The Delta Subunit of Rod-Specific Photoreceptor cGMP Phosphodiesterase (PDE6D) Contributes to Hepatocellular Carcinoma Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
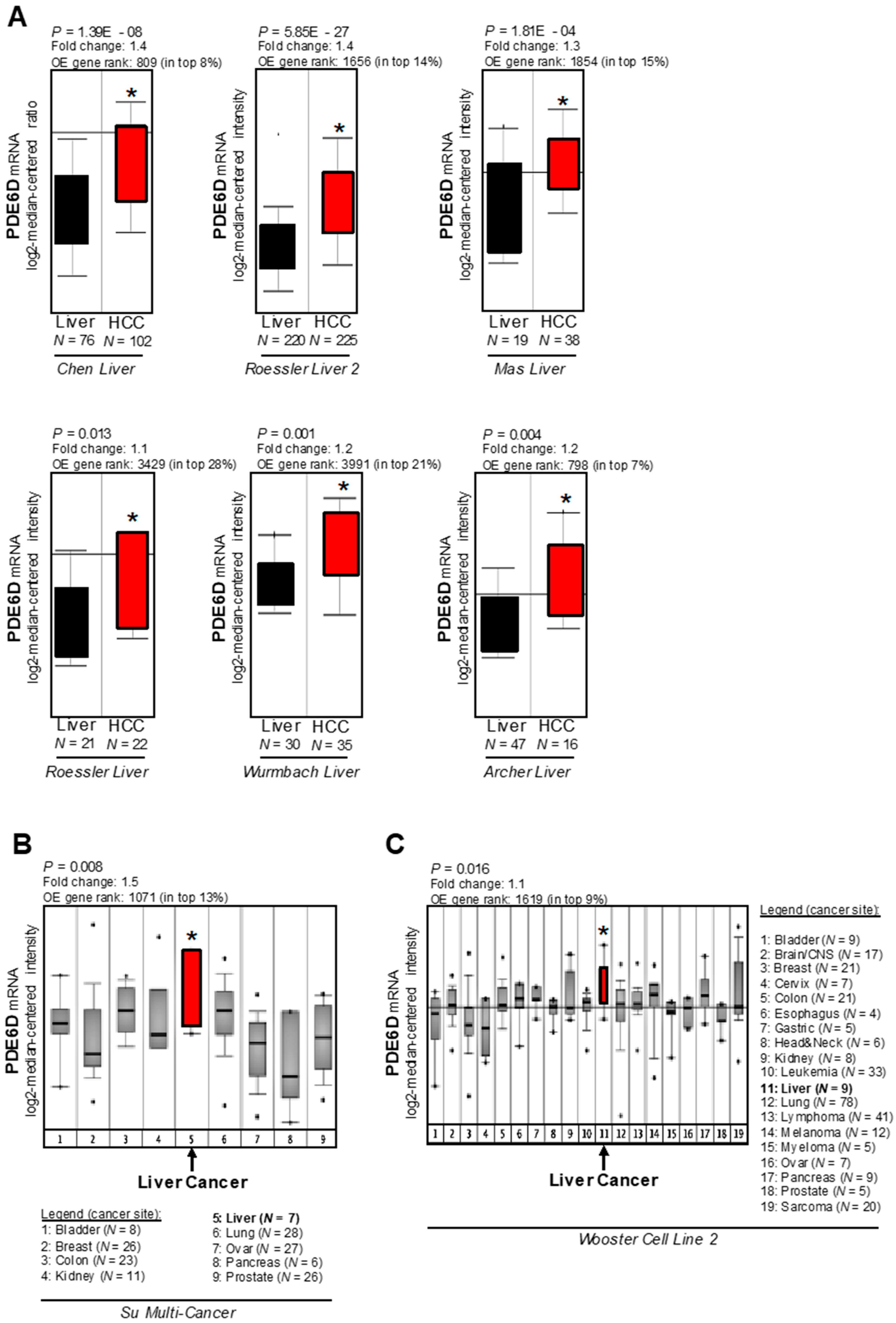
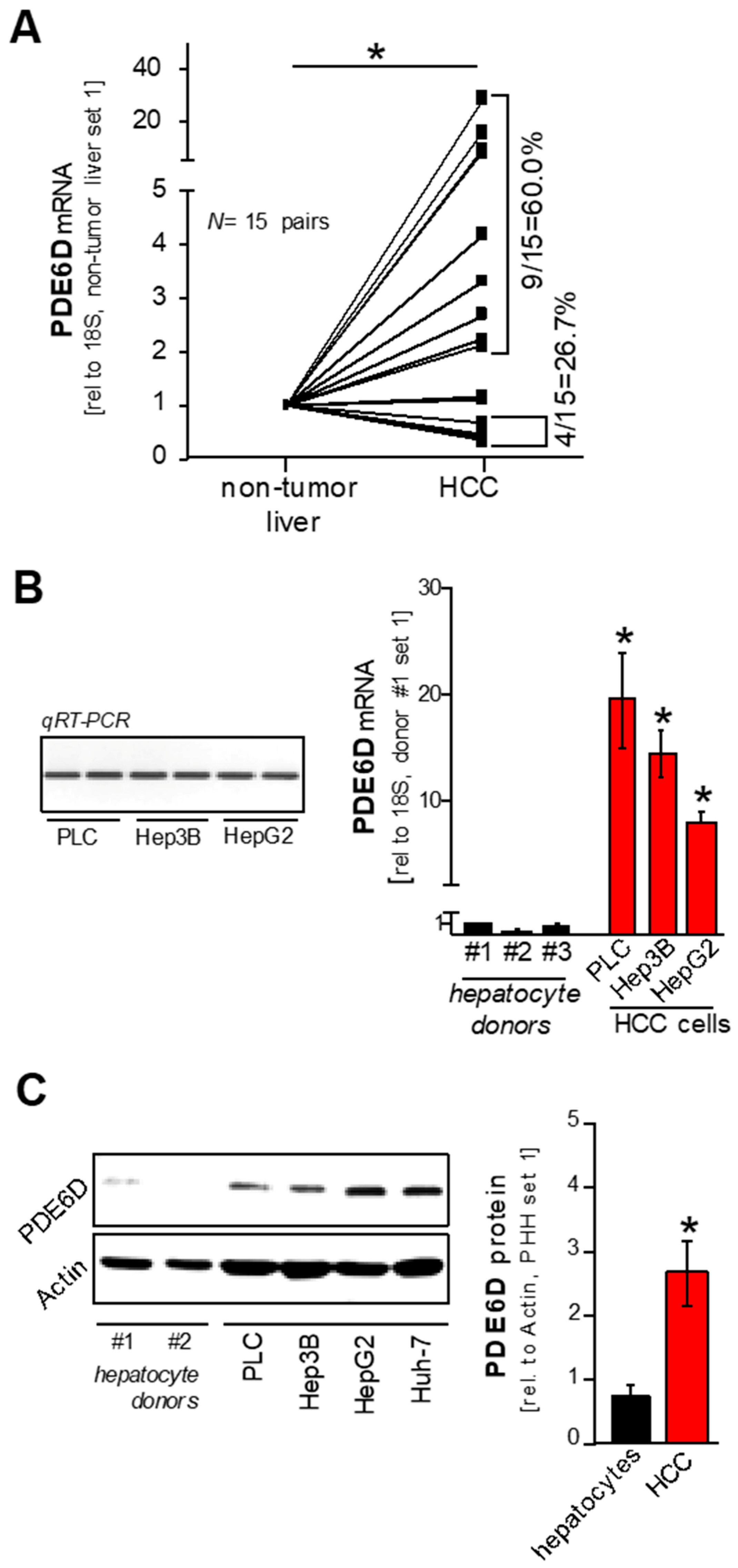
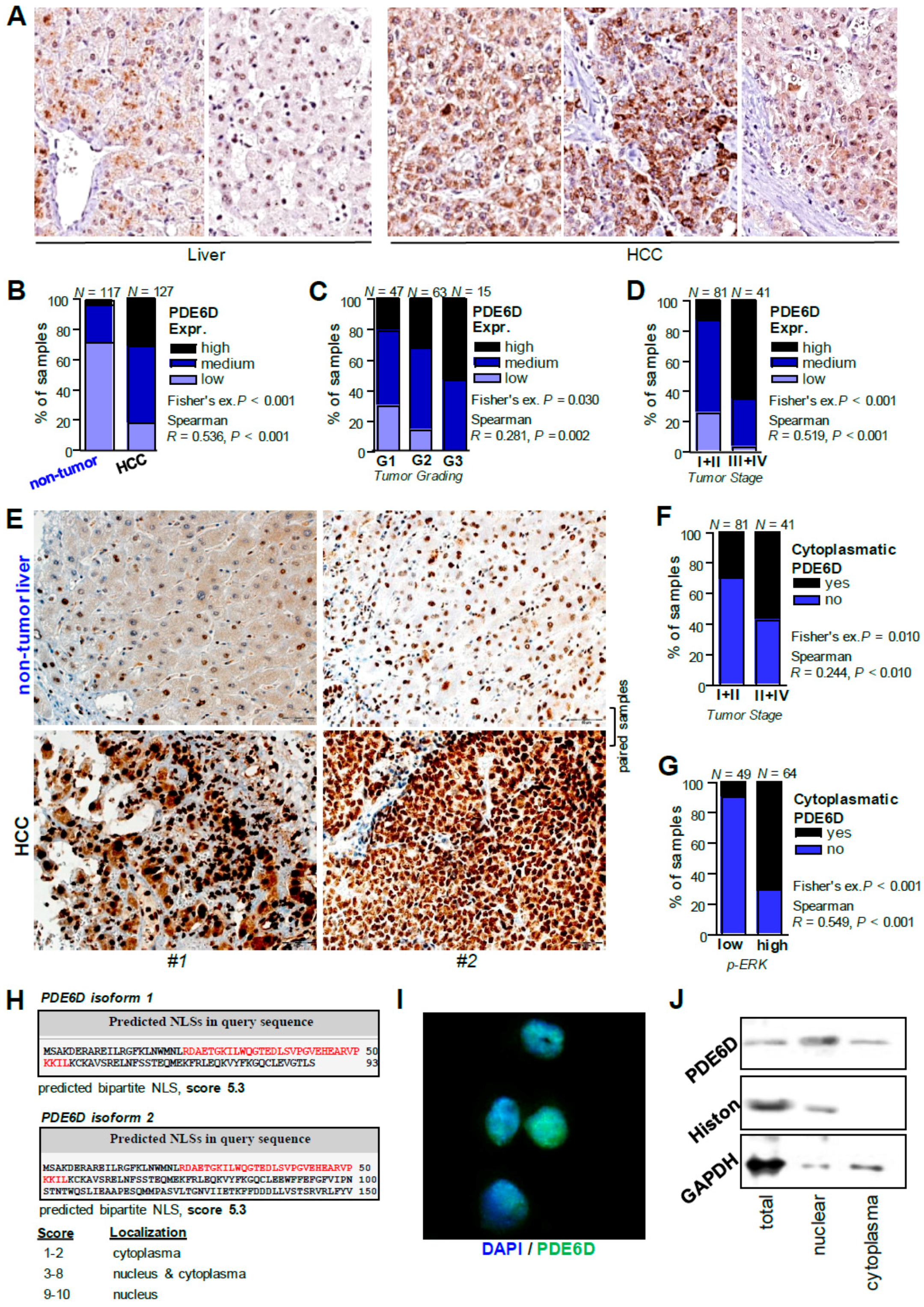
2.1. Expression of PDE6D in Hepatocellular Carcinoma
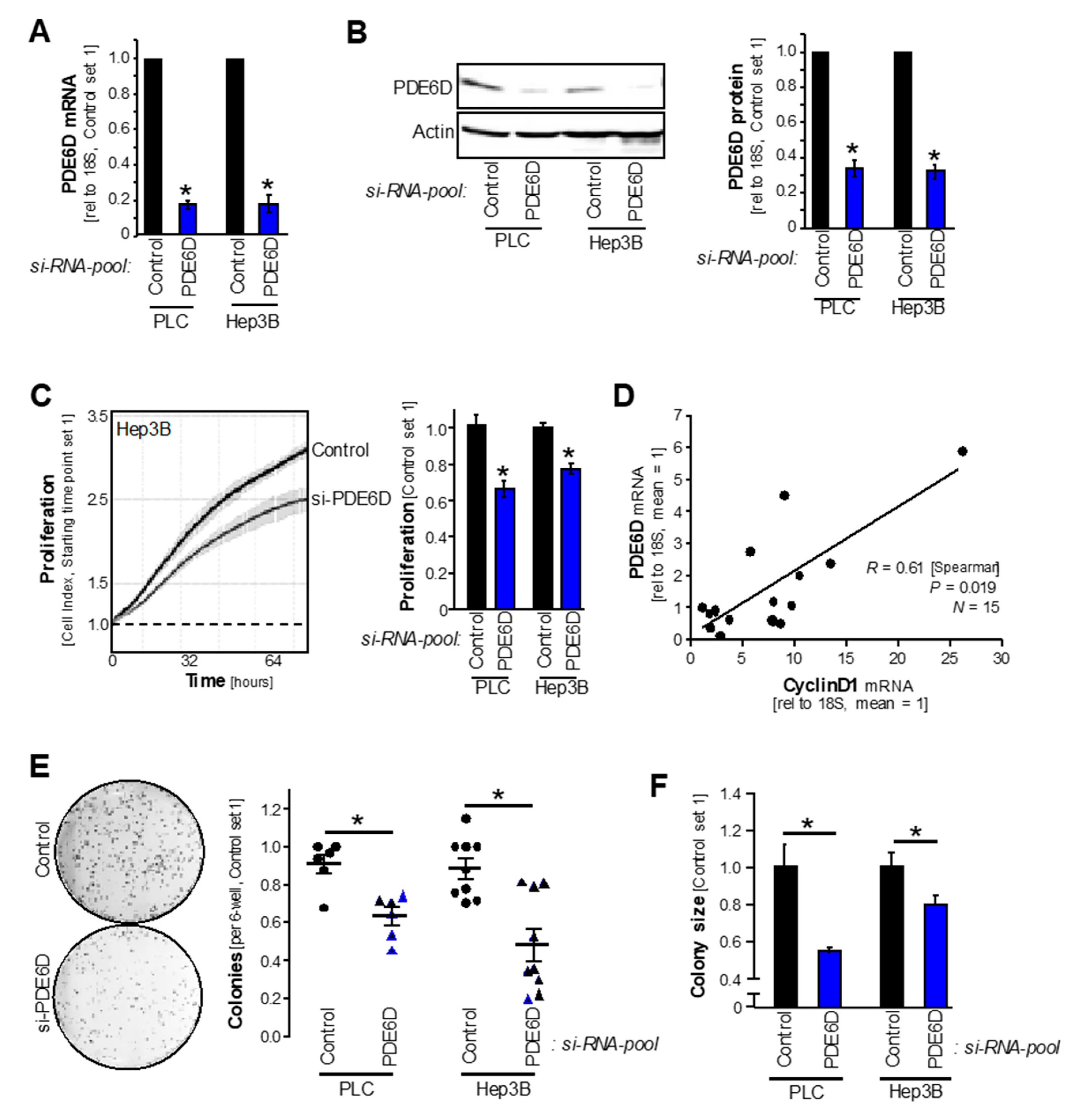
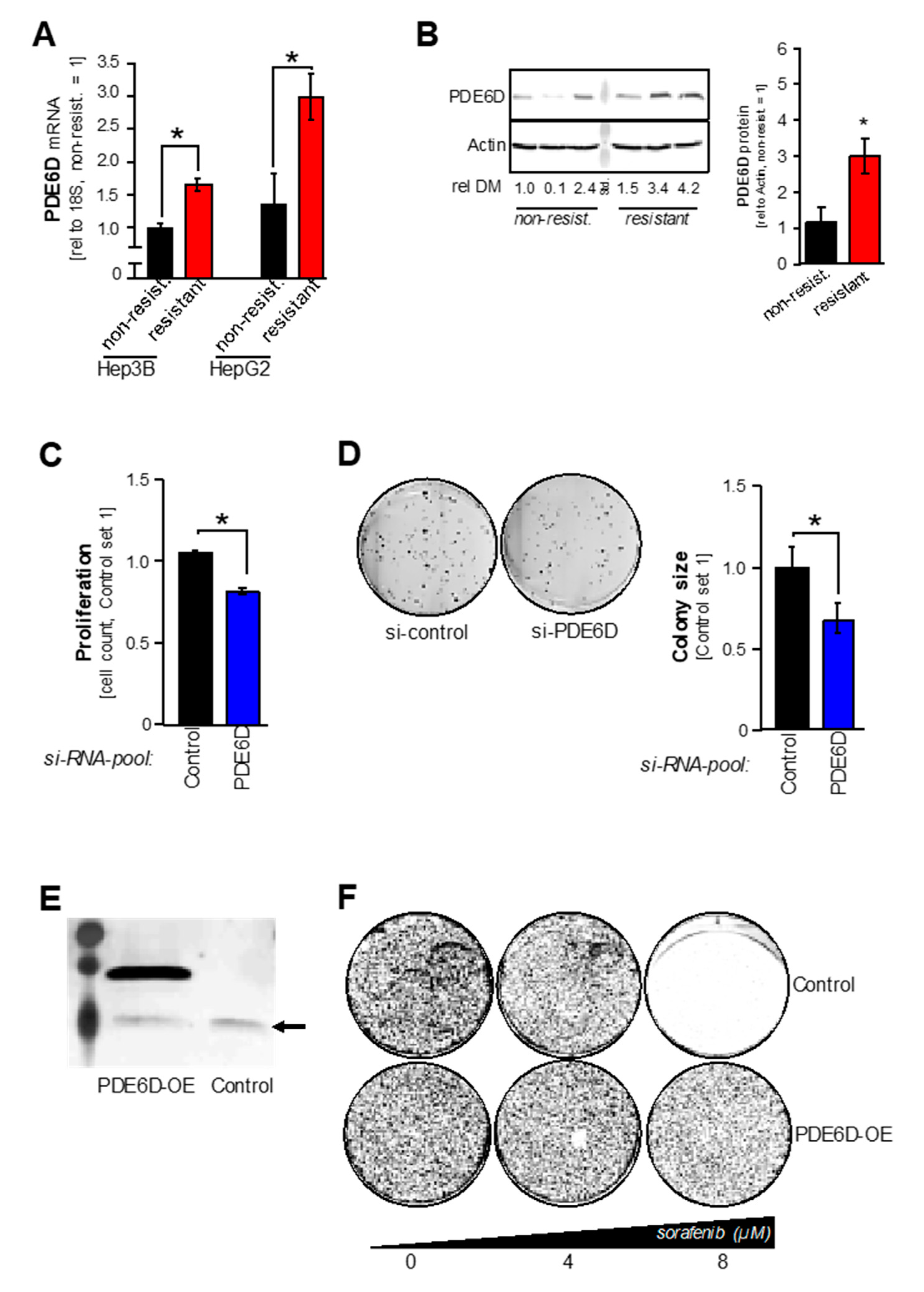
2.2. PDE6D Effects on HCC Proliferation, Clonogenicity and Sorafenib Resistance
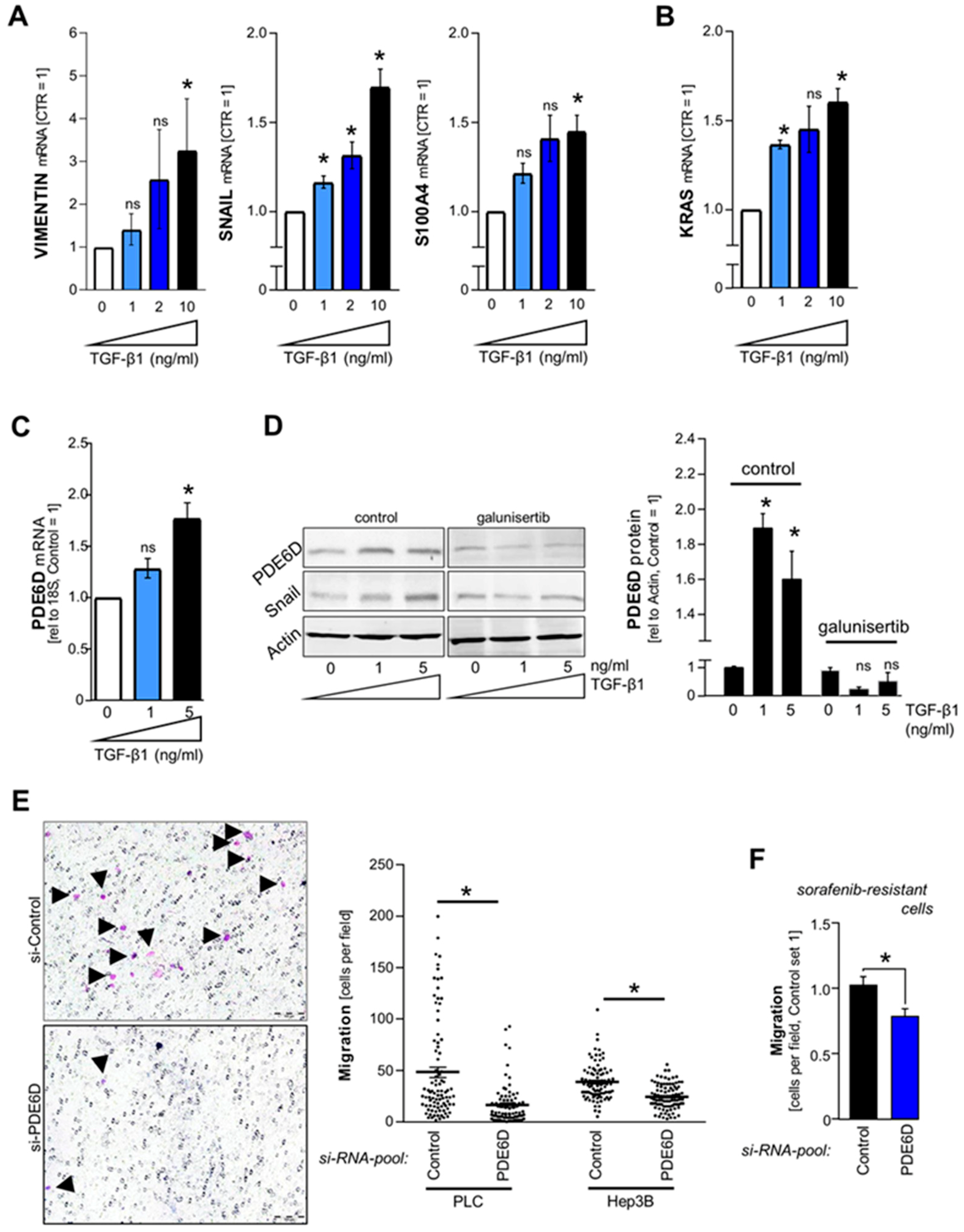
2.3. TGF-β-Mediated Regulation of PDE6D in HCC
2.4. Effect of PDE6D on HCC Cell Migration
2.5. Cellular Localization and Expression of PDE6D in HCC
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Human Material
4.3. Immunohistochemistry
4.4. Immunofluorescence
4.5. RNA Isolation and Reverse Transcription
4.6. Analysis of mRNA-Expression by Quantitative RT-PCR
4.7. Transfecting Cells with si-RNA-Pools and Plasmid DNA
4.8. Western Blotting
4.9. Nuclear and Cytoplasmatic Fractioning of HCC Cell Lysates
4.10. Functional Analysis of Proliferation, Clonogenicity, and Migration
4.11. In silico Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Steer, C.J.; Cressman, E.N. Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008, 48, 2047–2063. [Google Scholar] [CrossRef] [Green Version]
- Pascual, S.; Herrera, I.; Irurzun, J. New advances in hepatocellular carcinoma. World J. Hepatol. 2016, 8, 421–438. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug. Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Nishida, N.; Kitano, M.; Sakurai, T.; Kudo, M. Molecular Mechanism and Prediction of Sorafenib Chemoresistance in Human Hepatocellular Carcinoma. Dig. Dis. 2015, 33, 771–779. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Dietrich, P.; Koch, A.; Fritz, V.; Hartmann, A.; Bosserhoff, A.K.; Hellerbrand, C. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut 2018, 67, 1328–1341. [Google Scholar] [CrossRef]
- Dietrich, P.F.K.; Mahli, A.; Thasler, W.E.; Hellerbrand, C.; Bosserhoff, A.K. Combined effects of PLK1 and RAS in hepatocellular carcinoma reveal rigosertib as promising novel therapeutic “dual-hit” option. Oncotarget 2018, 9, 3605–3618. [Google Scholar] [CrossRef]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 2017, 37, 897. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. MicroRNA-622 is a novel mediator of tumorigenicity in melanoma by targeting Kirsten rat sarcoma. Pigment Cell Melanoma Res. 2018, 31, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.; Seo, S. RPGR, a prenylated retinal ciliopathy protein, is targeted to cilia in a prenylation- and PDE6D-dependent manner. Biol. Open 2016, 5, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, X.H.; Zhang, K.; Chen, C.K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cGMP phosphodiesterase delta subunit (PDEdelta) functions as a prenyl-binding protein. J. Biol. Chem. 2004, 279, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Nancy, V.; Callebaut, I.; El Marjou, A.; de Gunzburg, J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 2002, 277, 15076–15084. [Google Scholar] [CrossRef]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I.H. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Jang, H.; Gursoy, A.; Keskin, O.; Nussinov, R. PDEdelta Binding to Ras Isoforms Provides a Route to Proper Membrane Localization. J. Phys. Chem. B 2017, 121, 5917–5927. [Google Scholar] [CrossRef]
- Murarka, S.; Martin-Gago, P.; Schultz-Fademrecht, C.; Al Saabi, A.; Baumann, M.; Fansa, E.K.; Ismail, S.; Nussbaumer, P.; Wittinghofer, A.; Waldmann, H. Development of Pyridazinone Chemotypes Targeting the PDEdelta Prenyl Binding Site. Chemistry 2017, 23, 6083–6093. [Google Scholar] [CrossRef]
- Dharmaiah, S.; Bindu, L.; Tran, T.H.; Gillette, W.K.; Frank, P.H.; Ghirlando, R.; Nissley, D.V.; Esposito, D.; McCormick, F.; Stephen, A.G.; et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc. Natl. Acad. Sci. USA 2016, 113, E6766–E6775. [Google Scholar] [CrossRef]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martin-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Spiegel, J.; Cromm, P.M.; Zimmermann, G.; Grossmann, T.N.; Waldmann, H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. [Google Scholar] [CrossRef]
- Vasan, N.; Boyer, J.L.; Herbst, R.S. A RAS renaissance: Emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 3921–3930. [Google Scholar] [CrossRef]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef]
- Dong, H.; Claffey, K.P.; Brocke, S.; Epstein, P.M. Expression of phosphodiesterase 6 (PDE6) in human breast cancer cells. Springerplus 2013, 2, 680. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Yeh, Y.S.; Ma, C.J.; Huang, C.W.; Tsai, H.L.; Huang, M.Y.; Cheng, T.L.; Wang, J.Y. Optimization of a multigene biochip for detection of relapsed and early relapsed colorectal cancer. J. Surg. Res. 2017, 220, 427–437. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Roessler, S.; Jia, H.L.; Budhu, A.; Forgues, M.; Ye, Q.H.; Lee, J.S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.Y.; Qin, L.X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef]
- Wurmbach, E.; Chen, Y.B.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007, 45, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.O.; et al. Gene expression patterns in human liver cancers. Mol. Biol. Cell 2002, 13, 1929–1939. [Google Scholar] [CrossRef]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.; Kong, X.; Kulik, L.; Freise, C.E.; Olthoff, K.M.; Ghobrial, R.M.; McIver, P.; et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol. Med. 2009, 15, 85–94. [Google Scholar] [CrossRef]
- Archer, K.J.; Mas, V.R.; David, K.; Maluf, D.G.; Bornstein, K.; Fisher, R.A. Identifying genes for establishing a multigenic test for hepatocellular carcinoma surveillance in hepatitis C virus-positive cirrhotic patients. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2929–2932. [Google Scholar] [CrossRef]
- Su, A.I.; Welsh, J.B.; Sapinoso, L.M.; Kern, S.G.; Dimitrov, P.; Lapp, H.; Schultz, P.G.; Powell, S.M.; Moskaluk, C.A.; Frierson, H.F., Jr.; et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001, 61, 7388–7393. [Google Scholar]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFbeta signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol. Carcinog. 2017, 56, 1302–1311. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Giannelli, G.; Villa, E.; Lahn, M. Transforming growth factor-beta as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014, 74, 1890–1894. [Google Scholar] [CrossRef]
- Rani, B.; Malfettone, A.; Dituri, F.; Soukupova, J.; Lupo, L.; Mancarella, S.; Fabregat, I.; Giannelli, G. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018, 9, 373. [Google Scholar] [CrossRef] [Green Version]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef]
- Dzieran, J.; Fabian, J.; Feng, T.; Coulouarn, C.; Ilkavets, I.; Kyselova, A.; Breuhahn, K.; Dooley, S.; Meindl-Beinker, N.M. Comparative analysis of TGF-beta/Smad signaling dependent cytostasis in human hepatocellular carcinoma cell lines. PLoS ONE 2013, 8, e72252. [Google Scholar] [CrossRef]
- Bauer, R.; Valletta, D.; Bauer, K.; Thasler, W.E.; Hartmann, A.; Muller, M.; Reichert, T.E.; Hellerbrand, C. Downregulation of P-cadherin expression in hepatocellular carcinoma induces tumorigenicity. Int. J. Clin. Exp. Pathol. 2014, 7, 6125–6132. [Google Scholar] [CrossRef]
- Thasler, W.E.; Weiss, T.S.; Schillhorn, K.; Stoll, P.T.; Irrgang, B.; Jauch, K.W. Charitable State-Controlled Foundation Human Tissue and Cell Research: Ethic and Legal Aspects in the Supply of Surgically Removed Human Tissue for Research in the Academic and Commercial Sector in Germany. Cell Tissue Bank 2003, 4, 49–56. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016. [Google Scholar] [CrossRef]
- Kent, L.N.; Bae, S.; Tsai, S.Y.; Tang, X.; Srivastava, A.; Koivisto, C.; Martin, C.K.; Ridolfi, E.; Miller, G.C.; Zorko, S.M.; et al. Dosage-dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, L.; Jiang, M.; Huang, J.; Jiang, Z.; Feng, H.; Ji, Z. E2F1 interactive with BRCA1 pathway induces HCC two different small molecule metabolism or cell cycle regulation via mitochondrion or CD4+T to cytosol. J. Cell Physiol. 2018, 233, 1213–1221. [Google Scholar] [CrossRef]
- Wattanavanitchakorn, S.; Rojvirat, P.; Chavalit, T.; MacDonald, M.J.; Jitrapakdee, S. CCAAT-enhancer binding protein-alpha (C/EBPalpha) and hepatocyte nuclear factor 4alpha (HNF4alpha) regulate expression of the human fructose-1,6-bisphosphatase 1 (FBP1) gene in human hepatocellular carcinoma HepG2 cells. PLoS ONE 2018, 13, e0194252. [Google Scholar] [CrossRef]
- Desert, R.; Rohart, F.; Canal, F.; Sicard, M.; Desille, M.; Renaud, S.; Turlin, B.; Bellaud, P.; Perret, C.; Clement, B.; et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017, 66, 1502–1518. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Yang, F.; Yin, Y.; Wang, F.; Wang, Y.; Zhang, L.; Tang, Y.; Sun, S. miR-17-5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology 2010, 51, 1614–1623. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Cong, M.; Liu, T.; Xu, H.; Wang, L.; Sun, G.; Yang, A.; Zhang, D.; Huang, J.; Sun, Y.; et al. Inhibitory effects of HNF4alpha on migration/maltransformation of hepatic progenitors: HNF4alpha-overexpressing hepatic progenitors for liver repopulation. Stem Cell Res. 2017, 8, 183. [Google Scholar] [CrossRef]
- Lee, S.M.; Schelcher, C.; Demmel, M.; Hauner, M.; Thasler, W.E. Isolation of human hepatocytes by a two-step collagenase perfusion procedure. J. Vis. Exp. JOVE 2013. [Google Scholar] [CrossRef]
- Voller, D.; Reinders, J.; Meister, G.; Bosserhoff, A.K. Strong reduction of AGO2 expression in melanoma and cellular consequences. Br. J. Cancer 2013, 109, 3116–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannus, M.; Beitzinger, M.; Engelmann, J.C.; Weickert, M.T.; Spang, R.; Hannus, S.; Meister, G. siPools: Highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014, 42, 8049–8061. [Google Scholar] [CrossRef] [PubMed]
- Ruedel, A.; Dietrich, P.; Schubert, T.; Hofmeister, S.; Hellerbrand, C.; Bosserhoff, A.K. Expression and function of microRNA-188-5p in activated rheumatoid arthritis synovial fibroblasts. Int. J. Clin. Exp. Pathol. 2015, 8, 6607–6616. [Google Scholar] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dietrich, P.; Hellerbrand, C.; Bosserhoff, A. The Delta Subunit of Rod-Specific Photoreceptor cGMP Phosphodiesterase (PDE6D) Contributes to Hepatocellular Carcinoma Progression. Cancers 2019, 11, 398. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030398
Dietrich P, Hellerbrand C, Bosserhoff A. The Delta Subunit of Rod-Specific Photoreceptor cGMP Phosphodiesterase (PDE6D) Contributes to Hepatocellular Carcinoma Progression. Cancers. 2019; 11(3):398. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030398
Chicago/Turabian StyleDietrich, Peter, Claus Hellerbrand, and Anja Bosserhoff. 2019. "The Delta Subunit of Rod-Specific Photoreceptor cGMP Phosphodiesterase (PDE6D) Contributes to Hepatocellular Carcinoma Progression" Cancers 11, no. 3: 398. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030398