Huzhangoside A Suppresses Tumor Growth through Inhibition of Pyruvate Dehydrogenase Kinase Activity


Abstract
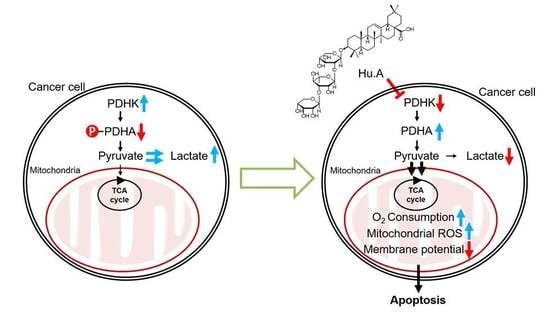
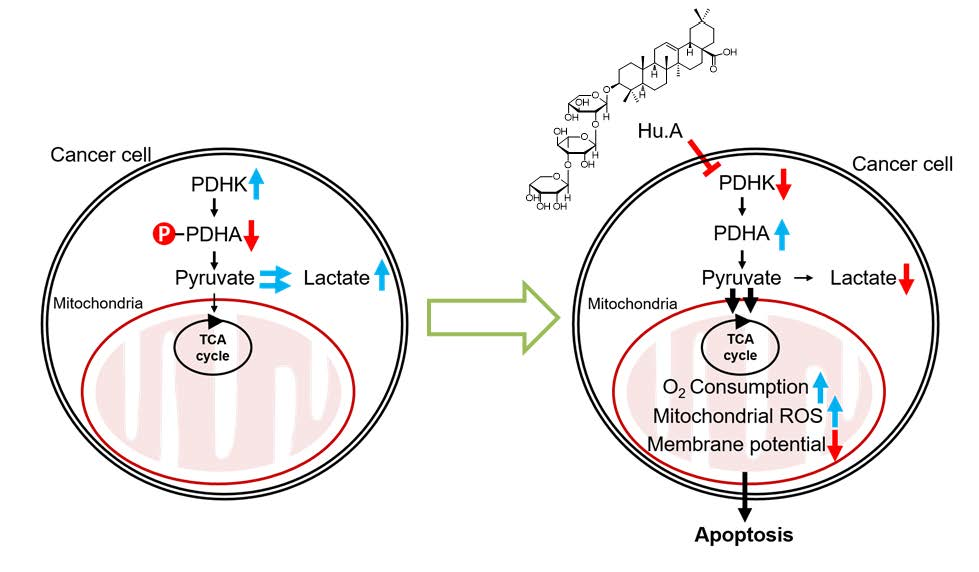
:
1. Introduction
2. Results
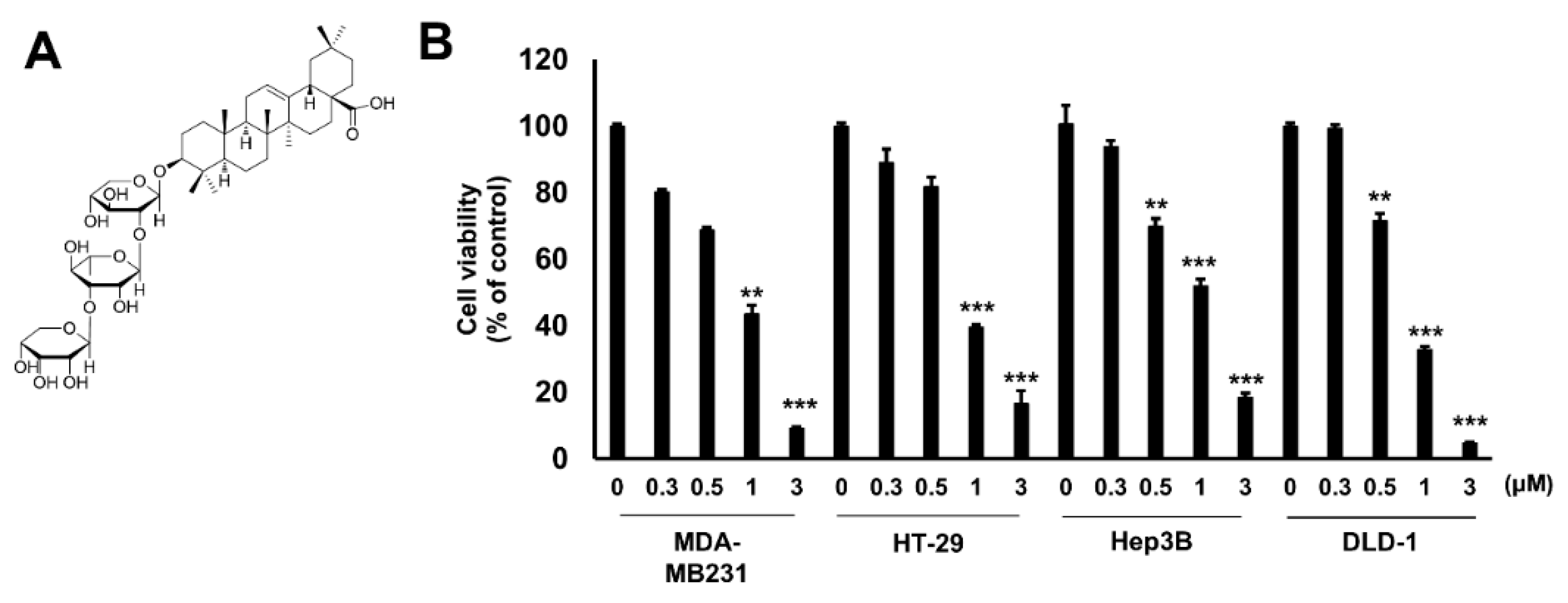
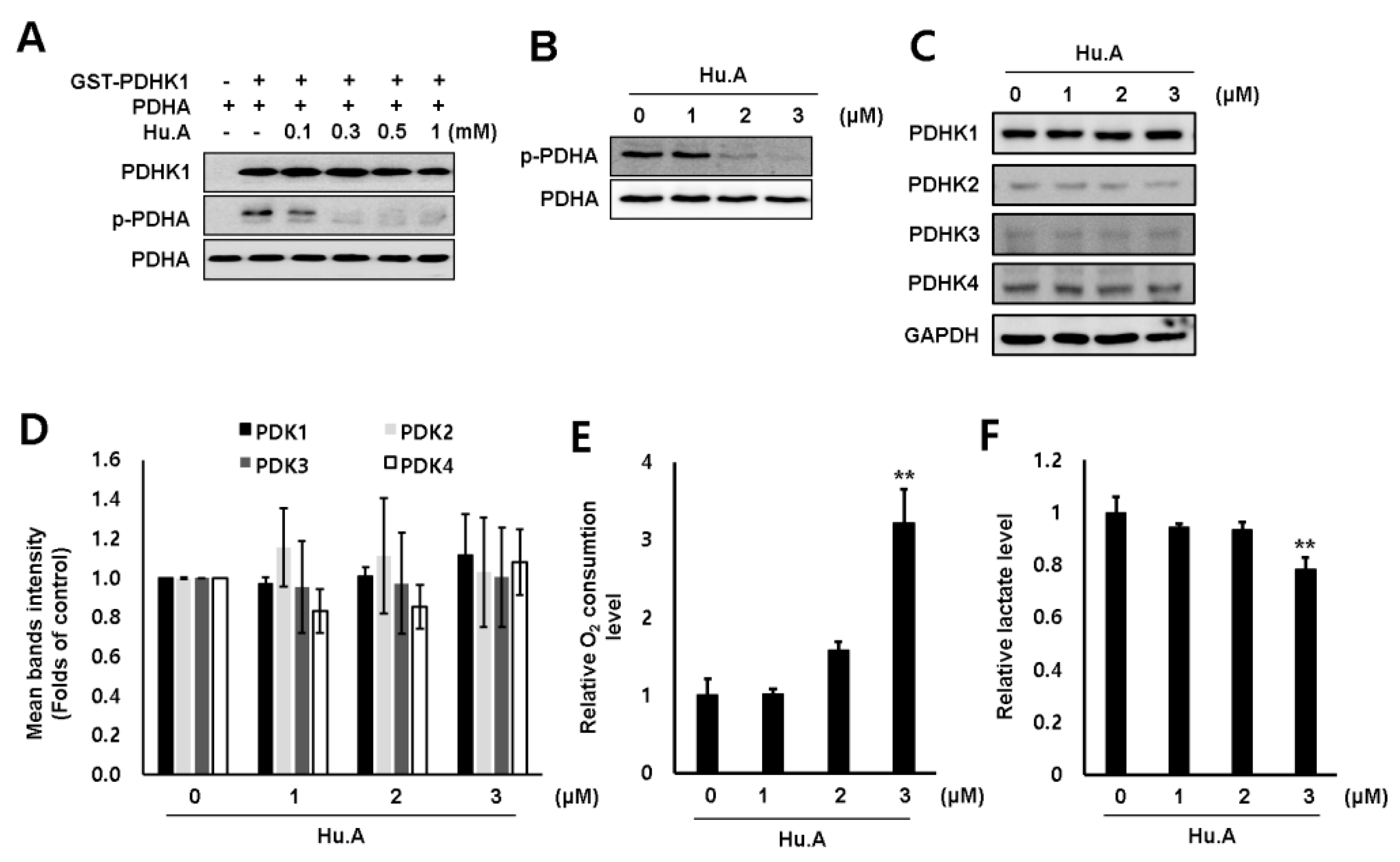
2.1. Hu.A Decreases Cancer Cell Viability and Inhibits PDHK Enzyme Activity
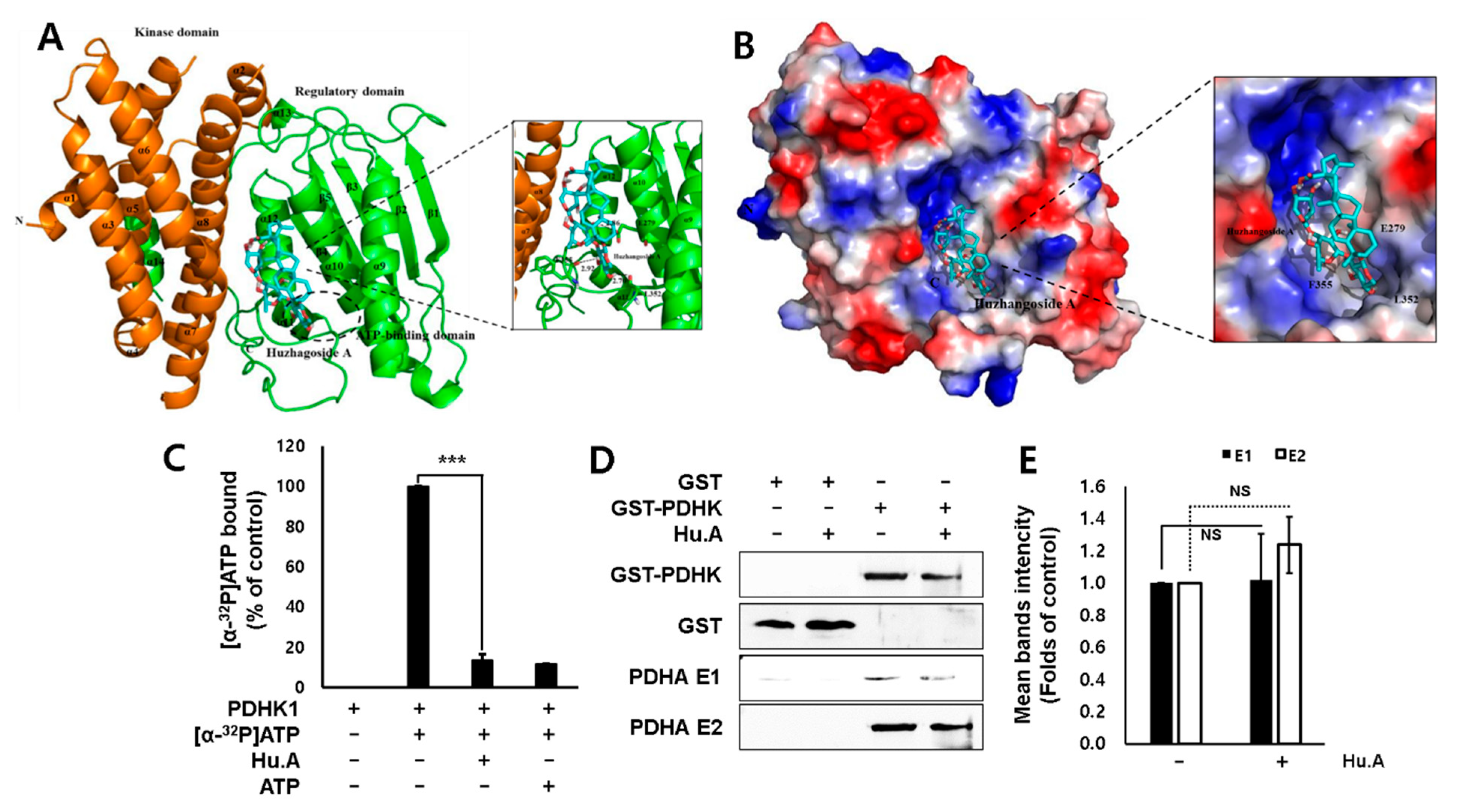
2.2. Hu.A Inhibits PDHK Enzyme Activity by Binding to the ATP-Binding Pocket of PDHK1
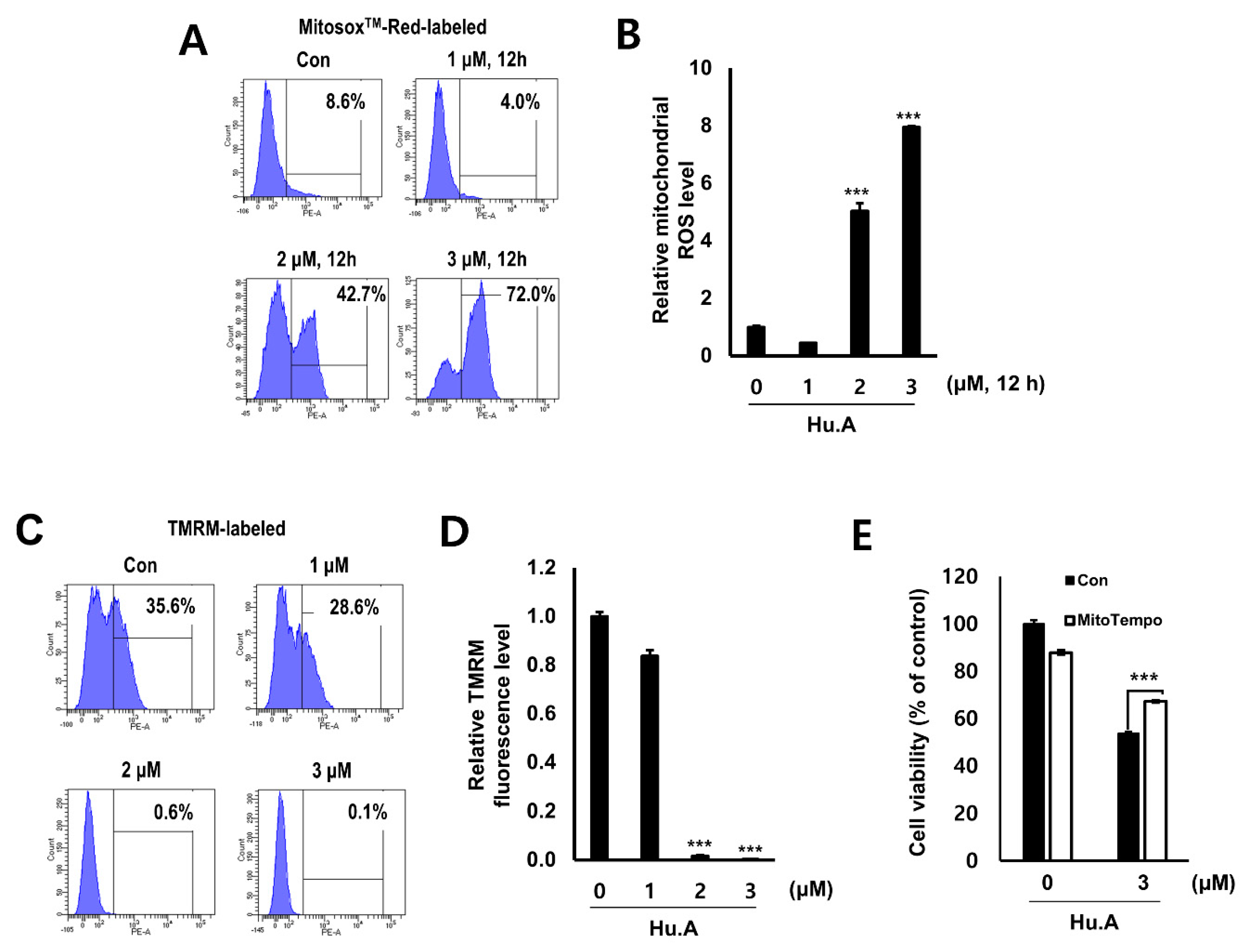
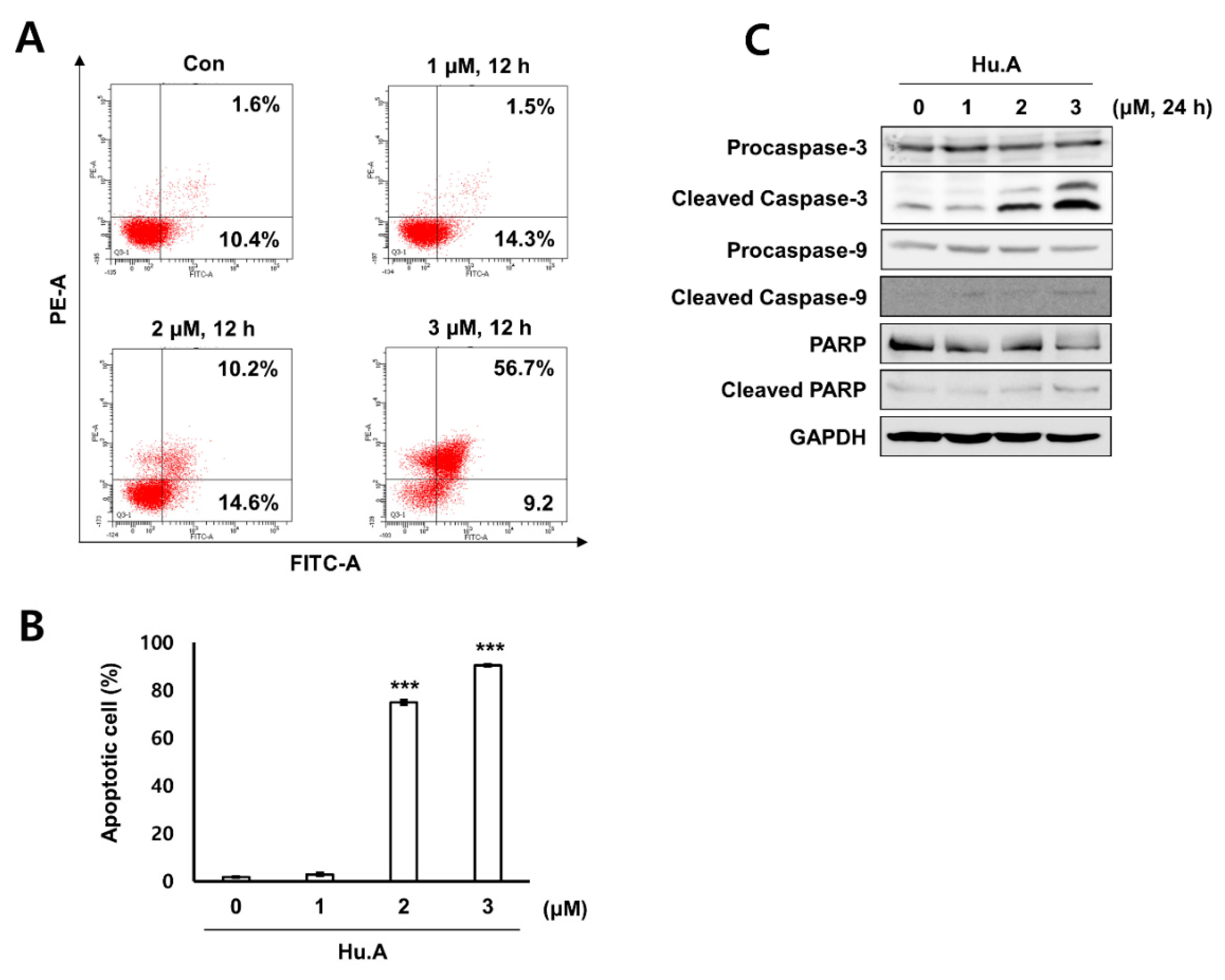
2.3. Hu.A Induces Mitochondrial ROS and Mitochondrial Damage in DLD-1 Cells
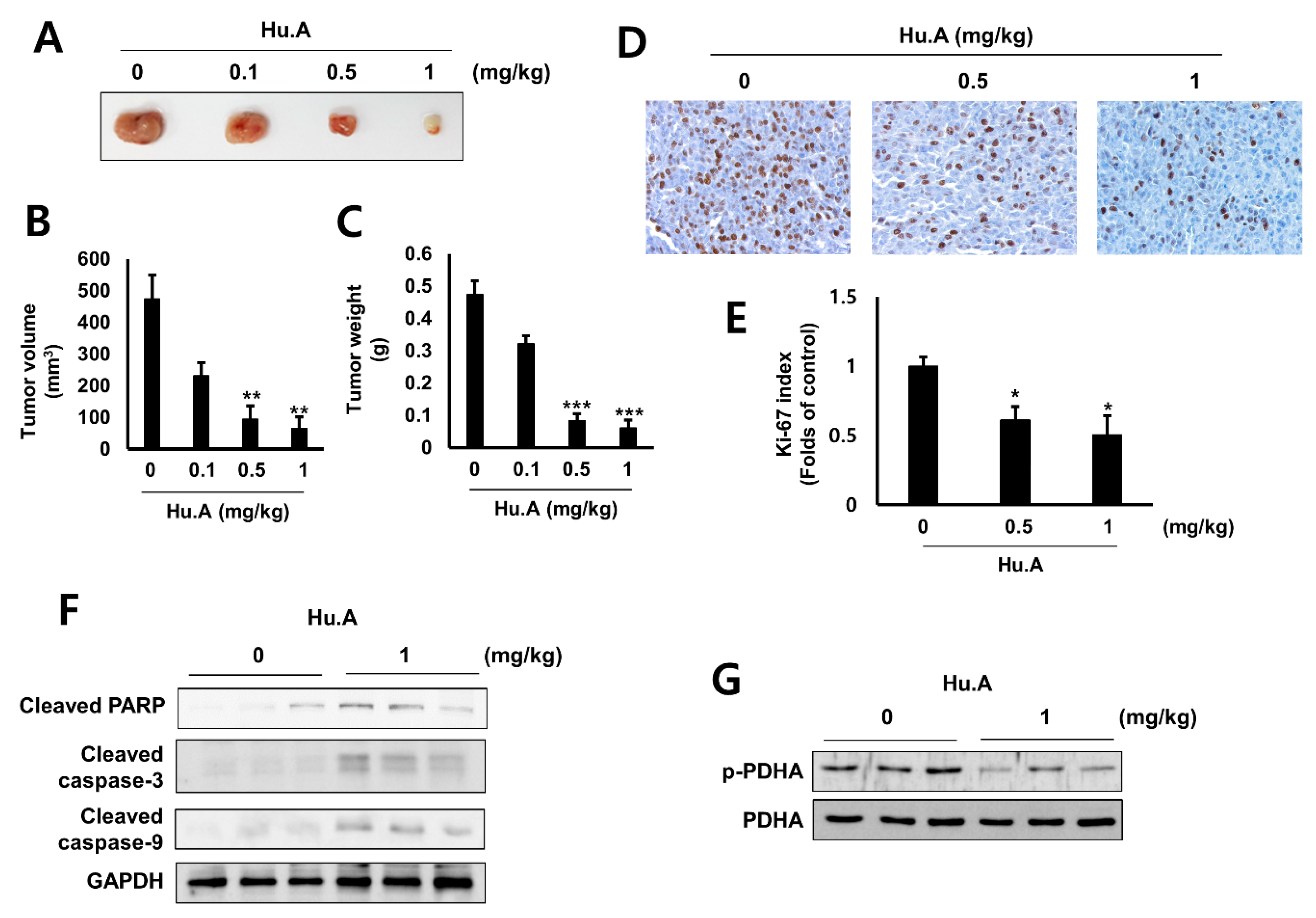
2.4. Hu.A Decreases Tumor Growth and Phospho-PDHA Levels In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Extraction and Isolation
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. In Vitro PDHK Kinase Assay
4.6. Western Blotting Assay
4.7. O2 Consumption Assay
4.8. Lactate Production Assay
4.9. Structural Prediction of PDHK1 and Hu.A interaction
4.10. ATP-Binding Assays
4.11. PDC Subunits and PDHK Binding Assay
4.12. Measurement of Mitochondrial Reactive Oxygen Species (ROS) and Mitochondrial Depolarization Assay
4.13. Annexin V and Propidium Iodide (PI) Staining
4.14. Animals
4.15. Tumor Allograft and Drug Treatment
4.16. Immunohistochemistry
4.17. Blood Biochemistry
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rardin, M.J.; Wiley, S.E.; Naviaux, R.K.; Murphy, A.N.; Dixon, J.E. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 2009, 389, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Popov, K.M.; Kedishvili, N.Y.; Zhao, Y.; Shimomura, Y.; Crabb, D.W.; Harris, R.A. Primary structure of pyruvate dehydrogenase kinase establishes a new family of eukaryotic protein kinases. J. Biol. Chem. 1993, 268, 26602–26606. [Google Scholar]
- Gudi, R.; Melissa, M.B.-K.; Kedishvili, N.Y.; Zhao, Y.; Popov, K.M. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J. Biol. Chem. 1995, 270, 28989–28994. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anti-Cancer Agents Med. Chem. 2008, 8, 305–312. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tso, S.-C.; Qi, X.; Gui, W.-J.; Wu, C.-Y.; Chuang, J.L.; Wernstedt-Asterholm, I.; Morlock, L.K.; Owens, K.R.; Scherer, P.E.; Williams, N.S. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 2014, 289, 4432–4443. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Su, J.; Sun, L. Computational study on new natural compound inhibitors of pyruvate dehydrogenase kinases. Int. J. Mol. Sci. 2016, 17, 340. [Google Scholar] [CrossRef]
- Sun, W.; Xie, Z.; Liu, Y.; Zhao, D.; Wu, Z.; Zhang, D.; Lv, H.; Tang, S.; Jin, N.; Jiang, H. JX06 selectively inhibits pyruvate dehydrogenase kinase PDK1 by a covalent cysteine modification. Cancer Res. 2015, 75, 4923–4936. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Ohtani, K.; Wei, J.-X.; Kasai, R.; Tanaka, O. Saponins from Anemone rivularis. Planta Med. 1984, 50, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-C.; Shao, J.-H.; Fan, J.-D. A new triterpenoid with antimicrobial activity from Anemone rivularis. Chem. Nat. Compd. 2012, 48, 803–805. [Google Scholar] [CrossRef]
- Yokosuka, A.; Sano, T.; Hashimoto, K.; Sakagami, H.; Mimaki, Y. Triterpene glycosides from the whole plant of Anemone hupehensis var. japonica and their cytotoxic activity. Chem. Pharm. Bull. 2009, 57, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.-W.; Lee, J.H.; Choi, H.-J.; Park, M.-J.; Kim, E.-Y.; Han, J.H.; Jang, S.B.; Lee, S.-O.; Lee, S.W.; Hang, J. Anemone rivularis inhibits pyruvate dehydrogenase kinase activity and tumor growth. J. Ethnopharmacol. 2017, 203, 47–54. [Google Scholar] [CrossRef]
- Hitosugi, T.; Fan, J.; Chung, T.W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; McConathy, J.; Couture, L.E.; Su, Y.; Massoumzadeh, P.; Leeds, H.S.; Chicoine, M.R.; Tran, D.D.; Huang, J.; Dahiya, S.; et al. Aerobic Glycolysis as a Marker of Tumor Aggressiveness: Preliminary Data in High Grade Human Brain Tumors. Dis. Markers 2015, 2015, 874904. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Kolobova, E.; Tuganova, A.; Boulatnikov, I.; Popov, K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 2001, 358, 69–77. [Google Scholar] [CrossRef]
- Jeoung, N.H. Pyruvate Dehydrogenase Kinases: Therapeutic Targets for Diabetes and Cancers. Diabetes Metab. J. 2015, 39, 188–197. [Google Scholar] [CrossRef]
- Roche, T.E.; Hiromasa, Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 2007, 64, 830–849. [Google Scholar] [CrossRef]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Zhang, S.; Perry, B.; Zhao, T.; Wang, Y.; Sun, C. Radicicol, a heat shock protein 90 inhibitor, inhibits differentiation and adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2013, 436, 169–174. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Porrata, L.F.; Adjei, A.A. The pharmacologic basis of high dose chemotherapy with haematopoietic stem cell support for solid tumours. Br. J. Cancer 2001, 85, 484–489. [Google Scholar] [CrossRef]
- Fan, J.; Hitosugi, T.; Chung, T.-W.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S. Tyrosine phosphorylation of lactate dehydrogenase a is important for NADH/NAD+ redox homeostasis in cancer cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Hu.A | ALT (U/L) | AST (U/L) | BUN (mg/dL) | Creatinine (mg/dL) |
|---|---|---|---|---|
| Control | 14.8 ± 2.5 | 79.8 ± 35.3 | 8.6 ± 0.8 | < 0.17 |
| 0.1 (mg/kg) | 16.7 ± 2.5 | 94.5 ± 41.0 | 8.8 ± 0.9 | < 0.17 |
| 0.5 (mg/kg) | 19.2 ± 5.6 | 87.5 ± 33.8 | 8.9 ± 0.6 | < 0.17 |
| 1 (mg/kg) | 19.6 ± 3.1 | 95.2 ± 43.1 | 8.4 ± 1.0 | < 0.17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwak, C.-H.; Lee, J.-H.; Kim, E.-Y.; Han, C.W.; Kim, K.-J.; Lee, H.; Cho, M.; Jang, S.B.; Kim, C.-H.; Chung, T.-W.; et al. Huzhangoside A Suppresses Tumor Growth through Inhibition of Pyruvate Dehydrogenase Kinase Activity. Cancers 2019, 11, 712. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050712
Kwak C-H, Lee J-H, Kim E-Y, Han CW, Kim K-J, Lee H, Cho M, Jang SB, Kim C-H, Chung T-W, et al. Huzhangoside A Suppresses Tumor Growth through Inhibition of Pyruvate Dehydrogenase Kinase Activity. Cancers. 2019; 11(5):712. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050712
Chicago/Turabian StyleKwak, Choong-Hwan, Jung-Hee Lee, Eun-Yeong Kim, Chang Woo Han, Keuk-Jun Kim, Hanna Lee, MyoungLae Cho, Se Bok Jang, Cheorl-Ho Kim, Tae-Wook Chung, and et al. 2019. "Huzhangoside A Suppresses Tumor Growth through Inhibition of Pyruvate Dehydrogenase Kinase Activity" Cancers 11, no. 5: 712. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050712