Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview


Abstract
:1. Introduction
2. Mammalian Target of Rapamycin Signaling as a Pharmacologic Target
3. Key Examples of Nanomaterials Used in Drug Products
4. Examples of Side Effects of Nanomaterials
5. Nanoparticles in the Modulation of Mammalian Target of Rapamycin Activity: Challenges in Finding Mechanisms
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef]
- Tukmachev, D.; Lunov, O.; Zablotskii, V.; Dejneka, A.; Babic, M.; Sykova, E.; Kubinova, S. An effective strategy of magnetic stem cell delivery for spinal cord injury therapy. Nanoscale 2015, 7, 3954–3958. [Google Scholar] [CrossRef]
- Schroeder, A.; Heller, D.A.; Winslow, M.M.; Dahlman, J.E.; Pratt, G.W.; Langer, R.; Jacks, T.; Anderson, D.G. Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 2011, 12, 39–50. [Google Scholar] [CrossRef]
- Uzhytchak, M.; Lynnyk, A.; Zablotskii, V.; Dempsey, N.M.; Dias, A.L.; Bonfim, M.; Lunova, M.; Jirsa, M.; Kubinova, S.; Lunov, O.; et al. The use of pulsed magnetic fields to increase the uptake of iron oxide nanoparticles by living cells. Appl. Phys. Lett. 2017, 111, 243703. [Google Scholar] [CrossRef]
- Buzea, C.; Pacheco, I.I.; Robbie, K. Nanomaterials and nanoparticles: Sources and toxicity. Biointerphases 2007, 2, Mr17–Mr71. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Rothenberg, M.L.; Carbone, D.R.; Johnson, D.H. Improving the evaluation of new cancer treatments: Challenges and opportunities. Nat. Rev. Cancer 2003, 3, 303–309. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Teachey, D.T.; Grupp, S.A.; Brown, V.I. Mammalian target of rapamycin inhibitors and their potential role in therapy in leukaemia and other haematological malignancies. Br. J. Haematol. 2009, 145, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Morad, S.A.; Schmid, M.; Buchele, B.; Siehl, H.U.; El Gafaary, M.; Lunov, O.; Syrovets, T.; Simmet, T. A novel semisynthetic inhibitor of the FRB domain of mammalian target of rapamycin blocks proliferation and triggers apoptosis in chemoresistant prostate cancer cells. Mol. Pharmacol. 2013, 83, 531–541. [Google Scholar] [CrossRef]
- Chiu, H.W.; Xia, T.; Lee, Y.H.; Chen, C.W.; Tsai, J.C.; Wang, Y.J. Cationic polystyrene nanospheres induce autophagic cell death through the induction of endoplasmic reticulum stress. Nanoscale 2015, 7, 736–746. [Google Scholar] [CrossRef]
- Loos, C.; Syrovets, T.; Musyanovych, A.; Mailander, V.; Landfester, K.; Simmet, T. Amino-functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials 2014, 35, 1944–1953. [Google Scholar] [CrossRef]
- Khan, M.I.; Mohammad, A.; Patil, G.; Naqvi, S.A.H.; Chauhan, L.K.S.; Ahmad, I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012, 33, 1477–1488. [Google Scholar] [CrossRef]
- Liu, H.L.; Zhang, Y.L.; Yang, N.; Zhang, Y.X.; Liu, X.Q.; Li, C.G.; Zhao, Y.; Wang, Y.G.; Zhang, G.G.; Yang, P.; et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis. 2011, 2, e159. [Google Scholar] [CrossRef]
- Park, K. Facing the truth about nanotechnology in drug delivery. ACS Nano 2013, 7, 7442–7447. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Torrice, M. Does nanomedicine have a delivery problem? ACS Cent. Sci. 2016, 2, 434–437. [Google Scholar] [CrossRef]
- McNeil, S.E. Evaluation of nanomedicines: Stick to the basics. Nat. Rev. Mater. 2016, 1, 16073. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Chan, W.C.W. Reply to “Evaluation of nanomedicines: Stick to the basics”. Nat. Rev. Mater. 2016, 1, 16074. [Google Scholar] [CrossRef]
- Hulea, L.; Markovic, Z.; Topisirovic, I.; Simmet, T.; Trajkovic, V. Biomedical potential of mTOR modulation by nanoparticles. Trends Biotechnol. 2016, 34, 349–353. [Google Scholar] [CrossRef]
- Xia, T.; Kovochich, M.; Liong, M.; Zink, J.I.; Nel, A.E. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano 2008, 2, 85–96. [Google Scholar] [CrossRef]
- Lunova, M.; Prokhorov, A.; Jirsa, M.; Hof, M.; Olzynska, A.; Jurkiewicz, P.; Kubinova, S.; Lunov, O.; Dejneka, A. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Sci. Rep. 2017, 7, 16049. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, L.M.P.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. The proton sponge hypothesis: Fable or fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190. [Google Scholar] [CrossRef]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
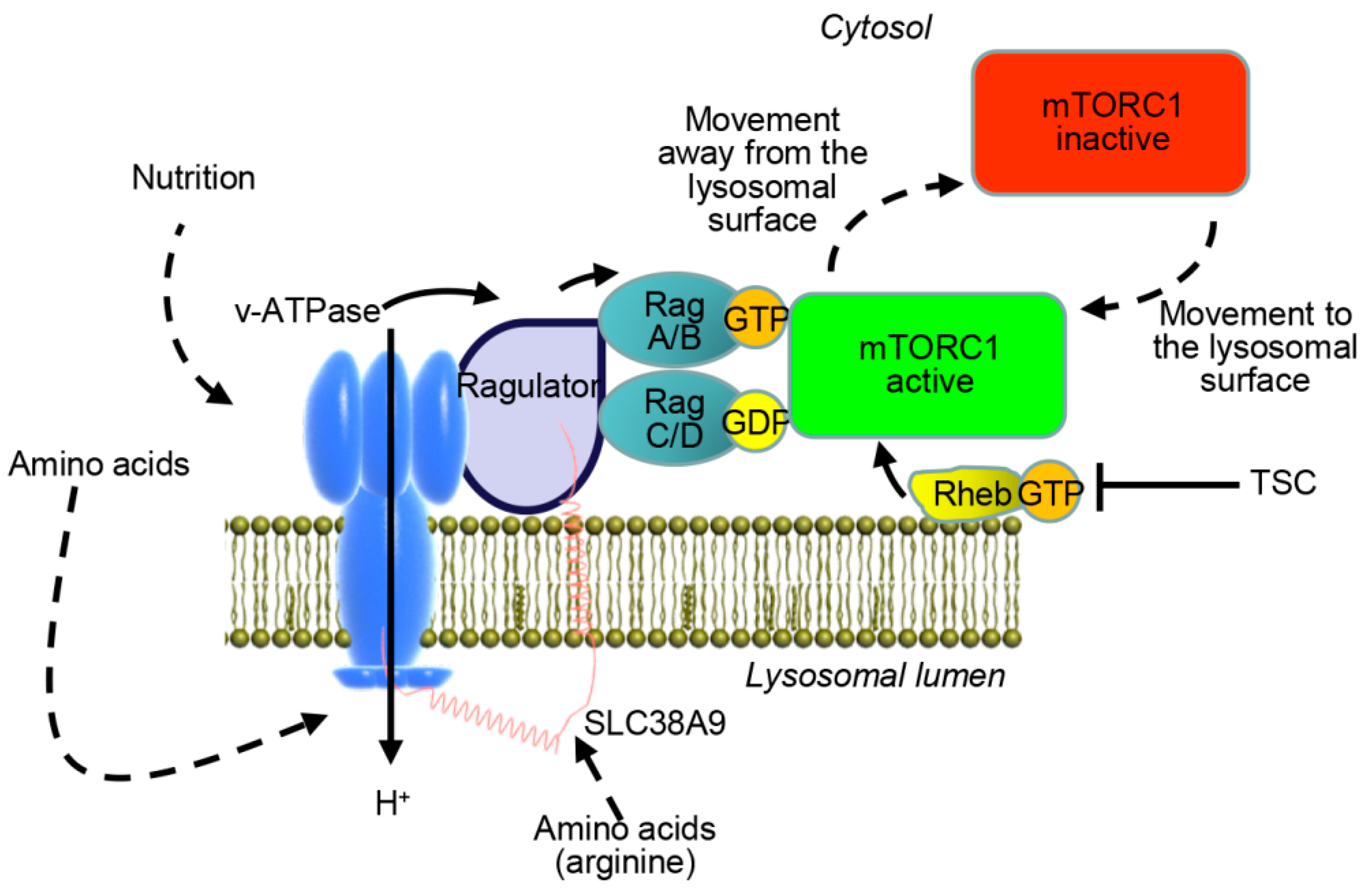
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef]
- Benjamin, D.; Hall, M.N. TSC on the peroxisome controls mTORC1. Nat. Cell Biol. 2013, 15, 1135–1136. [Google Scholar] [CrossRef]
- Zhang, J.W.; Kim, J.; Alexander, A.; Cai, S.L.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit crosstalk in the Rag GTPase heterodimer enables mTORC1 to respond rapidly to amino acid availability. Mol. Cell 2017, 68, 821. [Google Scholar] [CrossRef]
- Jung, J.; Genau, H.M.; Behrends, C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol. Cell. Biol. 2015, 35, 2479–2494. [Google Scholar] [CrossRef]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shimabukuro-Demoto, S.; Yoshida-Sugitani, R.; Furuyama-Tanaka, K.; Karyu, H.; Sugiura, Y.; Shimizu, Y.; Hosaka, T.; Goto, M.; Kato, N.; et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 2014, 41, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Ogmundsdottir, M.H.; Heublein, S.; Kazi, S.; Reynolds, B.; Visvalingam, S.M.; Shaw, M.K.; Goberdhan, D.C. Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS ONE 2012, 7, e36616. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, K.; Liu, P.; Zhang, X.; Dong, X.; Gao, J.; Liu, Q.; Barr, M.P.; Zhang, Q.; Hou, X.; et al. Bafilomycin A1 induces caspase-independent cell death in hepatocellular carcinoma cells via targeting of autophagy and MAPK pathways. Sci. Rep. 2016, 6, 37052. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Newton, P.T.; Vuppalapati, K.K.; Bouderlique, T.; Chagin, A.S. Pharmacological inhibition of lysosomes activates the MTORC1 signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy 2015, 11, 1594–1607. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, K.M.; Colby, A.H.; Zeng, J.; Las, G.; Feng, J.H.; Grinstaff, M.W.; Shirihai, O.S. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J. Cell Biol. 2016, 214, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Azoulay-Alfaguter, I.; Elya, R.; Avrahami, L.; Katz, A.; Eldar-Finkelman, H. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cell growth. Oncogene 2015, 34, 4613–4623. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Rad, E.; Murray, J.T.; Tee, A.R. Oncogenic signalling through mechanistic target of rapamycin (mTOR): A driver of metabolic transformation and cancer progression. Cancers 2018, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, E.; Manning, B.D. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef]
- Meng, L.H.; Zheng, X.F. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Canc. Res. 2010, 29, 44. [Google Scholar] [CrossRef]
- Lozupone, F.; Borghi, M.; Marzoli, F.; Azzarito, T.; Matarrese, P.; Iessi, E.; Venturi, G.; Meschini, S.; Canitano, A.; Bona, R.; et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene 2015, 34, 5163–5174. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Inoki, K.; Guan, K.L. mTOR pathway as a target in tissue hypertrophy. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 443–467. [Google Scholar] [CrossRef]
- Dy, G.K.; Adjei, A.A. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J. Clin. 2013, 63, 249–279. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Santoro, T.; Demartines, N.; Dormond, O. Evolving significance and future relevance of anti-angiogenic activity of mTOR inhibitors in cancer therapy. Cancers 2017, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR cross-talk in cancer and potential for combination therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef]
- D’Mello, S.R.; Cruz, C.N.; Chen, M.L.; Kapoor, M.; Lee, S.L.; Tyner, K.M. The evolving landscape of drug products containing nanomaterials in the United States. Nat. Nanotechnol. 2017, 12, 523–529. [Google Scholar] [CrossRef]
- Thorley, A.J.; Tetley, T.D. New perspectives in nanomedicine. Pharmacol. Ther. 2013, 140, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Pettinger, T.K.; Murdock, N. Nanopharmaceuticals (part 1): Products on the market. Int. J. Nanomed. 2014, 9, 4357–4373. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Hu, S.; Cho, M.H.; Hong, S.H.; Choi, Y.; Choi, H.S. Theranostic nanosystems for targeted cancer therapy. Nano Today 2018, 23, 59–72. [Google Scholar] [CrossRef]
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From bench to bedside and beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zheng, J. Clearance pathways and tumor targeting of imaging nanoparticles. ACS Nano 2015, 9, 6655–6674. [Google Scholar] [CrossRef]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer therapeutics in cancer: Current and future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Luehmann, H.; Xia, X.; Wan, D.; Cutler, C.; Xia, Y. Radioluminescent gold nanocages with controlled radioactivity for real-time in vivo imaging. Nano Lett. 2013, 13, 581–585. [Google Scholar] [CrossRef]
- Zhao, Y.; Sultan, D.; Detering, L.; Cho, S.; Sun, G.; Pierce, R.; Wooley, K.L.; Liu, Y. Copper-64-alloyed gold nanoparticles for cancer imaging: Improved radiolabel stability and diagnostic accuracy. Angew. Chem. Int. Ed. Engl. 2014, 53, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xing, G.; Chai, Z. Nanotoxicology: Are carbon nanotubes safe? Nat. Nanotechnol. 2008, 3, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.J.; Hutchison, G.R.; Christensen, F.M.; Peters, S.; Hankin, S.; Aschberger, K.; Stone, V. A critical review of the biological mechanisms underlying the in vivo and in vitro toxicity of carbon nanotubes: The contribution of physico-chemical characteristics. Nanotoxicology 2010, 4, 207–246. [Google Scholar] [CrossRef]
- Pan, Y.; Neuss, S.; Leifert, A.; Fischler, M.; Wen, F.; Simon, U.; Schmid, G.; Brandau, W.; Jahnen-Dechent, W. Size-dependent cytotoxicity of gold nanoparticles. Small 2007, 3, 1941–1949. [Google Scholar] [CrossRef]
- Malysheva, A.; Lombi, E.; Voelcker, N.H. Bridging the divide between human and environmental nanotoxicology. Nat. Nanotechnol. 2015, 10, 835–844. [Google Scholar] [CrossRef]
- Auffan, M.; Rose, J.; Bottero, J.Y.; Lowry, G.V.; Jolivet, J.P.; Wiesner, M.R. Towards a definition of inorganic nanoparticles from an environmental, health and safety perspective. Nat. Nanotechnol. 2009, 4, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Syrovets, T.; Loos, C.; Nienhaus, G.U.; Mailander, V.; Landfester, K.; Rouis, M.; Simmet, T. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano 2011, 5, 9648–9657. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.F.; Wu, N.; Porter, D.; Buford, M.; Wolfarth, M.; Holian, A. Particle length-dependent titanium dioxide nanomaterials toxicity and bioactivity. Part. Fibre Toxicol. 2009, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.S.; Duffin, R.; Howie, S.E.; Scotton, C.J.; Wallace, W.A.; Macnee, W.; Bradley, M.; Megson, I.L.; Donaldson, K. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn2+ dissolution inside lysosomes. Part. Fibre Toxicol. 2011, 8, 27. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Gonzalez-Fernandez, A.; Sadrieh, N.; Dobrovolskaia, M.A. Nanoparticles and the immune system. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Grabbe, S.; Landfester, K.; Schuppan, D.; Barz, M.; Zentel, R. Nanoparticles and the immune system: Challenges and opportunities. Nanomedicine 2016, 11, 2621–2624. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Zhang, L. Nanoparticle-based modulation of the immune system. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 305–326. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Battaglia, G. Exploiting endocytosis for nanomedicines. Cold Spring Harb. Perspect. Biol. 2013, 5, a016980. [Google Scholar] [CrossRef]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Holst, B.; Raby, A.C.; Hall, J.E.; Labeta, M.O. Complement takes its Toll: An inflammatory crosstalk between Toll-like receptors and the receptors for the complement anaphylatoxin C5a. Anaesthesia 2012, 67, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Farhangrazi, Z.S. Nanomedicine and the complement paradigm. Nanomedicine 2013, 9, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Al-Hanbali, O.; Hunter, A.C.; Rutt, K.J.; Andresen, T.L.; Moghimi, S.M. Distinct polymer architecture mediates switching of complement activation pathways at the nanosphere-serum interface: Implications for stealth nanoparticle engineering. ACS Nano 2010, 4, 6629–6638. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Hunter, A.C.; Moghimi, S.M. Complement monitoring of Pluronic 127 gel and micelles: Suppression of copolymer-mediated complement activation by elevated serum levels of HDL, LDL, and apolipoproteins AI and B-100. J. Control. Release 2013, 170, 167–174. [Google Scholar] [CrossRef]
- Roy, R.; Singh, S.K.; Chauhan, L.K.; Das, M.; Tripathi, A.; Dwivedi, P.D. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol. Lett. 2014, 227, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, H.; Li, Z.; Zhang, T.; Yang, Z. Nano-TiO2 induces autophagy to protect against cell death through antioxidative mechanism in podocytes. Cell Biol. Toxicol. 2016, 32, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Juan, J.; Cheng, L.; Shi, M.; Liu, Z.; Mao, X. Poly-(allylamine hydrochloride)-coated but not poly(acrylic acid)-coated upconversion nanoparticles induce autophagy and apoptosis in human blood cancer cells. J. Mater. Chem. B 2015, 3, 5769–5776. [Google Scholar] [CrossRef]
- Xue, X.; Wang, L.R.; Sato, Y.; Jiang, Y.; Berg, M.; Yang, D.S.; Nixon, R.A.; Liang, X.J. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano Lett. 2014, 14, 5110–5117. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, Y.; Lu, K.; Yang, M.; Li, Y.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int. J. Nanomed. 2017, 12, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, H.; Sun, Y.; Wang, H.; Guo, F.; Rao, S.; Deng, J.; Zhang, Y.; Miao, Y.; Guo, C.; et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J. Mol. Cell Biol. 2009, 1, 37–45. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, P.; He, X.; Wu, B.; Liu, Q.; Xu, Z.; Wu, H.; Liu, Z.; Qian, Y.; Wang, S.; et al. Etoposide loaded layered double hydroxide nanoparticles reversing chemoresistance and eradicating human glioma stem cells in vitro and in vivo. Nanoscale 2018, 10, 13106–13121. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, H.; Zhang, X.; Wang, Y.; Song, Z.; Zhao, J.; Shi, H.; Li, R.; Wang, Y.; Zhang, L.W. The protective role of autophagy in nephrotoxicity induced by bismuth nanoparticles through AMPK/mTOR pathway. Nanotoxicology 2018, 12, 586–601. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Wei, F.; Luo, Z.; Duan, Y. Combining autophagy-inducing peptides and brefeldin A delivered by perinuclear-localized mesoporous silica nanoparticles: A manipulation strategy for ER-phagy. Nanoscale 2018, 10, 8796–8805. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Zhang, C.; Cui, X.; Zhai, S.; Liu, Y.; Li, C.; Zhu, H.; Qu, G.; Jiang, G.; et al. Tuning cell autophagy by diversifying carbon nanotube surface chemistry. ACS Nano 2014, 8, 2087–2099. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Shack, L.A.; Naske, C.D.; Walters, K.B.; Nanduri, B. SILAC-based quantitative proteomic analysis of human lung cell response to copper oxide nanoparticles. PLoS ONE 2014, 9, e114390. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, N.; Wei, Y.; Li, J.; Sun, L.; Wu, J.; Huang, Q.; Liu, C.; Fan, C.; Song, H. Akt signaling-associated metabolic effects of dietary gold nanoparticles in Drosophila. Sci. Rep. 2012, 2, 563. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Li, S.; Yang, Z.; Zheng, W.; Le, W. Gold nanoparticles enhance the differentiation of embryonic stem cells into dopaminergic neurons via mTOR/p70S6K pathway. Nanomedicine 2017, 12, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Yang, Z.; Gao, Y.J.; Yuan, H.; Cui, K.; Shi, Y.; Wang, H.; Huang, X.; Wong, S.T.; Wang, Y.; et al. Treating triple-negative breast cancer by a combination of rapamycin and cyclophosphamide: An in vivo bioluminescence imaging study. Eur. J. Cancer 2010, 46, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Onyesom, I.; Lamprou, D.A.; Sygellou, L.; Owusu-Ware, S.K.; Antonijevic, M.; Chowdhry, B.Z.; Douroumis, D. Sirolimus encapsulated liposomes for cancer therapy: Physicochemical and mechanical characterization of sirolimus distribution within liposome bilayers. Mol. Pharmacol. 2013, 10, 4281–4293. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Frechet, J.M. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Wang, Z.; Qian, X.; Fan, J.; Zeng, X.; Sun, Y.; Song, P.; Feng, M.; Ju, D. Cationic poly(amidoamine) dendrimers induced cyto-protective autophagy in hepatocellular carcinoma cells. Nanotechnology 2014, 25, 365101. [Google Scholar] [CrossRef]
- Cirstea, D.; Hideshima, T.; Rodig, S.; Santo, L.; Pozzi, S.; Vallet, S.; Ikeda, H.; Perrone, G.; Gorgun, G.; Patel, K.; et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol. Cancer Ther. 2010, 9, 963–975. [Google Scholar] [CrossRef]
- Duan, J.; Yu, Y.; Yu, Y.; Li, Y.; Wang, J.; Geng, W.; Jiang, L.; Li, Q.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int. J. Nanomed. 2014, 9, 5131–5141. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, L.; Liu, J.; Liu, A.; Jia, C.; Ma, D.; Jiang, Y.; Bai, X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 2010, 22, 1469–1476. [Google Scholar] [CrossRef]
- Docter, D.; Westmeier, D.; Markiewicz, M.; Stolte, S.; Knauer, S.K.; Stauber, R.H. The nanoparticle biomolecule corona: Lessons learned—Challenge accepted? Chem. Soc. Rev. 2015, 44, 6094–6121. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, F.; Garry, D.; Monopoli, M.P.; Salvati, A.; Dawson, K.A. The intracellular destiny of the protein corona: A study on its cellular internalization and evolution. ACS Nano 2016, 10, 10471–10479. [Google Scholar] [CrossRef]
- Ke, P.C.; Lin, S.; Parak, W.J.; Davis, T.P.; Caruso, F. A decade of the protein corona. ACS Nano 2017, 11, 11773–11776. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Aberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Aberg, C.; Dawson, K.A.; Salvati, A. Effects of the presence or absence of a protein corona on silica nanoparticle uptake and impact on cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.J.; Liang, M.; Monteiro, M.; Toth, I.; Minchin, R.F. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat. Nanotechnol. 2011, 6, 39–44. [Google Scholar] [CrossRef]
- Fleischer, C.C.; Payne, C.K. Nanoparticle-cell interactions: Molecular structure of the protein corona and cellular outcomes. Acc. Chem. Res. 2014, 47, 2651–2659. [Google Scholar] [CrossRef]
- Wan, S.; Kelly, P.M.; Mahon, E.; Stöckmann, H.; Rudd, P.M.; Caruso, F.; Dawson, K.A.; Yan, Y.; Monopoli, M.P. The “sweet” side of the protein corona: Effects of glycosylation on nanoparticle–cell interactions. ACS Nano 2015, 9, 2157–2166. [Google Scholar] [CrossRef]
- Yan, Y.; Gause, K.T.; Kamphuis, M.M.; Ang, C.S.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Reynolds, E.C.; Nice, E.C.; Caruso, F. Differential roles of the protein corona in the cellular uptake of nanoporous polymer particles by monocyte and macrophage cell lines. ACS Nano 2013, 7, 10960–10970. [Google Scholar] [CrossRef]
- Shang, L.; Wang, Y.; Jiang, J.; Dong, S. pH-dependent protein conformational changes in albumin:gold nanoparticle bioconjugates: A spectroscopic study. Langmuir 2007, 23, 2714–2721. [Google Scholar] [CrossRef]
- Shang, W.; Nuffer, J.H.; Muniz-Papandrea, V.A.; Colon, W.; Siegel, R.W.; Dordick, J.S. Cytochrome C on silica nanoparticles: Influence of nanoparticle size on protein structure, stability, and activity. Small 2009, 5, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Nuffer, J.H.; Dordick, J.S.; Siegel, R.W. Unfolding of ribonuclease A on silica nanoparticle surfaces. Nano Lett. 2007, 7, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Cabaleiro-Lago, C.; Xue, W.F.; Lynch, I.; Lindman, S.; Thulin, E.; Radford, S.E.; Dawson, K.A. Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 8691–8696. [Google Scholar] [CrossRef] [Green Version]
- Mirshafiee, V.; Kim, R.; Park, S.; Mahmoudi, M.; Kraft, M.L. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials 2016, 75, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine 2016, 11, 81–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunov, O.; Syrovets, T.; Loos, C.; Beil, J.; Delecher, M.; Tron, K.; Nienhaus, G.U.; Musyanovych, A.; Mailander, V.; Landfester, K.; et al. Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano 2011, 5, 1657–1669. [Google Scholar] [CrossRef]
- Lee, Y.K.; Choi, E.J.; Webster, T.J.; Kim, S.H.; Khang, D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int. J. Nanomed. 2015, 10, 97–113. [Google Scholar]
- Juling, S.; Niedzwiecka, A.; Bohmert, L.; Lichtenstein, D.; Selve, S.; Braeuning, A.; Thunemann, A.F.; Krause, E.; Lampen, A. Protein corona analysis of silver nanoparticles links to their cellular effects. J. Proteome Res. 2017, 16, 4020–4034. [Google Scholar] [CrossRef]
- Yang, M.H.; Yuan, S.S.; Huang, Y.F.; Lin, P.C.; Lu, C.Y.; Chung, T.W.; Tyan, Y.C. A proteomic view to characterize the effect of chitosan nanoparticle to hepatic cells: Is chitosan nanoparticle an enhancer of PI3K/AKT1/mTOR pathway? Biomed. Res. Int. 2014, 2014, 789591. [Google Scholar] [CrossRef]
- Ma, X.; Hartmann, R.; Jimenez de Aberasturi, D.; Yang, F.; Soenen, S.J.H.; Manshian, B.B.; Franz, J.; Valdeperez, D.; Pelaz, B.; Feliu, N.; et al. Colloidal gold nanoparticles induce changes in cellular and subcellular morphology. ACS Nano 2017, 11, 7807–7820. [Google Scholar] [CrossRef]
- Yang, J.A.; Lohse, S.E.; Murphy, C.J. Tuning cellular response to nanoparticles via surface chemistry and aggregation. Small 2014, 10, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.P.; Kikuchi-Uehara, E.; Sonnemann, G.W.; Aymonier, C.; Hirao, M. Evaluating nanotechnology opportunities and risks through integration of life-cycle and risk assessment. Nat. Nanotechnol. 2017, 12, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Poland, C.A. Nanotoxicity: Challenging the myth of nano-specific toxicity. Curr. Opin. Biotechnol. 2013, 24, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell 2017, 67, 922–935. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Nanomaterials | Size Range | Clinical Indications |
|---|---|---|
| Polymer-based NPs | 5 nm–5 µm | Severe combined immunodeficiency disease (SCID) |
| Crohn’s disease | ||
| Rheumatoid arthritis | ||
| Psoriatic Arthritis | ||
| Ankylosing Spondylitis | ||
| Multiple Sclerosis (MS) | ||
| Prostate Cancer | ||
| Hepatitis B; Hepatitis C | ||
| Acute lymphoblastic leukemia | ||
| Chronic gout | ||
| Hemophilia | ||
| Liposome formulations | ≈100 nm | Pancreatic Cancer |
| Fungal/protozoal infections | ||
| Breast cancer | ||
| Cutaneous T-Cell lymphoma | ||
| Acute lymphoblastic leukemia | ||
| Kaposi’s Sarcoma | ||
| Ovarian cancer | ||
| Fungal infections | ||
| Micellar NPs | 10–200 nm | Antifungal |
| Menopausal therapy | ||
| Antineoplastic | ||
| Aneasthesia | ||
| Immunosuppressant | ||
| Anti-HIV | ||
| Protein NPs | 50–500 nm | Breast cancer |
| Pancreatic cancer | ||
| Cutaneous T-Cell lymphoma | ||
| Nanocrystals | 50–1000 nm | Antiemetic |
| Hyperlipidemia | ||
| Immunosuppressant | ||
| Anti-anorexic | ||
| Psychostimulant | ||
| Muscle relaxant | ||
| Inorganic and metallic NPs | 10–200 nm | Glioblastoma |
| Iron deficiency in chronic kidney disease | ||
| Iron deficiency in patients undergoing chronic hemodialysis | ||
| Iron deficiency anemia |
| NPs | Charge/Surface Modification | Size (nm) | Zeta Potential (mV) | Activity of mTOR | Ref. |
|---|---|---|---|---|---|
| PS 1 | Positive/NH2 | 62 nm | +34.97 in dH2O −12.33 in DMEM | inhibited | [22] |
| PS 1 | Positive/NH2 | 117 ± 17 nm | +54.4 in PBS | inhibited | [23] |
| Iron oxide | Negative/N.A. 2 | 51 nm | −39.3 in dH2O | inhibited | [24] |
| Zinc oxide | N.A. 2/N.A. 2 | N.A.2 | N.A. 2 | inhibited | [111] |
| PS 1 | Positive/NH2 | 30.6 ± 6.1 nm | +39.1 ± 6.5 in PBS | inhibited | [33] |
| nano-TiO2 | N.A. 2/N.A. 2 | 21 nm | N.A. 2 | inhibited | [112] |
| UCNP Upconversion NPs | Positive/poly-(allylamine hydrochloride) (PAH) | 110 nm | +35 in PBS | inhibited | [113] |
| SWCNT functionalized single-walled carbon nanotube | N.A. 2/COOH | N.A. 2 | N.A. 2 | inhibited | [25] |
| SWCNT functionalized single-walled carbon nanotube | N.A. 2/N.A. 2 | N.A. 2 | N.A. 2 | inhibited | [114] |
| Silica | N.A. 2/N.A. 2 | 62.1 ± 7.2 nm | −40 in dH2O | inhibited | [115] |
| PAMAM polyamidoamine dendrimers | N.A. 2/N.A. 2 | N.A. 2 | N.A. 2 | inhibited | [116] |
| Layered double hydroxide (LDH) NPs LDH-VP16 nanocomposites | Positive/Etoposide (VP16) | 105 nm | +39.9 in PBS | inhibited | [117] |
| Bismuth NPs (BiNP) | Negative/N.A. 2 | 63.72 nm in water 52.46 nm and 52.92 nm in PBS and DMEM | −27.43 ± 0.39 in dH2O −10.71 ± 0.53 in PBS −11.38 ± 0.5 in DMEM | inhibited | [118] |
| Mesoporous silica NPs (MSNs) | N.A. 2/BFA (Brefeldin A) | 72 nm | N.A. | inhibited | [119] |
| Multiwalled carbon nanotubes (MWCNTs) | Negative/COOH | ≈30–50 (outer), ≈5–12 (inner) | −30.5 ± 74.2 in ultrapure dH2O | inhibited | [120] |
| Silica | Positive/NH2 | 28.6 ± 4.2 nm | +36.9 ± 8.2 in PBS | activated | [33] |
| Silica | Negative/OH | 31.2 ± 5.5 nm | −40.3 ± 7.4 in PBS | activated | [33] |
| Copper Oxide | Negative/N.A. 2 | 56.2 ± 22.9 nm in media 85.6 ± 27.2 nm in water | −0.057 in dH2O | activated | [121] |
| Gold NPs | N.A. 2/N.A. 2 | 2 nm | N.A. 2 | activated | [122] |
| Gold NPs | N.A. 2/N.A. 2 | 30 nm | N.A. 2 | activated | [123] |
| PS | Negative/COOH | 119 ± 19 nm | −36.2 in PBS | activated | [23] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunova, M.; Smolková, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinová, Š.; Dejneka, A.; Lunov, O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers 2019, 11, 82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010082
Lunova M, Smolková B, Lynnyk A, Uzhytchak M, Jirsa M, Kubinová Š, Dejneka A, Lunov O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers. 2019; 11(1):82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010082
Chicago/Turabian StyleLunova, Mariia, Barbora Smolková, Anna Lynnyk, Mariia Uzhytchak, Milan Jirsa, Šárka Kubinová, Alexandr Dejneka, and Oleg Lunov. 2019. "Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview" Cancers 11, no. 1: 82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010082