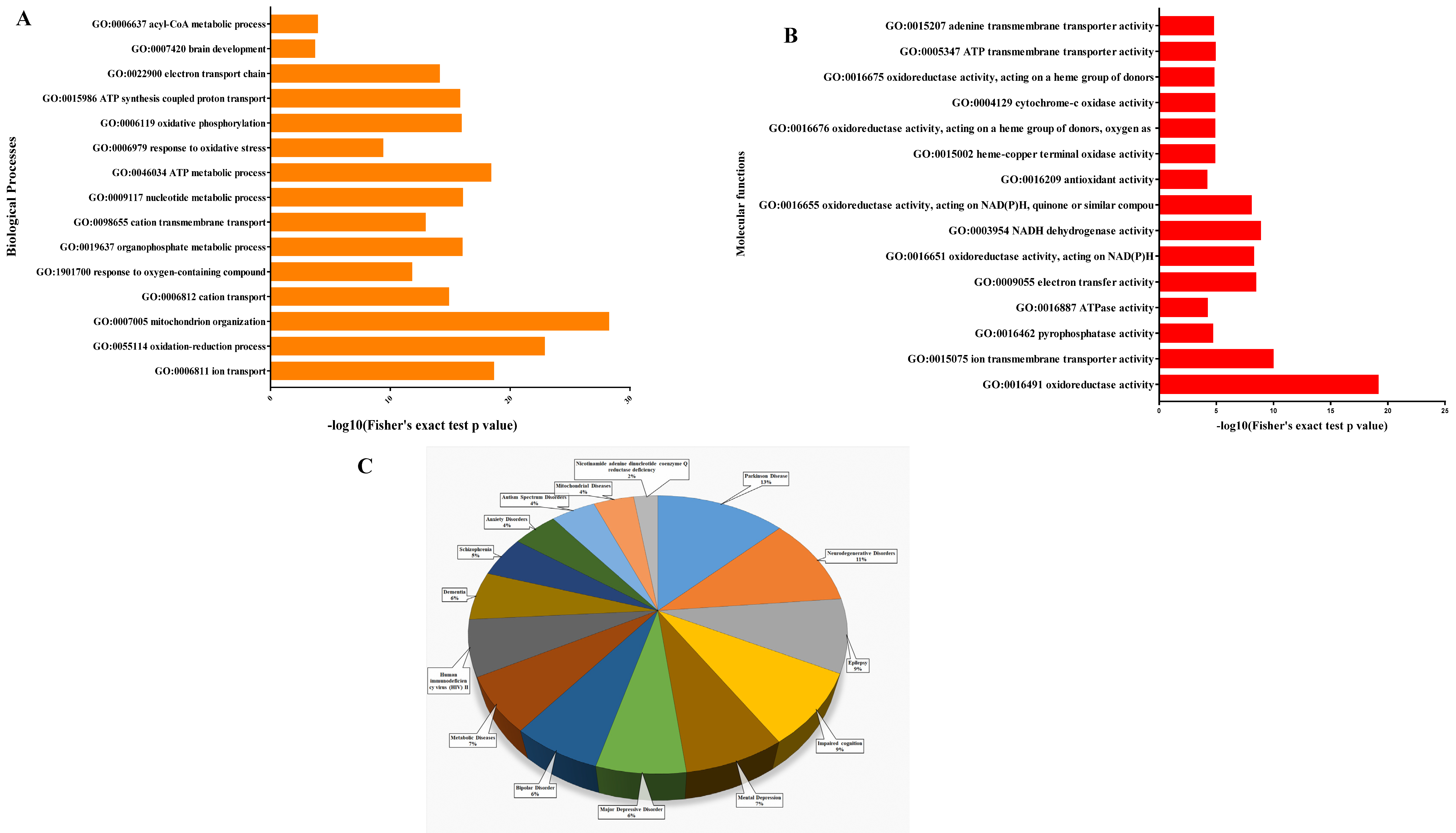
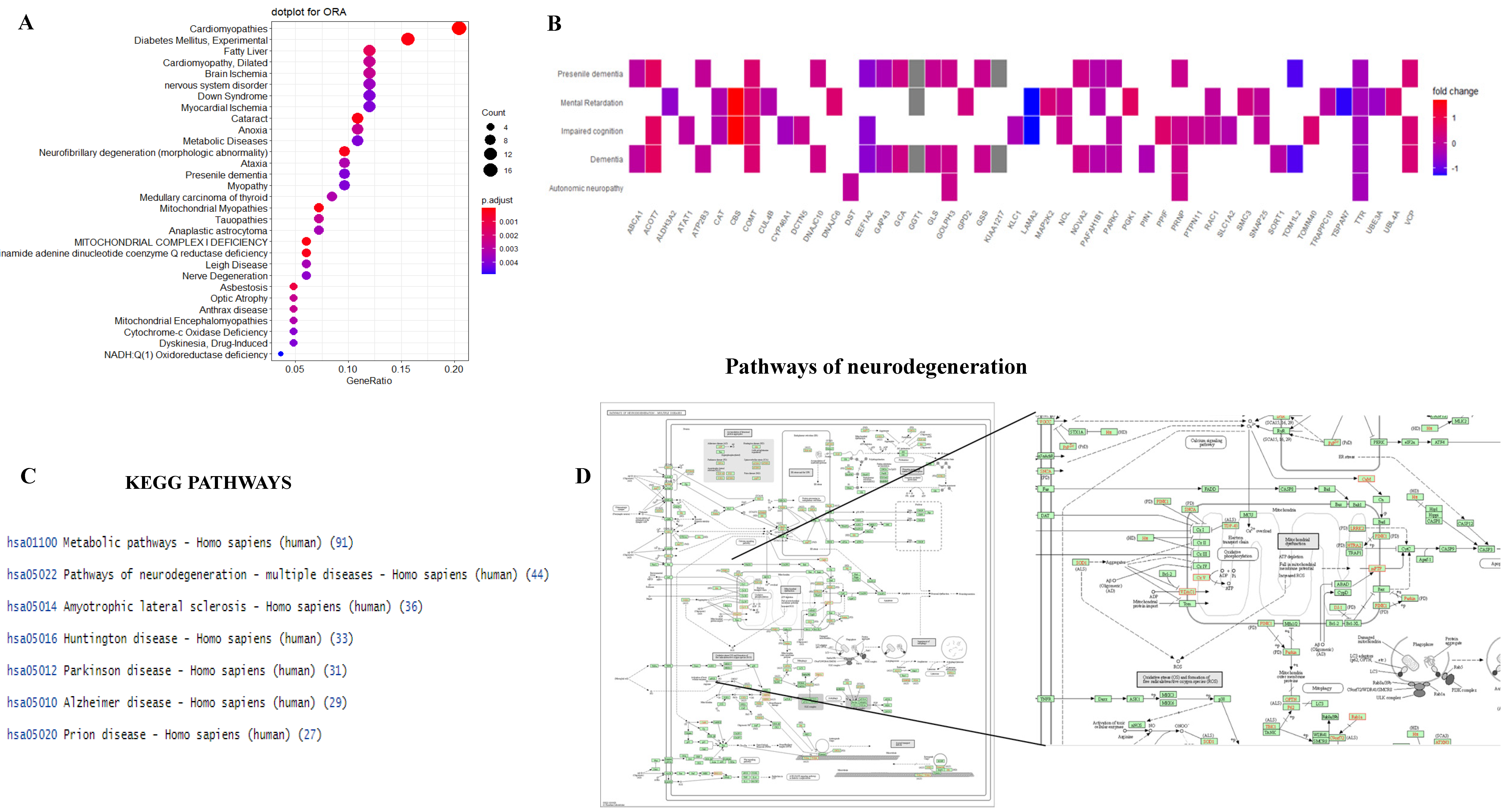
3.5. Functional Analysis of Proteins between HIV+ Brain Samples Compared to Normal Brain Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the brain reservoirs: Toward an HIV cure. Front. Immunol. 2016, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Thangavel, S.; Mulet, C.T.; Atluri, V.S.R.; Agudelo, M.; Rosenberg, R.; Devieux, J.G.; Nair, M.P.N. Oxidative Stress in HIV Infection and Alcohol Use: Role of Redox Signals in Modulation of Lipid Rafts and ATP-Binding Cassette Transporters. Antioxid. Redox Signal. 2018, 28, 324–337. [Google Scholar] [CrossRef]
- Pérez-Matute, P.; Pérez-Martínez, L.; Blanco, J.R.; Oteo, J.A. Role of mitochondria in HIV infection and associated metabolic disorders: Focus on nonalcoholic fatty liver disease and lipodystrophy syndrome. Oxidative Med. Cell. Longev. 2013, 2013, 493413. [Google Scholar] [CrossRef]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the depressed brain: Role of histone acetylation and methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, D. Fundamentals of Biochemistry: Donald Voet: Free Download, Borrow, and Streaming: Internet Archive. Available online: https://archive.org/details/fundamentalsofbi00voet_0 (accessed on 19 November 2020).
- Wu, Y.; Lyon, G.J. NAA10-related syndrome. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Støve, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myklebust, L.M.; Van Damme, P.; Støve, S.I.; Dörfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2014, 24, 1956–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, V.; Lima, A.; Alex Do Nascimento, L.; Eliezer, D.; Follmer, C. Role of Parkinson’s Disease-Linked Mutations and N-Terminal Acetylation on the Oligomerization of α-Synuclein Induced by 3,4-Dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. 2018, 10, 690–703. [Google Scholar] [CrossRef]
- Yu, X.; Gao, L.; Wang, H.; Yin, Z.; Fang, J.; Chen, J.; Li, Q.; Xu, H.; Gui, X. Neuroanatomical changes underlying vertical HIV infection in adolescents. Front. Immunol. 2019, 10, 814. [Google Scholar] [CrossRef] [PubMed]
- Tavano, B.; Tsipouri, V.; Hardy, G.A.D.; Royle, C.M.; Keegan, M.R.; Fuchs, D.; Patterson, S.; Almond, N.; Berry, N.; Ham, C.; et al. Immune responses in the central nervous system are anatomically segregated in a non-human primate model of human immunodeficiency virus infection. Front. Immunol. 2017, 8, 361. [Google Scholar] [CrossRef]
- NNTC NIMH » National NeuroAIDS Tissue Consortium. Available online: https://www.nimh.nih.gov/about/organization/dar/hiv-neuropathogenesis-genetics-and-therapeutics-branch/national-neuroaids-tissue-consortium (accessed on 25 June 2021).
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Aronow, B.J.; Jegga, A.G. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinform. 2009, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Nikolsky, Y.; Bryant, J. Protein networks and pathway analysis. Preface. Methods Mol. Biol. 2009, 563, 3–14. [Google Scholar]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis—Hadley Wickham; Springer: New York, NY, USA, 2009; ISBN 2197-5744. [Google Scholar]
- Yu, G. Using meshes for MeSH term enrichment and semantic analyses. Bioinformatics 2018, 34, 3766–3767. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni-Gosavi, P.; Makhoul, C.; Gleeson, P.A. Form and function of the Golgi apparatus: Scaffolds, cytoskeleton and signalling. FEBS Lett. 2019, 593, 2289–2305. [Google Scholar] [CrossRef] [Green Version]
- Buschman, M.D.; Xing, M.; Field, S.J. The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front. Neurosci. 2015, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Simons, J.P.; Al-Shawi, R.; Minogue, S.; Waugh, M.G.; Wiedemann, C.; Evangelou, S.; Loesch, A.; Sihra, T.S.; King, R.; Warner, T.T.; et al. Loss of phosphatidylinositol 4-kinase 2α activity causes late onset degeneration of spinal cord axons. Proc. Natl. Acad. Sci. USA. 2009, 106, 11535–11539. [Google Scholar] [CrossRef] [Green Version]
- Neuner, S.M.; Wilmott, L.A.; Hoffmann, B.R.; Mozhui, K.; Kaczorowski, C.C. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav. Brain Res. 2017, 322, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Puthiyedth, N.; Riveros, C.; Berretta, R.; Moscato, P. Identification of differentially expressed genes through integrated study of Alzheimer’s disease affected brain regions. PLoS ONE 2016, 11, e0152342. [Google Scholar] [CrossRef] [Green Version]
- Chassefeyre, R.; Martínez-Hernández, J.; Bertaso, F.; Bouquier, N.; Blot, B.; Laporte, M.; Fraboulet, S.; Couté, Y.; Devoy, A.; Isaacs, A.M.; et al. Regulation of postsynaptic function by the dementia-related ESCRT-III subunit CHMP2B. J. Neurosci. 2015, 35, 3155–3173. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, K.B.; Chaparala, S.; Lo, C.W.; Ganapathiraju, M.K. Cilia interactome with predicted protein–protein interactions reveals connections to Alzheimer’s disease, aging and other neuropsychiatric processes. Sci. Rep. 2020, 10, 15629. [Google Scholar] [CrossRef]
- Shigemizu, D.; Mori, T.; Akiyama, S.; Higaki, S.; Watanabe, H.; Sakurai, T.; Niida, S.; Ozaki, K. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimer’s Res. Ther. 2020, 12, 87. [Google Scholar] [CrossRef]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef] [Green Version]
- Smagin, D.A.; Kovalenko, I.L.; Galyamina, A.G.; Bragin, A.O.; Orlov, Y.L.; Kudryavtseva, N.N. Dysfunction in ribosomal gene expression in the hypothalamus and hippocampus following chronic social defeat stress in male mice as revealed by RNA-Seq. Neural Plast. 2016, 2016, 3289187. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.; Kino, Y.; Yanaizu, M.; Ishida, T.; Saito, Y. Microglia express GPNMB in the brains of Alzheimer’s disease and Nasu-Hakola disease. Intractable Rare Dis. Res. 2019, 8, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Hüttenrauch, M.; Ogorek, I.; Klafki, H.; Otto, M.; Stadelmann, C.; Weggen, S.; Wiltfang, J.; Wirths, O. Glycoprotein NMB: A novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aichholzer, F.; Klafki, H.W.; Ogorek, I.; Vogelgsang, J.; Wiltfang, J.; Scherbaum, N.; Weggen, S.; Wirths, O. Evaluation of cerebrospinal fluid glycoprotein NMB (GPNMB) as a potential biomarker for Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 94. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doke, M.; Ramasamy, T.; Sundar, V.; McLaughlin, J.P.; Samikkannu, T. Proteomics Profiling with SWATH-MS Quantitative Analysis of Changes in the Human Brain with HIV Infection Reveals a Differential Impact on the Frontal and Temporal Lobes. Brain Sci. 2021, 11, 1438. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111438
Doke M, Ramasamy T, Sundar V, McLaughlin JP, Samikkannu T. Proteomics Profiling with SWATH-MS Quantitative Analysis of Changes in the Human Brain with HIV Infection Reveals a Differential Impact on the Frontal and Temporal Lobes. Brain Sciences. 2021; 11(11):1438. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111438
Chicago/Turabian StyleDoke, Mayur, Tamizhselvi Ramasamy, Vaishnavi Sundar, Jay P. McLaughlin, and Thangavel Samikkannu. 2021. "Proteomics Profiling with SWATH-MS Quantitative Analysis of Changes in the Human Brain with HIV Infection Reveals a Differential Impact on the Frontal and Temporal Lobes" Brain Sciences 11, no. 11: 1438. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111438