Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
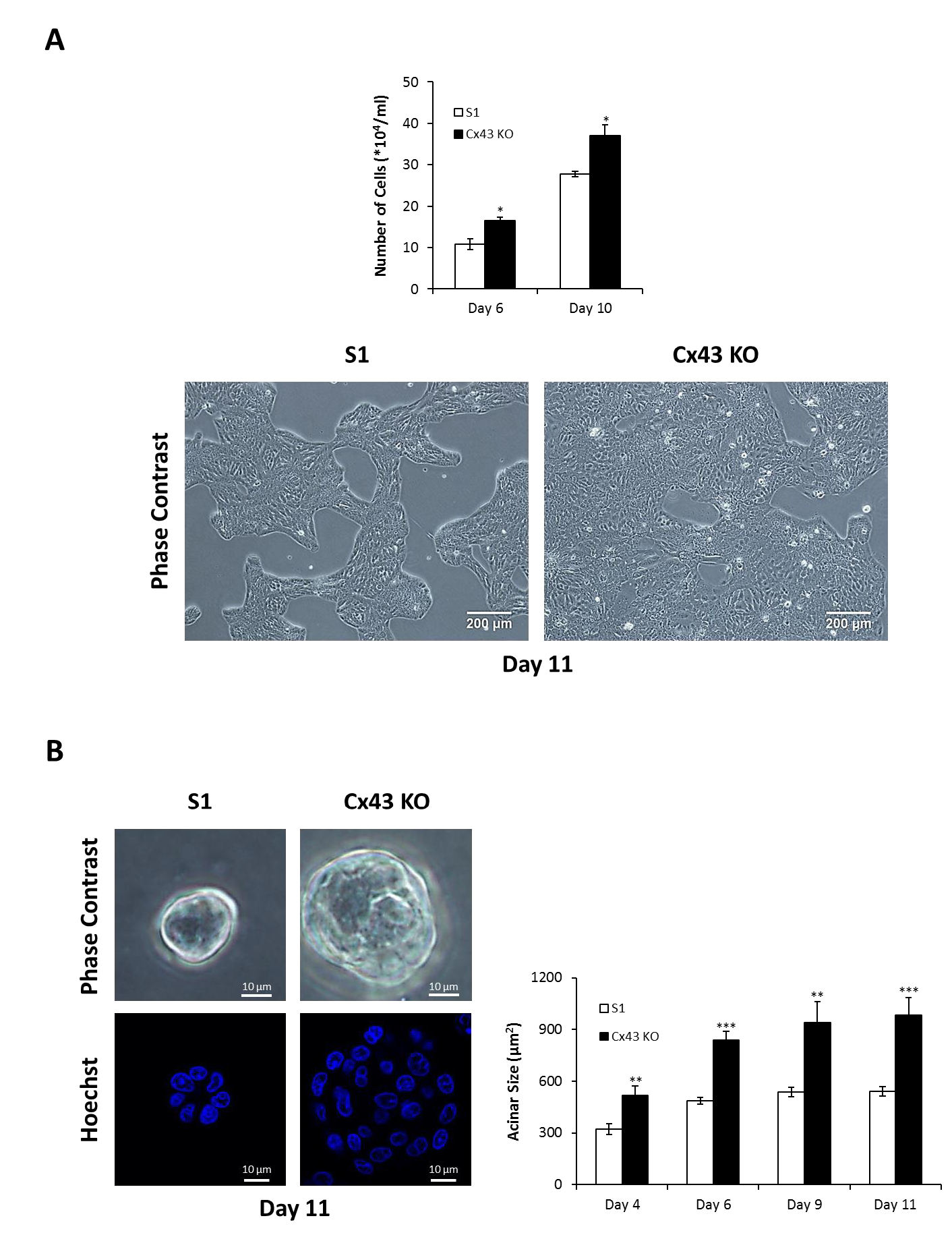
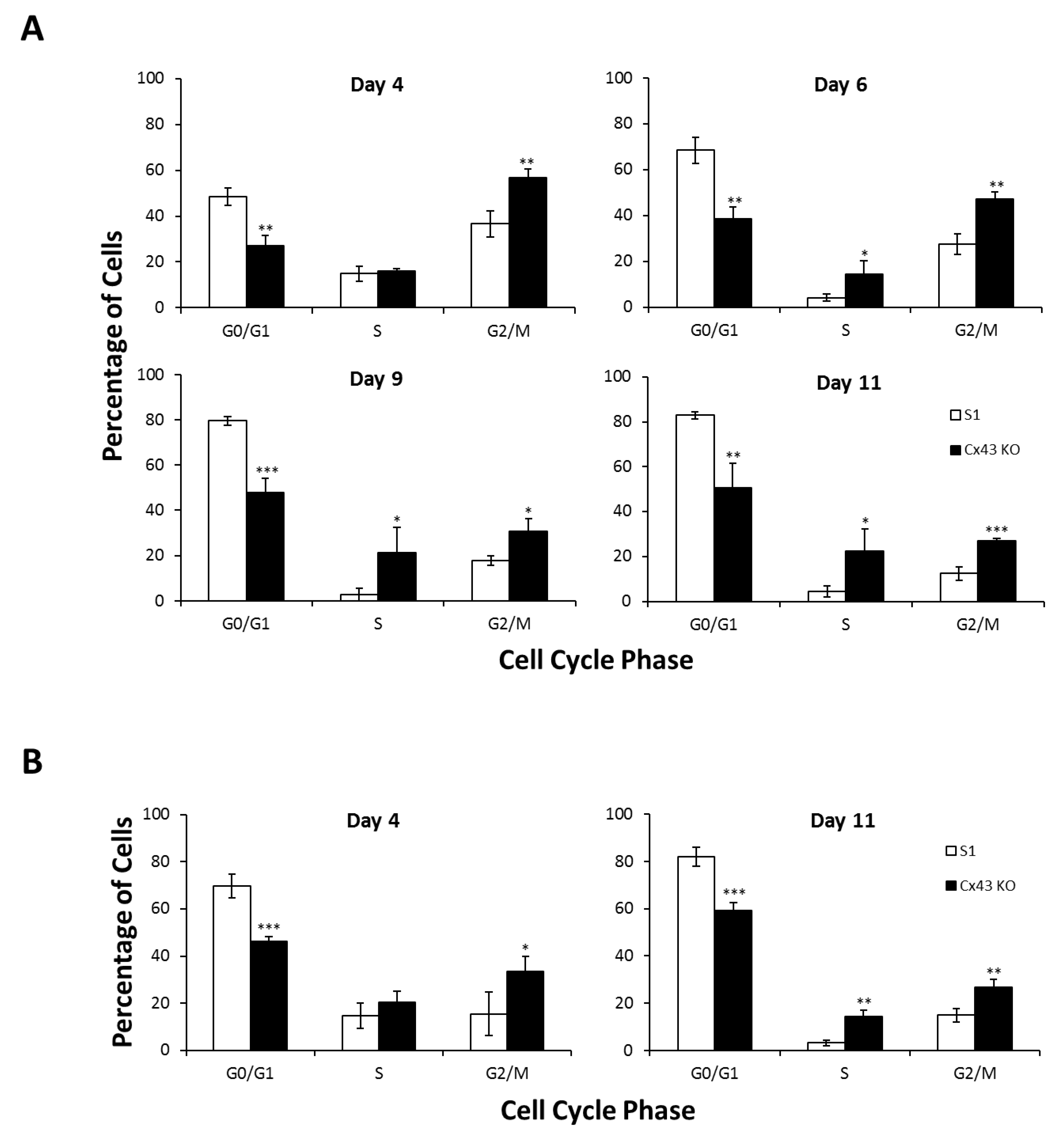
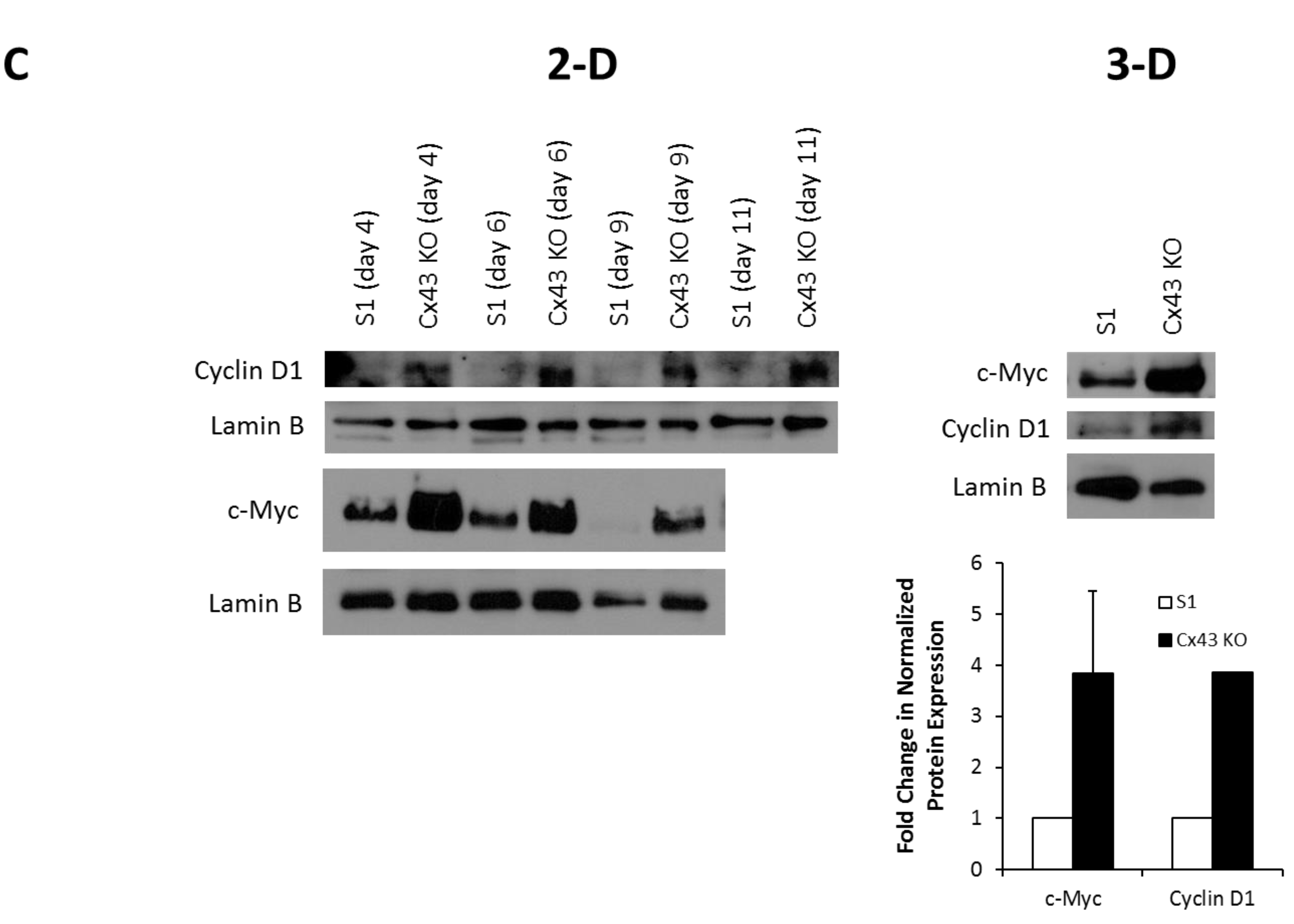
2.1. Silencing Cx43 Triggers Cell Cycle Entry and Enhances the Proliferation Rate
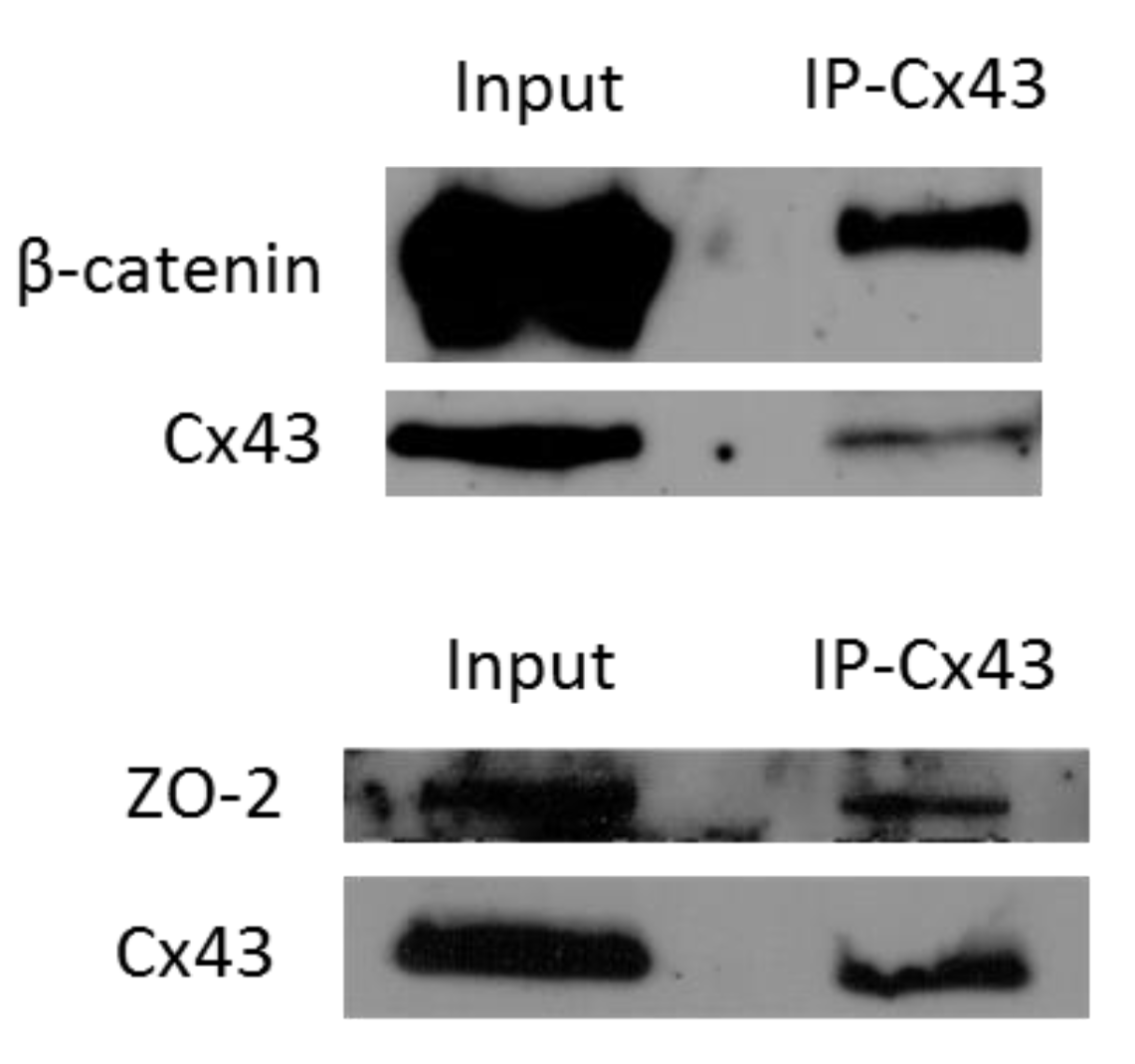
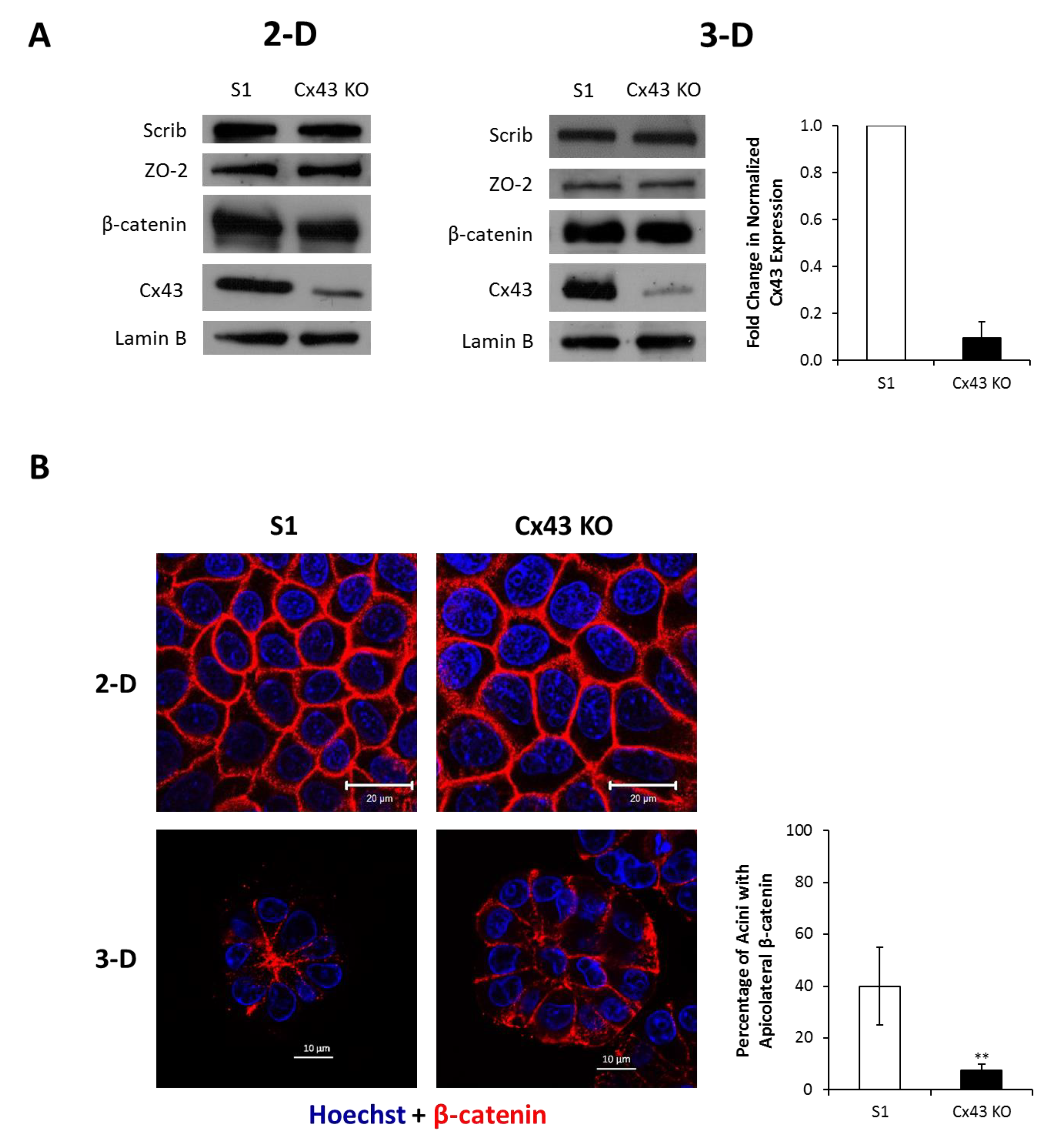
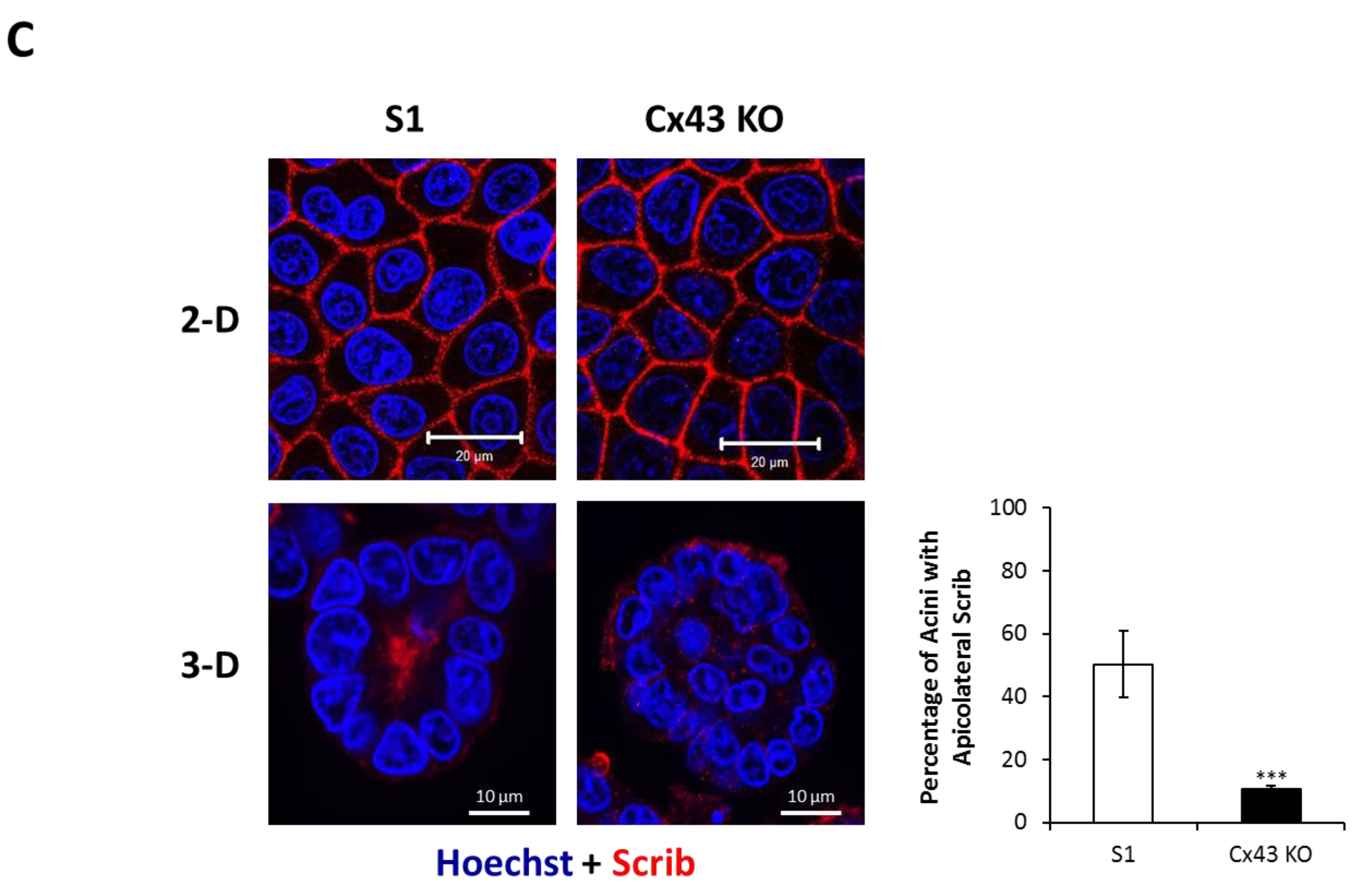
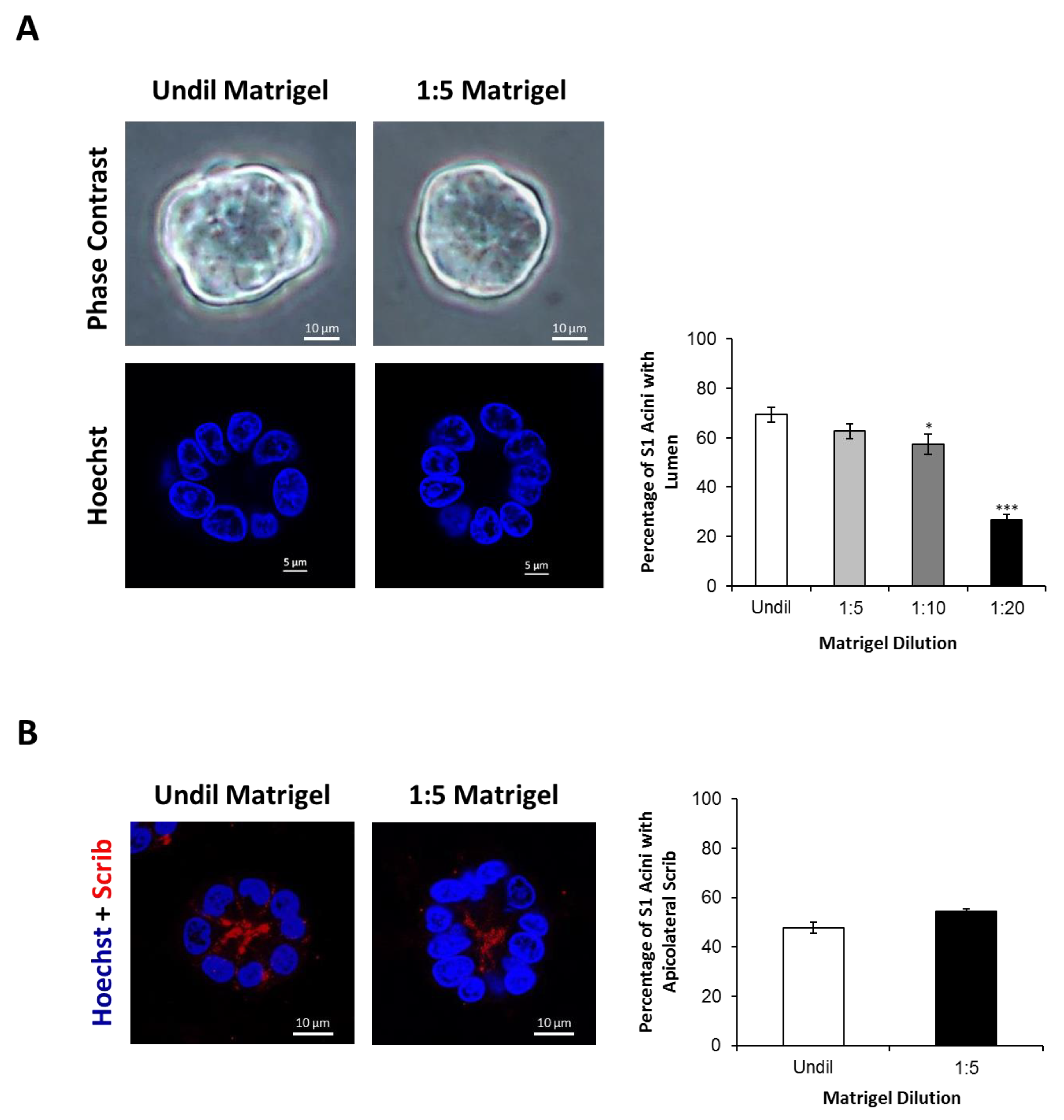
2.2. Silencing Cx43 Alters the Localization of Junctional and Polarity Proteins
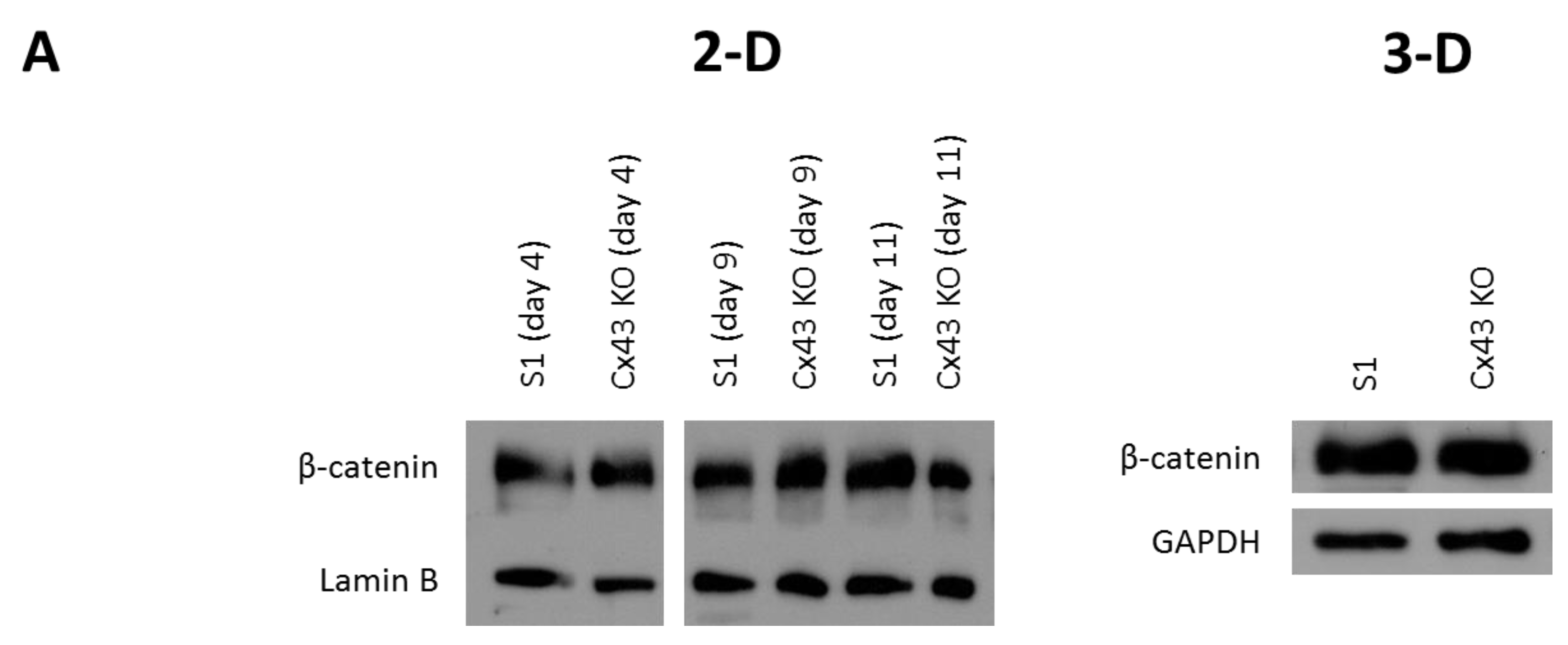
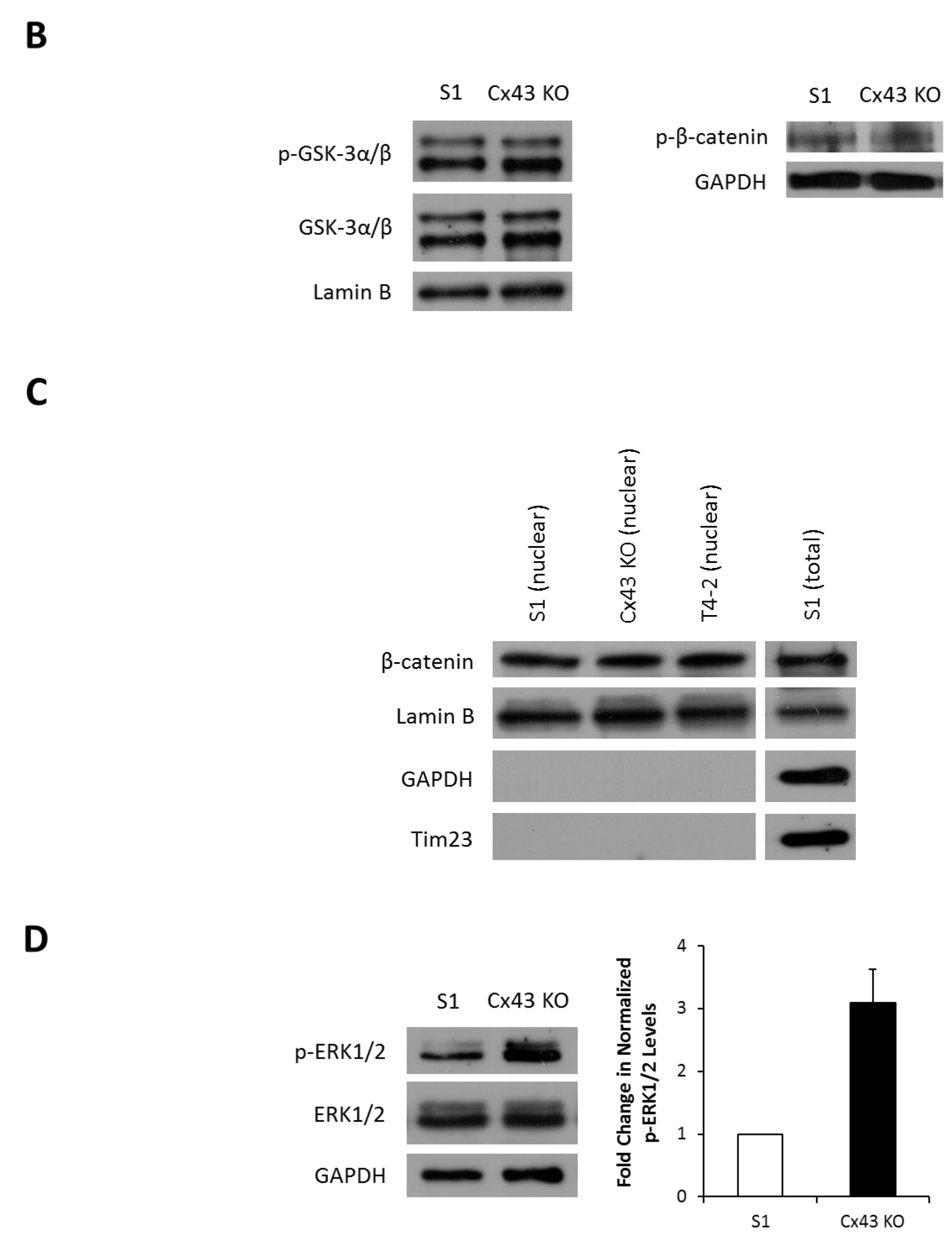
2.3. Silencing Cx43 Activates MAPK but Not Wnt/β-Catenin Signaling
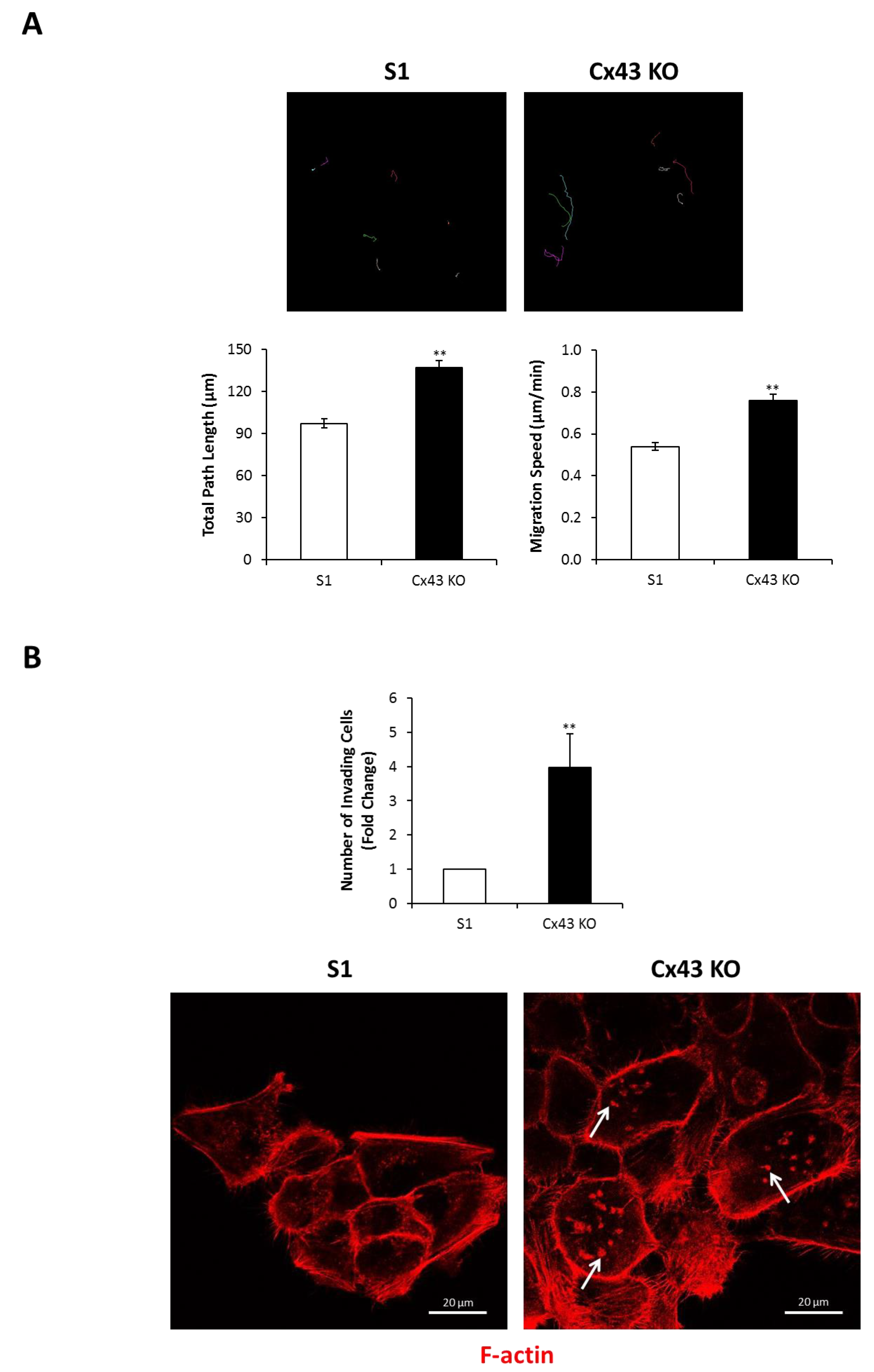
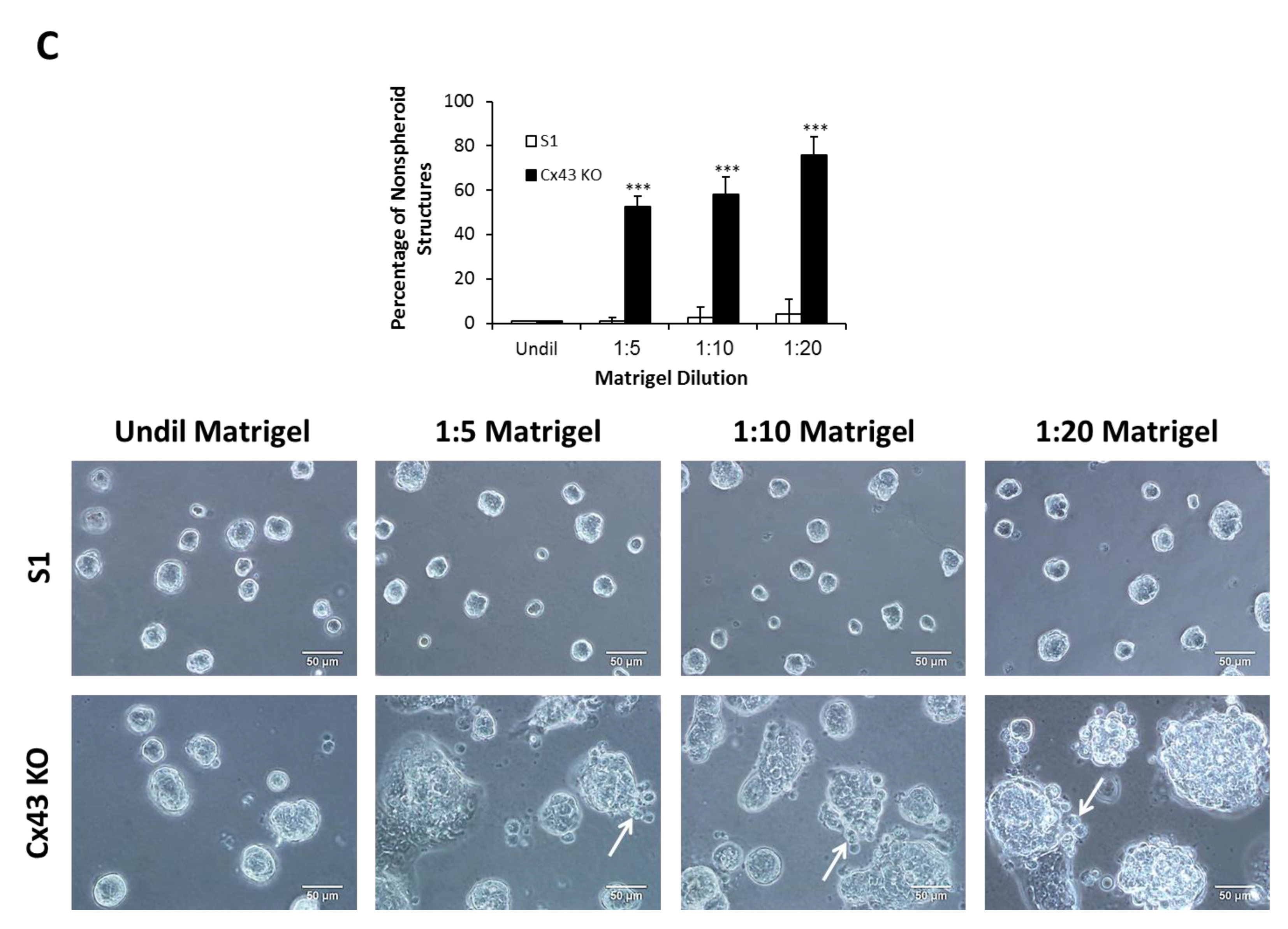
2.4. Silencing Cx43 Induces Motility and Invasion
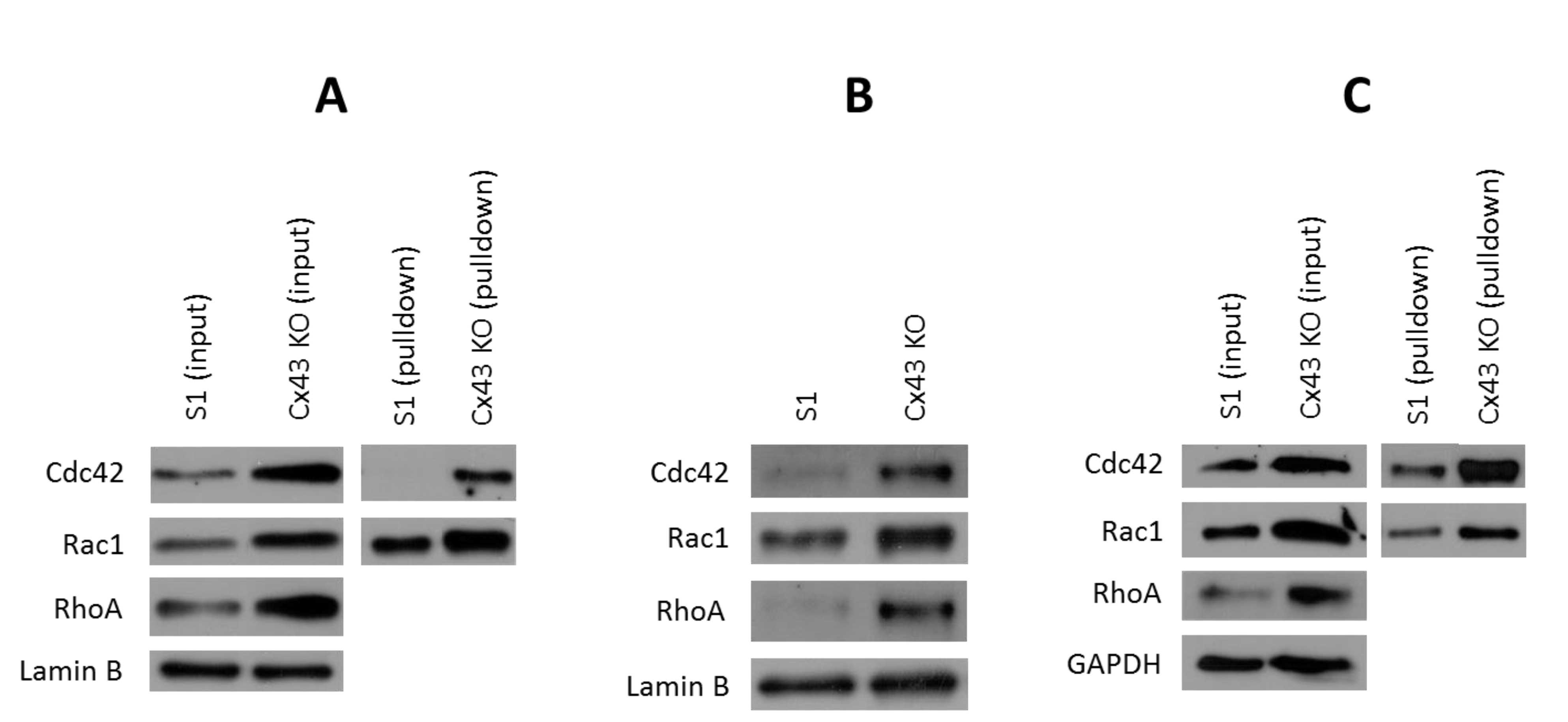
2.5. Silencing Cx43 Activates Rho GTPase Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection and Infection Protocols
4.3. Trypan Blue Exclusion Method
4.4. Cell Cycle Analysis
4.5. Time-Lapse Imaging
4.6. Transwell Cell Invasion Assay
4.7. Immunofluorescence
4.8. Preparation of Whole Cell Protein Extracts and Western Blot Analysis
4.9. Coimmunoprecipitation
4.10. Pulldown Assay
4.11. Subcellular Fractionation
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Monaghan, P.; Perusinghe, N.; Carlile, G.; Evans, W.H. Rapid modulation of gap junction expression in mouse mammary gland during pregnancy, lactation, and involution. J. Histochem. Cytochem. 1994, 42, 931–938. [Google Scholar] [CrossRef]
- Locke, D.; Perusinghe, N.; Newman, T.; Jayatilake, H.; Evans, W.H.; Monaghan, P. Developmental expression and assembly of connexins into homomeric and heteromeric gap junction hemichannels in the mouse mammary gland. J. Cell. Physiol. 2000, 183, 228–237. [Google Scholar] [CrossRef]
- Locke, D.; Stein, T.; Davies, C.; Morris, J.; Harris, A.L.; Evans, W.H.; Monaghan, P.; Gusterson, B. Altered permeability and modulatory character of connexin channels during mammary gland development. Exp. Cell Res. 2004, 298, 643–660. [Google Scholar] [CrossRef]
- Locke, D.; Jamieson, S.; Stein, T.; Liu, J.; Hodgins, M.B.; Harris, A.L.; Gusterson, B. Nature of Cx30-containing channels in the adult mouse mammary gland. Cell Tissue Res. 2007, 328, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, R.S.; Elble, R.C.; Bassam, R.; Daher, M.; Sfeir, A.; Mosleh, L.A.; El-Khoury, H.; Hamoui, S.; Pauli, B.U.; El-Sabban, M.E. Developmental expression patterns and regulation of connexins in the mouse mammary gland: Expression of connexin30 in lactogenesis. Cell Tissue Res. 2005, 319, 49–59. [Google Scholar] [CrossRef]
- Dianati, E.; Poiraud, J.; Weber-Ouellette, A.; Plante, I. Connexins, E-cadherin, Claudin-7 and β-catenin transiently form junctional nexuses during the post-natal mammary gland development. Dev. Biol. 2016, 416, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Clarke, C.; Perusinghe, N.P.; Moss, D.W.; Chen, X.-Y.; Evans, W.H. Gap junction distribution and connexin expression in human breast. Exp. Cell Res. 1996, 223, 29–38. [Google Scholar] [CrossRef]
- Jamieson, S.; Going, J.J.; D’Arcy, R.; George, W.D. Expression of gap junction proteins connexin 26 and connexin 43 in normal human breast and in breast tumours. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 1998, 184, 37–43. [Google Scholar] [CrossRef]
- Laird, D.W.; Fistouris, P.; Batist, G.; Alpert, L.; Huynh, H.T.; Carystinos, G.D.; Alaoui-Jamali, M.A. Deficiency of connexin43 gap junctions is an independent marker for breast tumors. Cancer Res. 1999, 59, 4104–4110. [Google Scholar]
- Singal, R.; Tu, Z.; Vanwert, J.; Ginder, G.; Kiang, D. Modulation of the connexin26 tumor suppressor gene expression through methylation in human mammary epithelial cell lines. Anticancer Res. 2000, 20, 59–64. [Google Scholar]
- Kańczuga-Koda, L.; Sulkowska, M.; Koda, M.; Reszeć, J.; Famulski, W.; Baltaziak, M.; Sulkowski, S. Expression of connexin 43 in breast cancer in comparison with mammary dysplasia and the normal mammary gland. Folia Morphol. 2003, 62, 439–442. [Google Scholar]
- Yeh, E.S.; Williams, C.J.; Williams, C.B.; Bonilla, I.V.; Klauber-DeMore, N.; Phillips, S.L. Dysregulated connexin 43 in HER2-positive drug resistant breast cancer cells enhances proliferation and migration. Oncotarget 2017, 8, 109358. [Google Scholar] [CrossRef] [PubMed]
- Kanczuga-Koda, L.; Sulkowski, S.; Tomaszewski, J.; Koda, M.; Sulkowska, M.; Przystupa, W.; Golaszewska, J.; Baltaziak, M. Connexins 26 and 43 correlate with Bak, but not with Bcl-2 protein in breast cancer. Oncol. Rep. 2005, 14, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Kanczuga-Koda, L.; Sulkowski, S.; Lenczewski, A.; Koda, M.; Wincewicz, A.; Baltaziak, M.; Sulkowska, M. Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J. Clin. Pathol. 2006, 59, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L.; Dulski, K.M.; Trosko, J.E. Loss of intercellular junctional communication correlates with metastatic potential in mammary adenocarcinoma cells. Proc. Natl. Acad. Sci. USA 1988, 85, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.; Lichtner, R.; Trosko, J. Cytoskeletal and junctional heterogeneity in mammary tumor cells and their possible significance in tumor progression. Adv. Exp. Med. Biol. 1988, 233, 21–26. [Google Scholar] [PubMed]
- Plante, I.; Laird, D.W. Decreased levels of connexin43 result in impaired development of the mammary gland in a mouse model of oculodentodigital dysplasia. Dev. Biol. 2008, 318, 312–322. [Google Scholar] [CrossRef]
- Plante, I.; Wallis, A.; Shao, Q.; Laird, D.W. Milk secretion and ejection are impaired in the mammary gland of mice harboring a Cx43 mutant while expression and localization of tight and adherens junction proteins remain unchanged. Biol. Reprod. 2010, 82, 837–847. [Google Scholar] [CrossRef]
- Mroue, R.; Inman, J.; Mott, J.; Budunova, I.; Bissell, M.J. Asymmetric expression of connexins between luminal epithelial-and myoepithelial-cells is essential for contractile function of the mammary gland. Dev. Biol. 2015, 399, 15–26. [Google Scholar] [CrossRef]
- Teleki, I.; Szasz, A.M.; Maros, M.E.; Gyorffy, B.; Kulka, J.; Meggyeshazi, N.; Kiszner, G.; Balla, P.; Samu, A.; Krenacs, T. Correlations of differentially expressed gap junction connexins Cx26, Cx30, Cx32, Cx43 and Cx46 with breast cancer progression and prognosis. PLoS ONE 2014, 9, e112541. [Google Scholar] [CrossRef]
- Chasampalioti, M.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Jackson, A.M.; Spendlove, I.; Ramage, J.M. Connexin 43 is an independent predictor of patient outcome in breast cancer patients. Breast Cancer Res. Treat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, K.K.; Xu, C.; Tsukamoto, T.; Sager, R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. Publ. Am. Assoc. Cancer Res. 1996, 7, 861–870. [Google Scholar]
- Qin, H.; Shao, Q.; Curtis, H.; Galipeau, J.; Belliveau, D.J.; Wang, T.; Alaoui-Jamali, M.A.; Laird, D.W. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J. Biol. Chem. 2002, 277, 29132–29138. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, R.S.; Fares, M.-B.; Rahme, G.J.; Hariri, H.H.; Rayess, T.; Dbouk, H.A.; Bazzoun, D.; Al-Labban, D.; El-Sabban, M.E. Context dependent reversion of tumor phenotype by connexin-43 expression in MDA-MB231 cells and MCF-7 cells: Role of β-catenin/connexin43 association. Exp. Cell Res. 2013, 319, 3065–3080. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, Z.; Welch, D.R.; Donahue, H.J. Expressing connexin 43 in breast cancer cells reduces their metastasis to lungs. Clin. Exp. Metastasis 2008, 25, 893–901. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Donahue, H.J. Alterations in Cx43 and OB-cadherin affect breast cancer cell metastatic potential. Clin. Exp. Metastasis 2008, 25, 265–272. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.; Shao, Q.; Wang, H.-l.; Langlois, S.; Laird, D.W. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006, 66, 9886–9894. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Wang, H.; McLachlan, E.; Veitch, G.I.; Laird, D.W. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res. 2005, 65, 2705–2711. [Google Scholar] [CrossRef]
- Plante, I.; Stewart, M.; Barr, K.; Allan, A.; Laird, D. Cx43 suppresses mammary tumor metastasis to the lung in a Cx43 mutant mouse model of human disease. Oncogene 2011, 30, 1681. [Google Scholar] [CrossRef]
- Kalra, J.; Shao, Q.; Qin, H.; Thomas, T.; Alaoui-Jamali, M.A.; Laird, D.W. Cx26 inhibits breast MDA-MB-435 cell tumorigenic properties by a gap junctional intercellular communication-independent mechanism. Carcinogenesis 2006, 27, 2528–2537. [Google Scholar] [CrossRef]
- Fostok, S.F.; El-Sibai, M.; El-Sabban, M.; Talhouk, R.S. Gap Junctions and Wnt Signaling in the Mammary Gland: A Cross-Talk? J. Mammary Gland Biol. Neoplasia 2018. [Google Scholar] [CrossRef]
- Van Genderen, C.; Okamura, R.M.; Farinas, I.; Quo, R.-G.; Parslow, T.G.; Bruhn, L.; Grosschedl, R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994, 8, 2691–2703. [Google Scholar] [CrossRef]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.-C. β-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Imbert, A.; Eelkema, R.; Jordan, S.; Feiner, H.; Cowin, P. ΔN89β-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J. Cell Biol. 2001, 153, 555–568. [Google Scholar] [CrossRef]
- Milovanovic, T.; Planutis, K.; Nguyen, A.; Marsh, J.L.; Lin, F.; Hope, C.; Holcombe, R.F. Expression of Wnt genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int. J. Oncol. 2004, 25, 1337–1342. [Google Scholar] [CrossRef]
- Ewald, A.J.; Brenot, A.; Duong, M.; Chan, B.S.; Werb, Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 2008, 14, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.P.; Mirza, S.; Sharma, G.; Prashad, R.; DattaGupta, S.; Rath, G.; Ralhan, R. Epigenetic alterations of CDH1 and APC genes: Relationship with activation of Wnt/β-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008, 83, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, C.; Zylstra, C.R.; Evans, N.; West, R.A.; Dykema, K.; Furge, K.A.; Williams, B.O. The Wnt co-receptor Lrp6 is required for normal mouse mammary gland development. PLoS ONE 2009, 4, e5813. [Google Scholar] [CrossRef]
- Raymond, K.; Cagnet, S.; Kreft, M.; Janssen, H.; Sonnenberg, A.; Glukhova, M.A. Control of mammary myoepithelial cell contractile function by α3β1 integrin signalling. Embo J. 2011, 30, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Bray, K.; Gillette, M.; Young, J.; Loughran, E.; Hwang, M.; Sears, J.C.; Vargo-Gogola, T. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013, 15, R91. [Google Scholar] [CrossRef] [PubMed]
- Bagci, H.; Laurin, M.; Huber, J.; Muller, W.; Cote, J. Impaired cell death and mammary gland involution in the absence of Dock1 and Rac1 signaling. Cell Death Dis. 2014, 5, e1375. [Google Scholar] [CrossRef]
- Van der Heyden, M.; Rook, M.B.; Hermans, M.; Rijksen, G.; Boonstra, J.; Defize, L.; Destree, O. Identification of connexin43 as a functional target for Wnt signalling. J. Cell Sci. 1998, 111, 1741–1749. [Google Scholar] [PubMed]
- Constantinou, T.; Baumann, F.; Lacher, M.D.; Saurer, S.; Friis, R.; Dharmarajan, A. SFRP-4 abrogates Wnt-3a-induced β-catenin and Akt/PKB signalling and reverses a Wnt-3a-imposed inhibition of in vitro mammary differentiation. J. Mol. Signal. 2008, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Baxley, S.E.; Jiang, W.; Serra, R. Misexpression of wingless-related MMTV integration site 5A in mouse mammary gland inhibits the milk ejection response and regulates connexin43 phosphorylation. Biol. Reprod. 2011, 85, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D. Connexin’s connection in breast cancer growth and progression. Int. J. Cell Biol. 2016, 2016. [Google Scholar] [CrossRef]
- Carystinos, G.D.; Bier, A.; Batist, G. The role of connexin-mediated cell–cell communication in breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 2001, 6, 431–440. [Google Scholar] [CrossRef]
- McLachlan, E.; Shao, Q.; Laird, D.W. Connexins and gap junctions in mammary gland development and breast cancer progression. J. Membr. Biol. 2007, 218, 107–121. [Google Scholar] [CrossRef]
- Grek, C.L.; Rhett, J.M.; Bruce, J.S.; Ghatnekar, G.S.; Yeh, E.S. Connexin 43, breast cancer tumor suppressor: Missed connections? Cancer Lett. 2016, 374, 117–126. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Elzarrad, M.K.; Haroon, A.; Willecke, K.; Dobrowolski, R.; Gillespie, M.N.; Al-Mehdi, A.-B. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Kanczuga-Koda, L.; Sulkowska, M.; Koda, M.; Rutkowski, R.; Sulkowski, S. Increased expression of gap junction protein—Connexin 32 in lymph node metastases of human ductal breast cancer. Folia Histochem. Cytobiol. 2007, 45, 175–180. [Google Scholar]
- Naoi, Y.; Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Arai, T.; Tamaki, Y.; Noguchi, S. Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2007, 106, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, P.S.; Sinyuk, M.; Turaga, S.M.; Mulkearns-Hubert, E.E.; Hale, J.S.; Rao, V.; Demelash, A.; Saygin, C.; China, A.; Alban, T.J. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, M.-A.; Shao, Q.; Laird, D.W.; Sandig, M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005, 7, R522. [Google Scholar] [CrossRef] [PubMed]
- Insua-Rodriguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Briand, P.; Petersen, O.; Van Deurs, B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell. Dev. Biol. 1987, 23, 181–188. [Google Scholar] [CrossRef]
- Plachot, C.; Lelièvre, S.A. DNA methylation control of tissue polarity and cellular differentiation in the mammary epithelium. Exp. Cell Res. 2004, 298, 122–132. [Google Scholar] [CrossRef]
- Petersen, O.W.; Rønnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, R.S.; Mroue, R.; Mokalled, M.; Abi-Mosleh, L.; Nehme, R.; Ismail, A.; Khalil, A.; Zaatari, M.; El-Sabban, M.E. Heterocellular interaction enhances recruitment of α and β-catenins and ZO-2 into functional gap-junction complexes and induces gap junction-dependant differentiation of mammary epithelial cells. Exp. Cell Res. 2008, 314, 3275–3291. [Google Scholar] [CrossRef] [PubMed]
- El-Sabban, M.E.; Sfeir, A.J.; Daher, M.H.; Kalaany, N.Y.; Bassam, R.A.; Talhouk, R.S. ECM-induced gap junctional communication enhances mammary epithelial cell differentiation. J. Cell Sci. 2003, 116, 3531–3541. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, S.I.; Iden, S.; Collard, J.G. Cell polarity proteins and cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2012; pp. 208–215. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zardawi, S.J.; O’Toole, S.A.; Sutherland, R.L.; Musgrove, E.A. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol. Histopathol. 2009, 24, 385–398. [Google Scholar] [PubMed]
- Rao, T.P.; KUhl, M. An updated overview on Wnt signaling pathways: A prelude for more. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Chambard, J.-C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Grill, C.; Gheyas, F.; Dayananth, P.; Weihong, J.; Wei, D.; Ping, Q.; Luquan, W.; Jessie, M. Analysis of the ERK1, 2 transcriptome in mammary epithelial cells. Biochem. J. 2004, 381, 635–644. [Google Scholar] [CrossRef]
- Whyte, J.; Bergin, O.; Bianchi, A.; McNally, S.; Martin, F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009, 11, 209. [Google Scholar] [CrossRef]
- Stupack, D.G.; Cho, S.Y.; Klemke, R.L. Molecular signaling mechanisms of cell migration and invasion. Immunol. Res. 2000, 21, 83–88. [Google Scholar] [CrossRef]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, P.G.; Nebreda, A.R. Deconstructing ERK signaling in tumorigenesis. Mol. Cell 2010, 38, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, Y.; Jorgens, D.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.; Weaver, V. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8, 026013. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, L.; Miroshnikova, Y.A.; Ou, G.; Lakins, J.; Weaver, V.M. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J. Biotechnol. 2015, 193, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.; Liphardt, J.; Hwang, E.; Weaver, V. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef]
- Haage, A.; Schneider, I.C. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014, 28, 3589–3599. [Google Scholar] [CrossRef]
- Ansardamavandi, A.; Tafazzoli-Shadpour, M.; Shokrgozar, M.A. Behavioral remodeling of normal and cancerous epithelial cell lines with differing invasion potential induced by substrate elastic modulus. Cell Adhes. Migr. 2018. [Google Scholar] [CrossRef]
- Zaman, M.H.; Trapani, L.M.; Sieminski, A.L.; MacKellar, D.; Gong, H.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Matsudaira, P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 10889–10894. [Google Scholar] [CrossRef]
- Burbelo, P.; Wellstein, A.; Pestell, R.G. Altered Rho GTPase signaling pathways in breast cancer cells. Breast Cancer Res. Treat. 2004, 84, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Oh, W.; Ulu, A.; Frost, J.A. Minireview: Mouse Models of Rho GTPase Function in Mammary Gland Development, Tumorigenesis, and Metastasis. Mol. Endocrinol. 2016, 30, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK-and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- El-Sibai, M.; Nalbant, P.; Pang, H.; Flinn, R.J.; Sarmiento, C.; Macaluso, F.; Cammer, M.; Condeelis, J.S.; Hahn, K.M.; Backer, J.M. Cdc42 is required for EGF-stimulated protrusion and motility in MTLn3 carcinoma cells. J. Cell Sci. 2007, 120, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.-C.; El-Sibai, M.; Coniglio, S.J.; Mouneimne, G.; Eddy, R.J.; Drees, B.E.; Neilsen, P.O.; Goswami, S.; Symons, M.; Condeelis, J.S. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J. Cell Sci. 2007, 120, 3138–3146. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-H.; Lee, S.-W.; Li, C.-F.; Wang, J.; Yang, W.-L.; Wu, C.-Y.; Wu, J.; Nakayama, K.I.; Kang, H.-Y.; Huang, H.-Y. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010, 12, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Oser, M.; Chen, X.; Eddy, R.; Hodgson, L.; Condeelis, J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 2011, 21, 635–644. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Sharma, V.P.; Roh-Johnson, M.; Chen, X.; Eddy, R.; Condeelis, J.; Hodgson, L. Spatial regulation of RhoC activity defines protrusion formation in migrating cells. J. Cell Sci. 2013, 126, 3356–3369. [Google Scholar] [CrossRef]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef]
- Moshfegh, Y.; Bravo-Cordero, J.J.; Miskolci, V.; Condeelis, J.; Hodgson, L. A Trio–Rac1–Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol. 2014, 16, 571–583. [Google Scholar] [CrossRef] [PubMed]
- El-Sibai, M.; Pertz, O.; Pang, H.; Yip, S.-C.; Lorenz, M.; Symons, M.; Condeelis, J.S.; Hahn, K.M.; Backer, J.M. RhoA/ROCK-mediated switching between Cdc42-and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp. Cell Res. 2008, 314, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Tcherkezian, J.; Triki, I.; Stenne, R.; Danek, E.I.; Lamarche-Vane, N. The human orthologue of CdGAP is a phosphoprotein and a GTPase-activating protein for Cdc42 and Rac1 but not RhoA. Biol. Cell 2006, 98, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Tcherkezian, J.; Danek, E.I.; Jenna, S.; Triki, I.; Lamarche-Vane, N. Extracellular signal-regulated kinase 1 interacts with and phosphorylates CdGAP at an important regulatory site. Mol. Cell. Biol. 2005, 25, 6314–6329. [Google Scholar] [CrossRef] [PubMed]
- Almog, T.; Lazar, S.; Reiss, N.; Etkovitz, N.; Milch, E.; Rahamim, N.; Dobkin-Bekman, M.; Rotem, R.; Kalina, M.; Ramon, J. Identification of extracellular signal-regulated kinase 1/2 and p38 MAPK as regulators of human sperm motility and acrosome reaction and as predictors of poor spermatozoan quality. J. Biol. Chem. 2008, 283, 14479–14489. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, S.-h.; Tanimura, S.; Mure, S.; Kashimoto, Y.; Watanabe, K.; Kohno, M. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 2008, 368, 162–167. [Google Scholar] [CrossRef]
- Demirkan, B. The roles of epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-epithelial transition (MET) in breast cancer bone metastasis: Potential targets for prevention and treatment. J. Clin. Med. 2013, 2, 264–282. [Google Scholar] [CrossRef]
- Nakaya, Y.; Sheng, G. EMT in developmental morphogenesis. Cancer Lett. 2013, 341, 9–15. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Momiyama, M.; Omori, Y.; Ishizaki, Y.; Nishikawa, Y.; Tokairin, T.; Ogawa, J.; Enomoto, K. Connexin26-mediated gap junctional communication reverses the malignant phenotype of MCF-7 breast cancer cells. Cancer Sci. 2003, 94, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Akshinthala, S.D.; Araki, K.; Rosenberg, A.Z.; Muthuswamy, L.B.; Martin, B.; Lehmann, B.D.; Berman, H.K.; Pietenpol, J.A.; Cardiff, R.D. Mislocalization of the cell polarity protein scribble promotes mammary tumorigenesis and is associated with basal breast cancer. Cancer Res. 2014, 74, 3180–3194. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.J.; McCaffrey, L. Emerging role of cell polarity proteins in breast cancer progression and metastasis. Breast Cancer Targets Ther. 2014, 6, 15. [Google Scholar]
- Chandramouly, G.; Abad, P.C.; Knowles, D.W.; Lelièvre, S.A. The control of tissue architecture over nuclear organization is crucial for epithelial cell fate. J. Cell Sci. 2007, 120, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Perez-Mancera, P.A.; Dow, L.E.; Ryan, A.; Tennstedt, P.; Bogani, D.; Elsum, I.; Greenfield, A.; Tuveson, D.A.; Simon, R. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J. Clin. Investig. 2011, 121, 4257–4267. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Sharma, B.K.; Patil, M.; Elattar, S.; Yuan, J.; Hou, S.X.; Kolhe, R.; Satyanarayana, A. The cell polarity protein Scrib functions as a tumor suppressor in liver cancer. Oncotarget 2017, 8, 26515. [Google Scholar] [CrossRef]
- Giepmans, B.N.; van IJzendoorn, S.C. Epithelial cell–cell junctions and plasma membrane domains. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 820–831. [Google Scholar] [CrossRef]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef]
- Scholz, M.; Nowak, P.; Blaheta, R.; Schuller, A.; Menon, S.; Cinatl, J.; Wimmer-Greinecker, G.; Moritz, A. Relocalization of endothelial cell β-catenin after coculture with activated neutrophils from patients undergoing cardiac surgery with cardiopulmonary bypass. J. Investig. Surg. 2004, 17, 143–149. [Google Scholar] [CrossRef]
- Scholz, M.; Wimmer-Greinecker, G.; Simon, A.; Dzemali, O.; Chang, H.-Y.; Kleine, P.; Matheis, G.; Moritz, A. Perioperative elastase activity in cardiac surgery and its role in endothelial leakage. Inflamm. Res. 2003, 52, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Buck, V.U.; Windoffer, R.; Leube, R.E.; Classen-Linke, I. Redistribution of adhering junctions in human endometrial epithelial cells during the implantation window of the menstrual cycle. Histochem. Cell Biol. 2012, 137, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Zheng, G.-Z.; Xie, P.; Zhang, Q.-H.; Lin, F.-X.; Chang, B.; Hu, Q.-X.; Du, S.-X.; Li, X.-D. Antitumor activity of resveratrol against human osteosarcoma cells: A key role of Cx43 and Wnt/β-catenin signaling pathway. Oncotarget 2017, 8, 111419. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Ravisankar, S.; Vanisree, A.J. Implication of connexin30 on the stemness of glioma: connexin30 reverses the malignant phenotype of glioma by modulating IGF-1R, CD133 and cMyc. J. Neuro-Oncol. 2017, 135, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhao, W.; Wang, Y.; Xu, Y.; Xu, J.; Tang, K.; Zhang, S.; Yin, Z.; Wu, Q.; Wang, X. Connexin32 regulates hepatoma cell metastasis and proliferation via the p53 and Akt pathways. Oncotarget 2015, 6, 10116. [Google Scholar] [CrossRef] [PubMed]
- Koffler, L.; Roshong, S.; Kyu Park, I.; Cesen-Cummings, K.; Thompson, D.C.; Dwyer-Nield, L.D.; Rice, P.; Mamay, C.; Malkinson, A.M.; Ruch, R.J. Growth inhibition in G1 and altered expression of cyclin D1 and p27kip-1 after forced connexin expression in lung and liver carcinoma cells. J. Cell. Biochem. 2000, 79, 347–354. [Google Scholar] [CrossRef]
- Jönsson, M.; Borg, Å.; Nilbert, M.; Andersson, T. Involvement of adenomatous polyposis coli (APC)/β-catenin signalling in human breast cancer. Eur. J. Cancer 2000, 36, 242–248. [Google Scholar] [CrossRef]
- Ozaki, S.; Ikeda, S.; Ishizaki, Y.; Kurihara, T.; Tokumoto, N.; Iseki, M.; Arihiro, K.; Kataoka, T.; Okajima, M.; Asahara, T. Alterations and correlations of the components in the Wnt signaling pathway and its target genes in breast cancer. Oncol. Rep. 2005, 14, 1437–1443. [Google Scholar] [CrossRef]
- Khan, Z.; Arafah, M.; Shaik, J.P.; Mahale, A.; Alanazi, M.S. High-frequency deregulated expression of Wnt signaling pathway members in breast carcinomas. Oncotargets Ther. 2018, 11, 323. [Google Scholar] [CrossRef]
- Lim, S.-C.; Lee, M.-S. Significance of E-cadherin/β-catenin complex and cyclin D1 in breast cancer. Oncol. Rep. 2002, 9, 915–928. [Google Scholar] [CrossRef]
- King, T.J.; Lampe, P.D. Mice deficient for the gap junction protein Connexin32 exhibit increased radiation-induced tumorigenesis associated with elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2) activation. Carcinogenesis 2004, 25, 669–680. [Google Scholar] [CrossRef] [PubMed]
- King, T.J.; Gurley, K.E.; Prunty, J.; Shin, J.-L.; Kemp, C.J.; Lampe, P.D. Deficiency in the gap junction protein connexin32 alters p27 Kip1 tumor suppression and MAPK activation in a tissue-specific manner. Oncogene 2005, 24, 1718. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Vanisree, A.J.; Ravisankar, S. Connexin 30 downregulates Insulin-like growth factor receptor-1, abolishes Erk and potentiates effects of an IGF-R inhibitor in a glioma cell line. Brain Res. 2016, 1643, 80–90. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, X.; Gao, K.; Zhang, Z.; Huang, Y.; Yoda, R.; Yao, J. The pivotal role of extracellular signal-regulated kinase in gap junction-mediated regulation of TXNIP. Cell. Signal. 2017, 38, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.F. Rho GTPases as key transducers of proliferative signals in G 1 cell cycle regulation. Breast Cancer Res. Treat. 2004, 84, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef]
- Hanna, S.; Khalil, B.; Nasrallah, A.; Saykali, B.A.; Sobh, R.; Nasser, S.; El-Sibai, M. StarD13 is a tumor suppressor in breast cancer that regulates cell motility and invasion. Int. J. Oncol. 2014, 44, 1499–1511. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Hu, R.; O’Neill, S.; Wang, C.M.; Thrasivoulou, C.; Power, K.T.; White, A.; Serena, T. Targeting Cx43 and N-cadherin, which are abnormally upregulated in venous leg ulcers, influences migration, adhesion and activation of Rho GTPases. PLoS ONE 2012, 7, e37374. [Google Scholar] [CrossRef]
- Asnaghi, L.; Vass, W.; Quadri, R.; Day, P.; Qian, X.; Braverman, R.; Papageorge, A.; Lowy, D. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: Role of Rho GTPases. Oncogene 2010, 29, 2760. [Google Scholar] [CrossRef]
- Zebda, N.; Tian, Y.; Tian, X.; Gawlak, G.; Higginbotham, K.; Reynolds, A.B.; Birukova, A.A.; Birukov, K.G. Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J. Biol. Chem. 2013, 288, 18290–18299. [Google Scholar] [CrossRef]
- Cáceres, M.; Guerrero, J.; Martínez, J. Overexpression of RhoA-GTP induces activation of the Epidermal Growth Factor Receptor, dephosphorylation of focal adhesion kinase and increased motility in breast cancer cells. Exp. Cell Res. 2005, 309, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Ramaswami, S.A.; Bouchard, R.J.; Aktories, K.; Linseman, D.A. Neuronal apoptosis induced by selective inhibition of Rac GTPase versus global suppression of Rho family GTPases is mediated by alterations in distinct mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 2015, 290, 9363–9376. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.I.; Kang, I.; You, W.-K.; McDonald, D.M.; Weaver, V.M. In situ force mapping of mammary gland transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Plodinec, M.; Loparic, M.; Monnier, C.A.; Obermann, E.C.; Zanetti-Dallenbach, R.; Oertle, P.; Hyotyla, J.T.; Aebi, U.; Bentires-Alj, M.; Lim, R.Y. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 2012, 7, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.E.; Jin, Y.-S.; Rao, J.; Gimzewski, J.K. Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol. 2007, 2, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Fenner, J.; Stacer, A.C.; Winterroth, F.; Johnson, T.D.; Luker, K.E.; Luker, G.D. Macroscopic stiffness of breast tumors predicts metastasis. Sci. Rep. 2014, 4, 5512. [Google Scholar] [CrossRef]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef]
- Blaschke, R.J.; Howlett, A.R.; Desprez, P.-Y.; Petersen, O.W.; Bissell, M.J. [25] Cell differentiation by extracellular matrix components. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 245, pp. 535–556. [Google Scholar]
- Vidi, P.-A.; Bissell, M.J.; Lelièvre, S.A. Three-dimensional culture of human breast epithelial cells: The how and the why. In Epithelial Cell Culture Protocols; Springer: Berlin/Heidelberg, Germany, 2012; pp. 193–219. [Google Scholar]
- Khalil, B.D.; Hanna, S.; Saykali, B.A.; El-Sitt, S.; Nasrallah, A.; Marston, D.; El-Sabban, M.; Hahn, K.M.; Symons, M.; El-Sibai, M. The regulation of RhoA at focal adhesions by StarD13 is important for astrocytoma cell motility. Exp. Cell Res. 2014, 321, 109–122. [Google Scholar] [CrossRef]
- Lelièvre, S.A.; Weaver, V.M.; Nickerson, J.A.; Larabell, C.A.; Bhaumik, A.; Petersen, O.W.; Bissell, M.J. Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc. Natl. Acad. Sci. USA 1998, 95, 14711–14716. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fostok, S.; El-Sibai, M.; Bazzoun, D.; Lelièvre, S.; Talhouk, R. Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling. Cancers 2019, 11, 339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030339
Fostok S, El-Sibai M, Bazzoun D, Lelièvre S, Talhouk R. Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling. Cancers. 2019; 11(3):339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030339
Chicago/Turabian StyleFostok, Sabreen, Mirvat El-Sibai, Dana Bazzoun, Sophie Lelièvre, and Rabih Talhouk. 2019. "Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling" Cancers 11, no. 3: 339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030339