RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
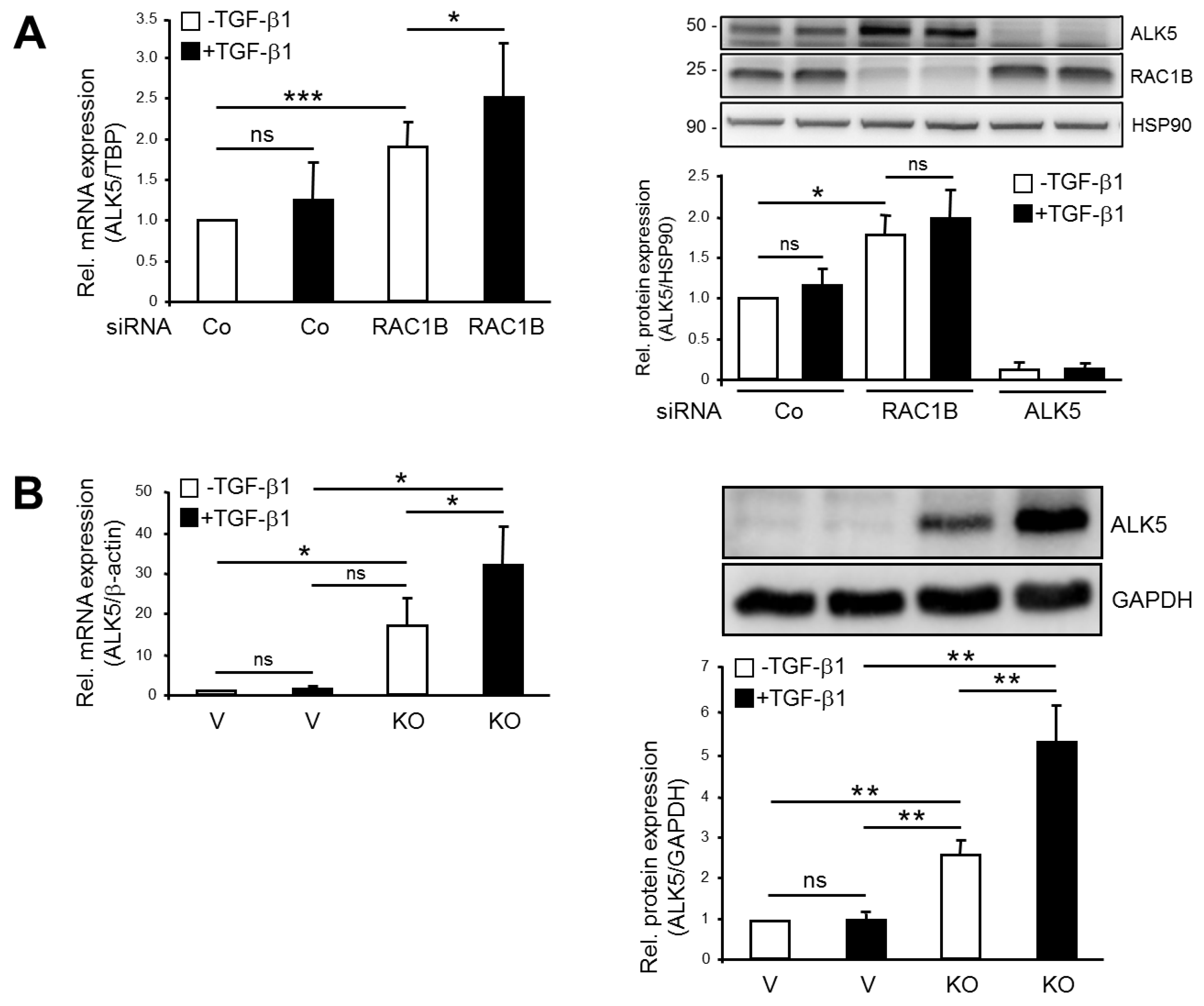
2.1. Knockout (KO) and Knockdown (KD) of RAC1B Increased Expression of ALK5
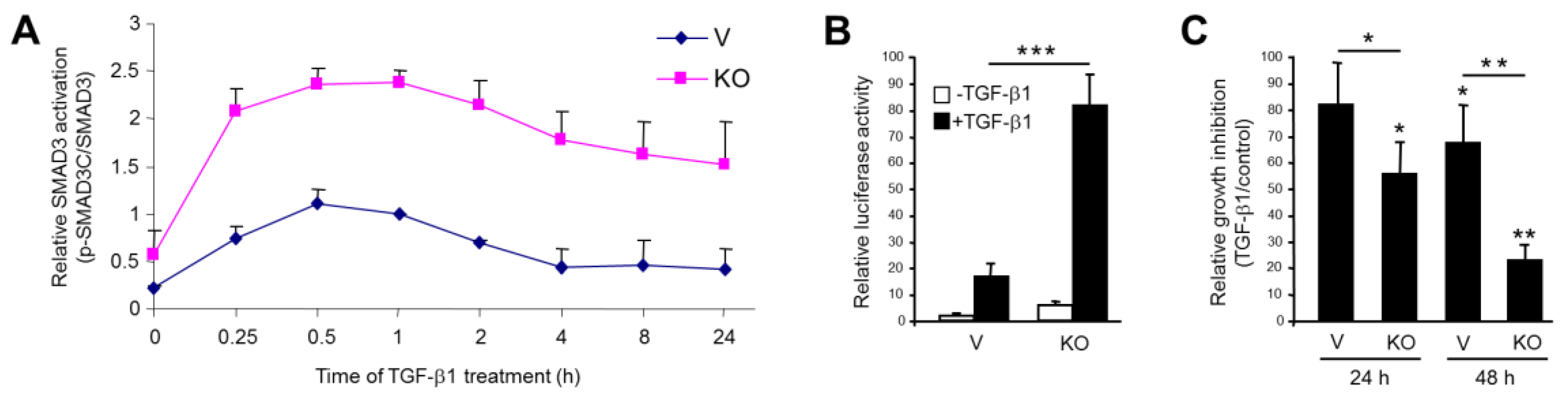
2.2. Stable KO of RAC1B Enhanced TGF-β1-Induced SMAD3 Activation and SMAD3-Dependent Responses
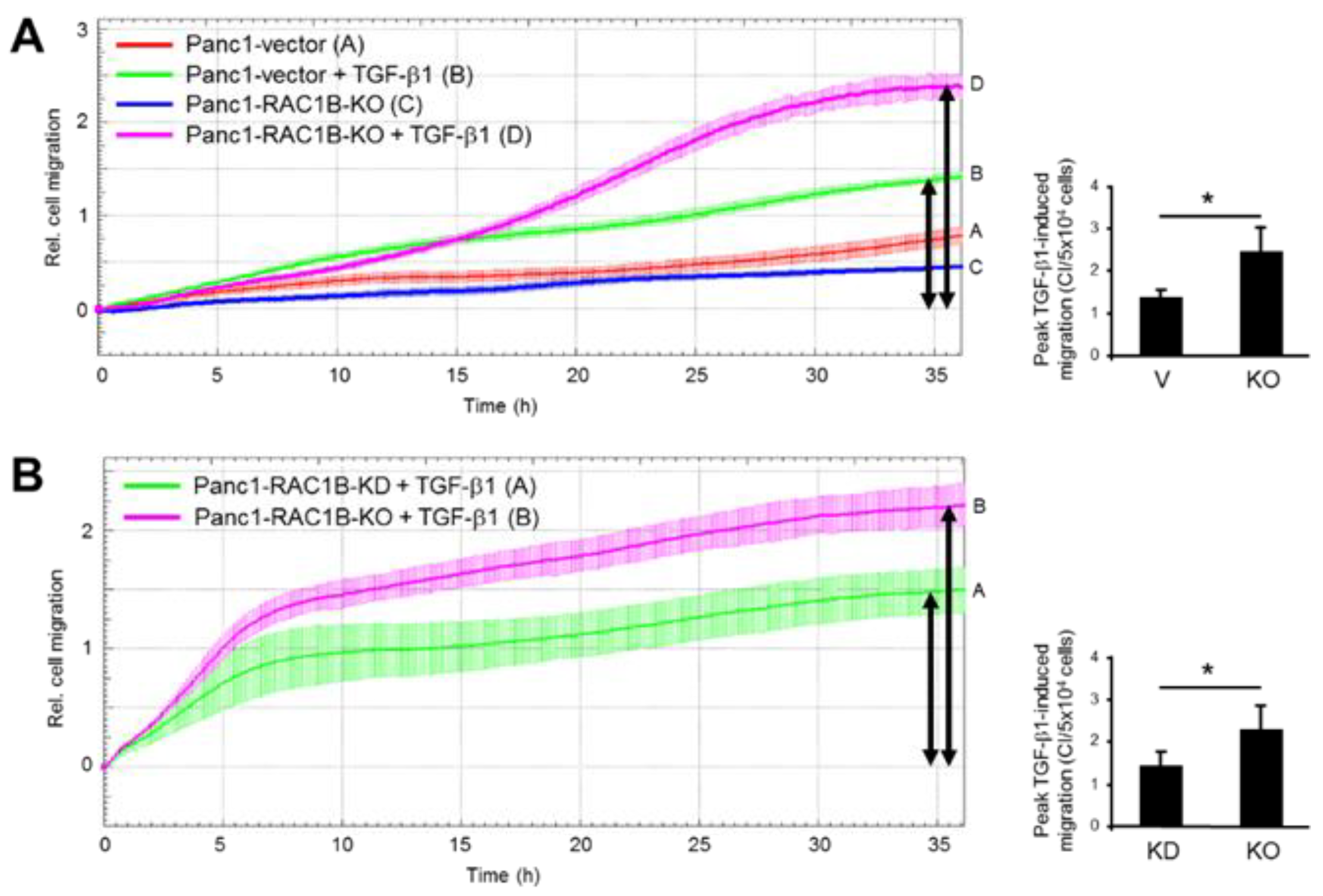
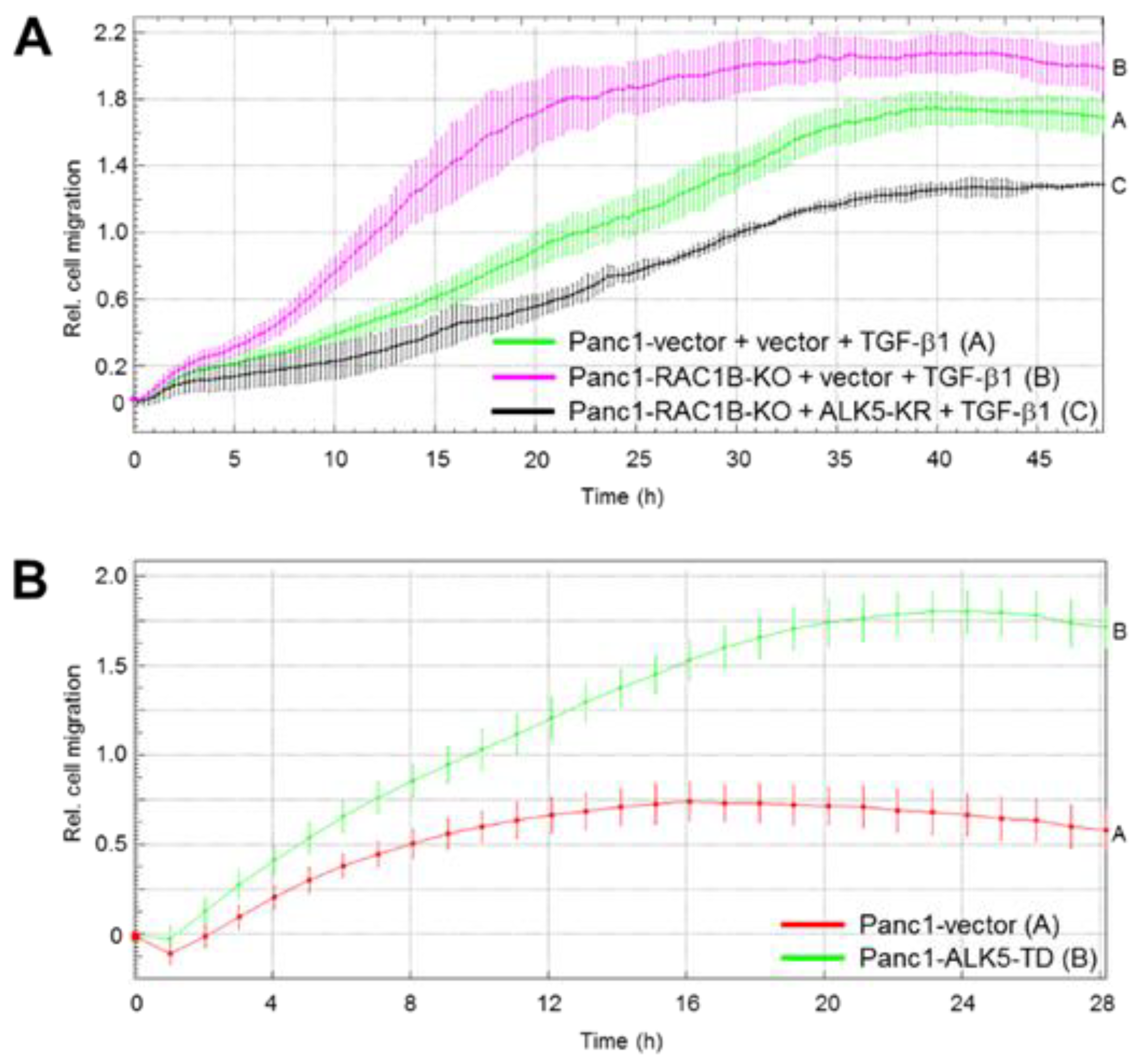
2.3. Stable KO of RAC1B Enhanced TGF-β1-Dependent Migration
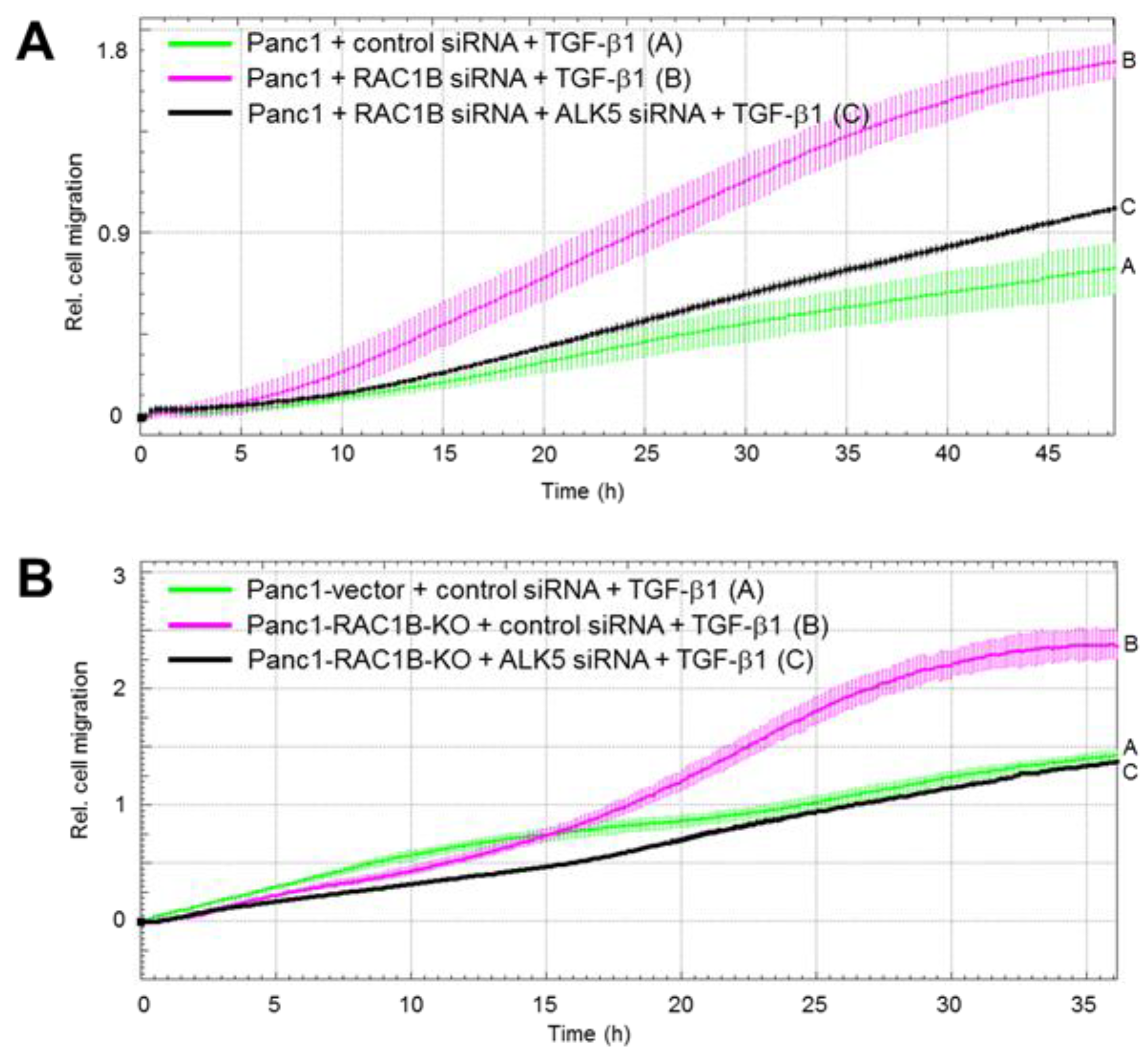
2.4. Downregulation of the Protein Abundance of ALK5 Suppressed TGF-β1-Induced Cell Migration Increase by RAC1B Deletion
2.5. ALK5 Kinase Activity is Required for RAC1B KD-Mediated Increase in TGF-β1-Induced Cell Migration
3. Discussion
4. Material and Methods
4.1. Antibodies and Reagents
4.2. Cells
4.3. Generation of RAC1 Exon 3b-Deleted Panc1 Cells by CRISPR/Cas9 Technology
4.4. QPCR Analysis
4.5. Transient Transfection of siRNAs and Plasmid Vectors
4.6. Cell Lysis and Immunoblotting
4.7. Dual Luciferase Assays
4.8. Real-Time Cell Migration Assays
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with versatile functions in malignant transformation and tumor progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R.; Ungefroren, H. TGF-beta-dependent growth arrest and cell migration in benign and malignant breast epithelial cells are antagonistically controlled by Rac1 and Rac1b. Int. J. Mol. Sci. 2017, 18, 1574. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, A.; Moepert, K.; Dames, S.; Sternberger, M.; Kaufmann, J.; Klippel, A. Differential regulation of TGF-beta signaling through Smad2, Smad3 and Smad4. Oncogene 2003, 22, 6748–6763. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Groth, S.; Sebens Müerköster, S.; Sipos, B.; Schäfer, H.; Kalthoff, H.; Fändrich, F.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Xu, P.; Lamouille, S.; Xu, J.; Derynck, R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell. 2009, 35, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of negative controls on TGF-β1 signaling in tumor progression. Cancers 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, H.; Li, H.; Gao, Z.; Song, K. Atrial overexpression of microRNA-27b attenuates angiotensin II-induced atrial fibrosis and fibrillation by targeting ALK5. Hum. Cell. 2018, 31, 251–260. [Google Scholar] [CrossRef]
- Fu, G.; Ye, G.; Nadeem, L.; Ji, L.; Manchanda, T.; Wang, Y.; Zhao, Y.; Qiao, J.; Wang, Y.L.; Lye, S.; et al. MicroRNA-376c impairs transforming growth factor-β and nodal signaling to promote trophoblast cell proliferation and invasion. Hypertension 2013, 61, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Mihara, K.; Ramachandran, R.; Saifeddine, M.; Hansen, K.K.; Renaux, B.; Polley, D.; Gibson, S.; Vanderboor, C.; Hollenberg, M.D. Thrombin-mediated direct activation of proteinase-activated Receptor-2: Another target for thrombin signaling. Mol. Pharmacol. 2016, 89, 606–614. [Google Scholar] [CrossRef]
- E-CRISP Home Page. Available online: http://www.e-crisp.org/E-CRISP/index.html (accessed on 7 April 2018).
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Witte, D.; Mihara, K.; Rauch, B.H.; Henklein, P.; Jöhren, O.; Bonni, S.; Settmacher, U.; Lehnert, H.; Hollenberg, M.D.; et al. Transforming growth factor-β1/Activin receptor-like kinase 5-mediated cell migration is dependent on the protein proteinase-activated Receptor 2 but not on proteinase-activated Receptor 2-stimulated Gq-calcium signaling. Mol. Pharmacol. 2017, 92, 519–532. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, 691. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050691
Ungefroren H, Otterbein H, Fiedler C, Mihara K, Hollenberg MD, Gieseler F, Lehnert H, Witte D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers. 2019; 11(5):691. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050691
Chicago/Turabian StyleUngefroren, Hendrik, Hannah Otterbein, Christian Fiedler, Koichiro Mihara, Morley D. Hollenberg, Frank Gieseler, Hendrik Lehnert, and David Witte. 2019. "RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5" Cancers 11, no. 5: 691. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050691