Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
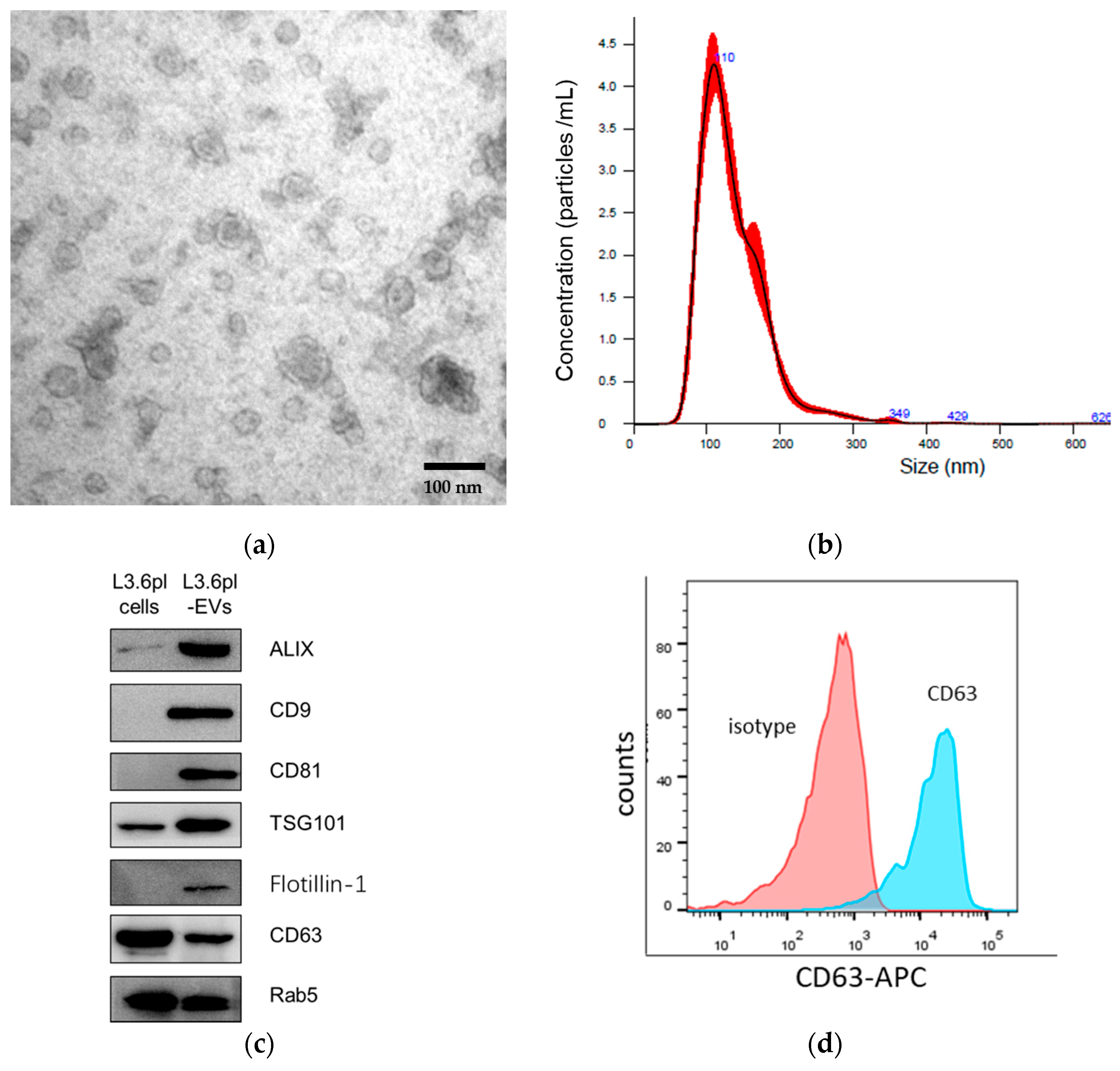
2.1. Characterization of Pancreatic Cancer-Derived EVs
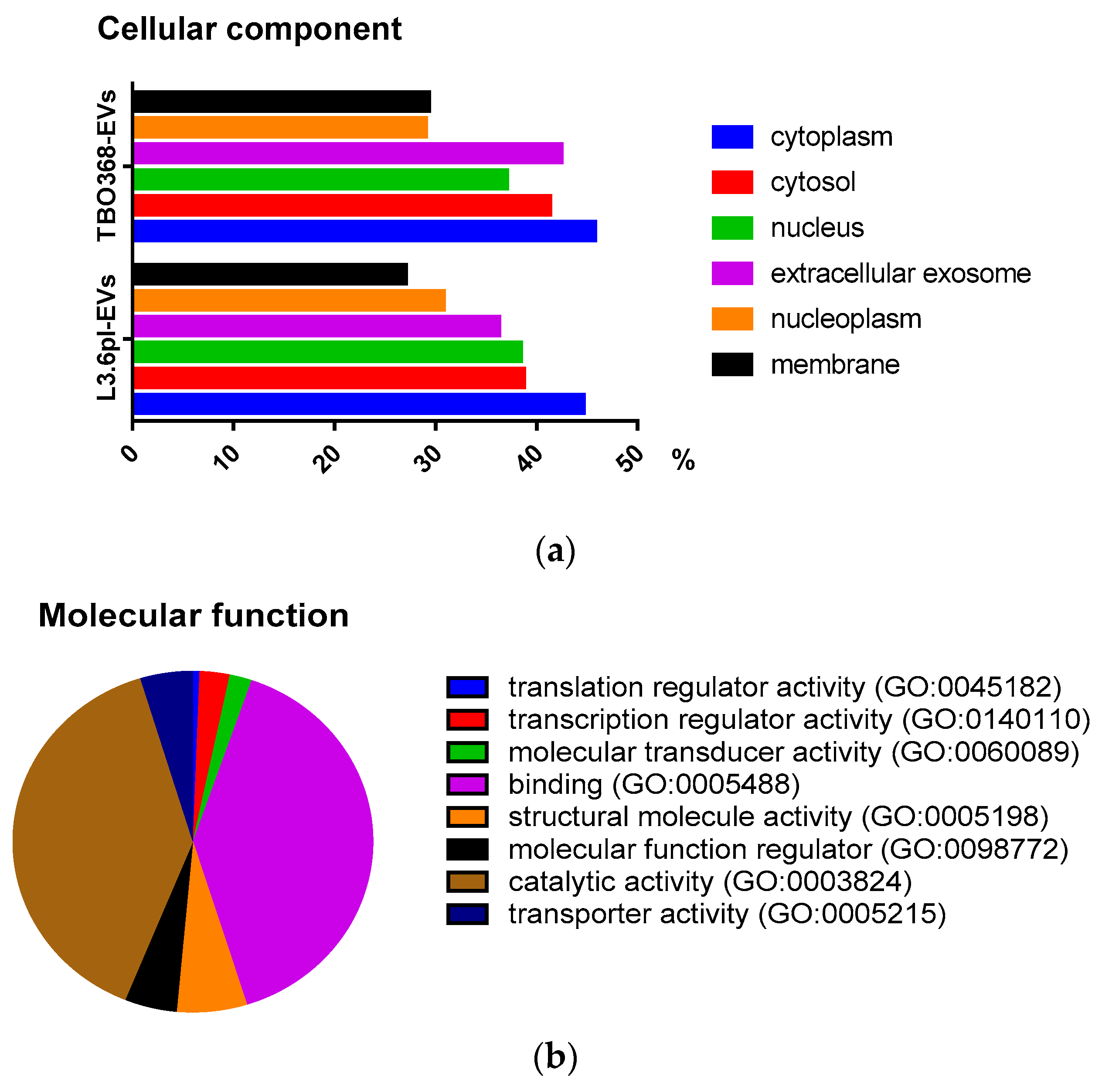
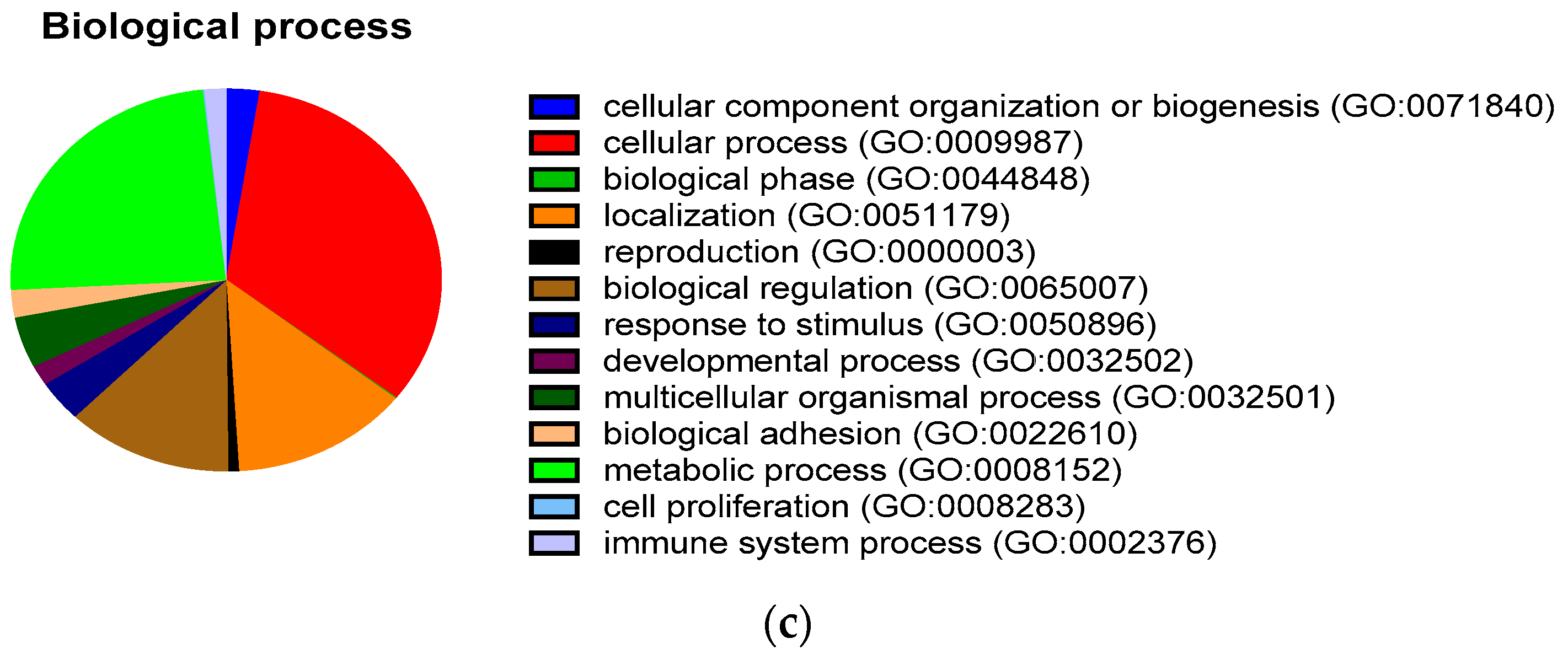
2.2. Comprehensive Proteomic Analysis of Pancreatic Cancer-Derived EVs
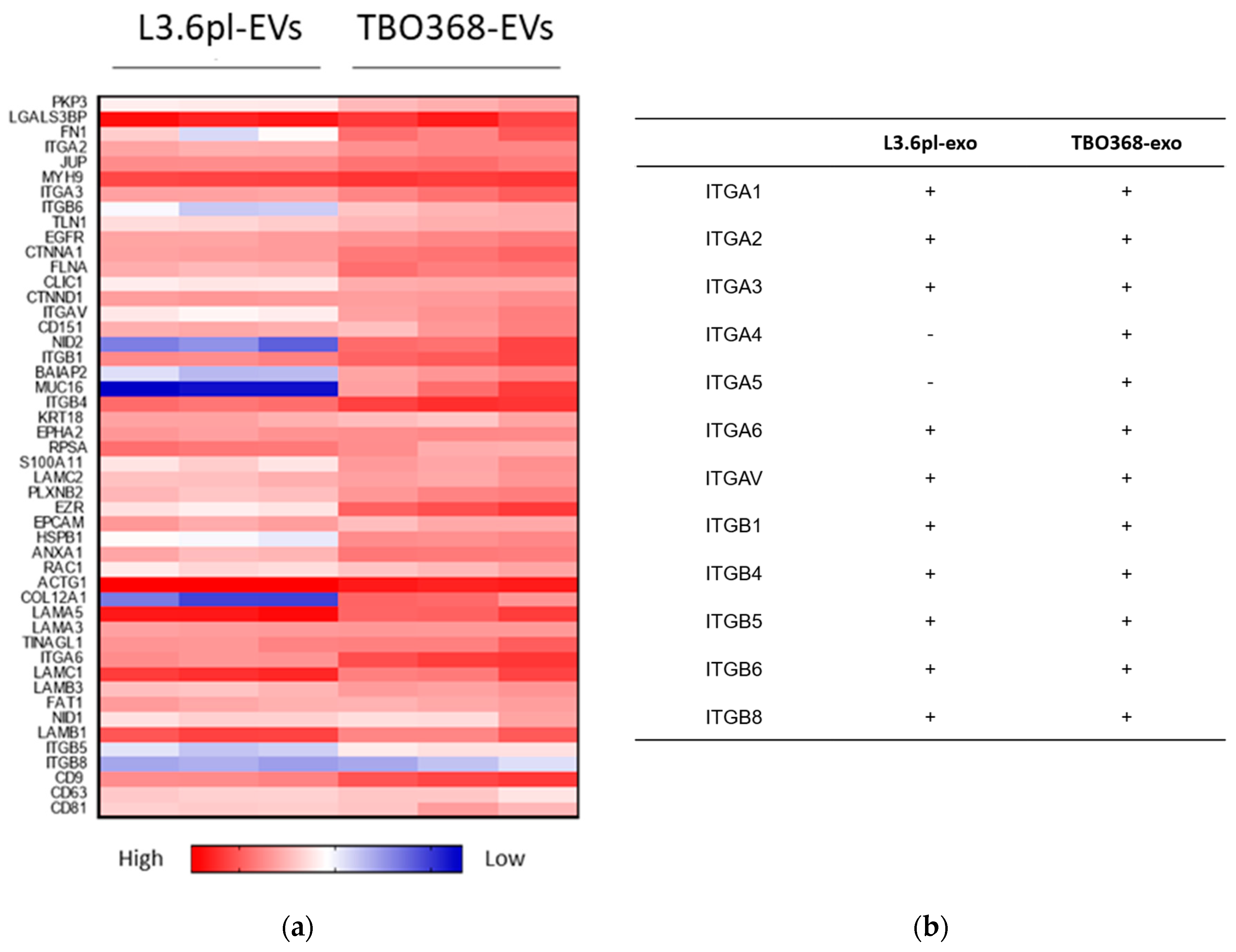
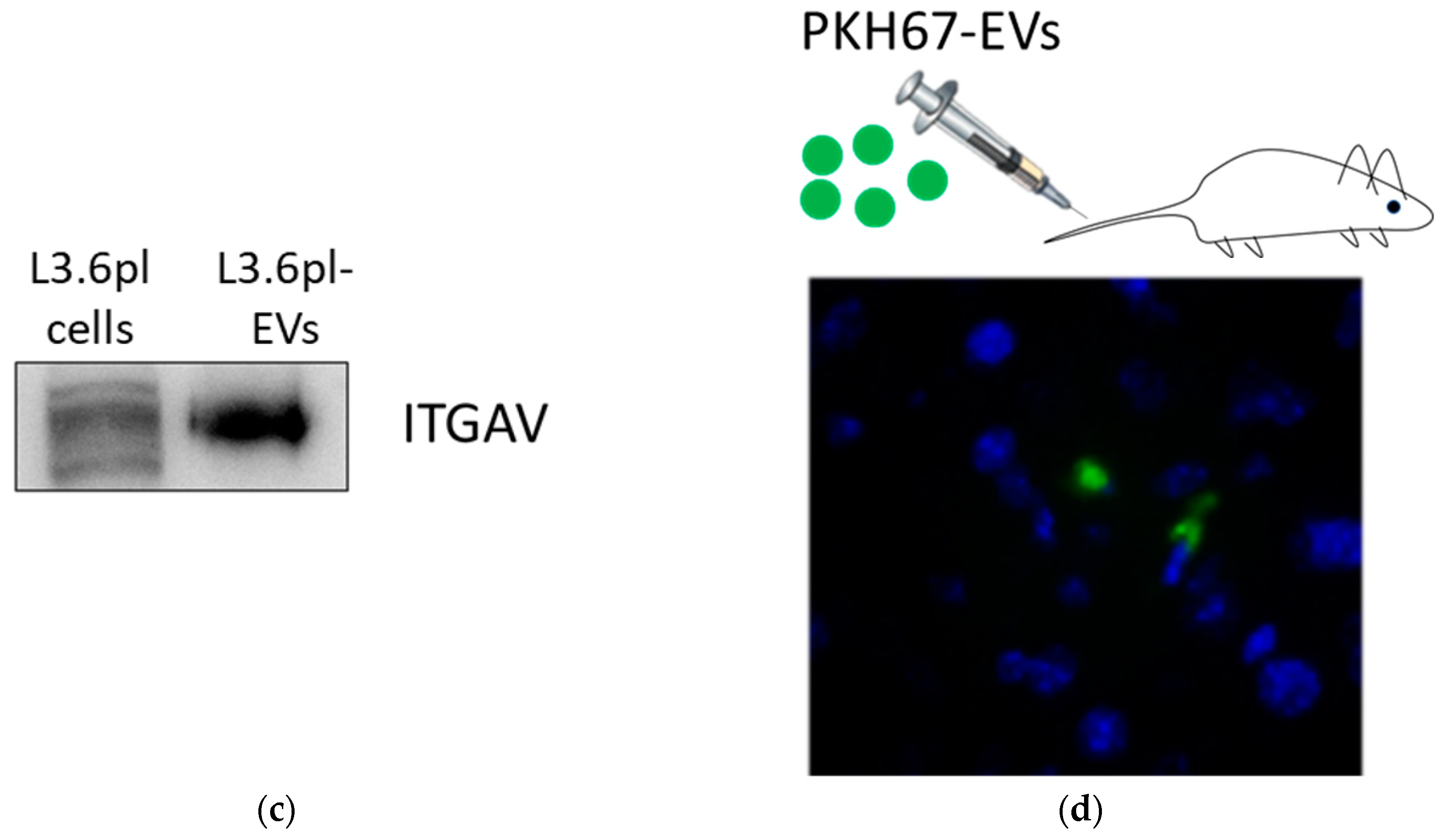
2.3. Pancreatic Cancer-Derived EVs Carry Adhesion Molecules
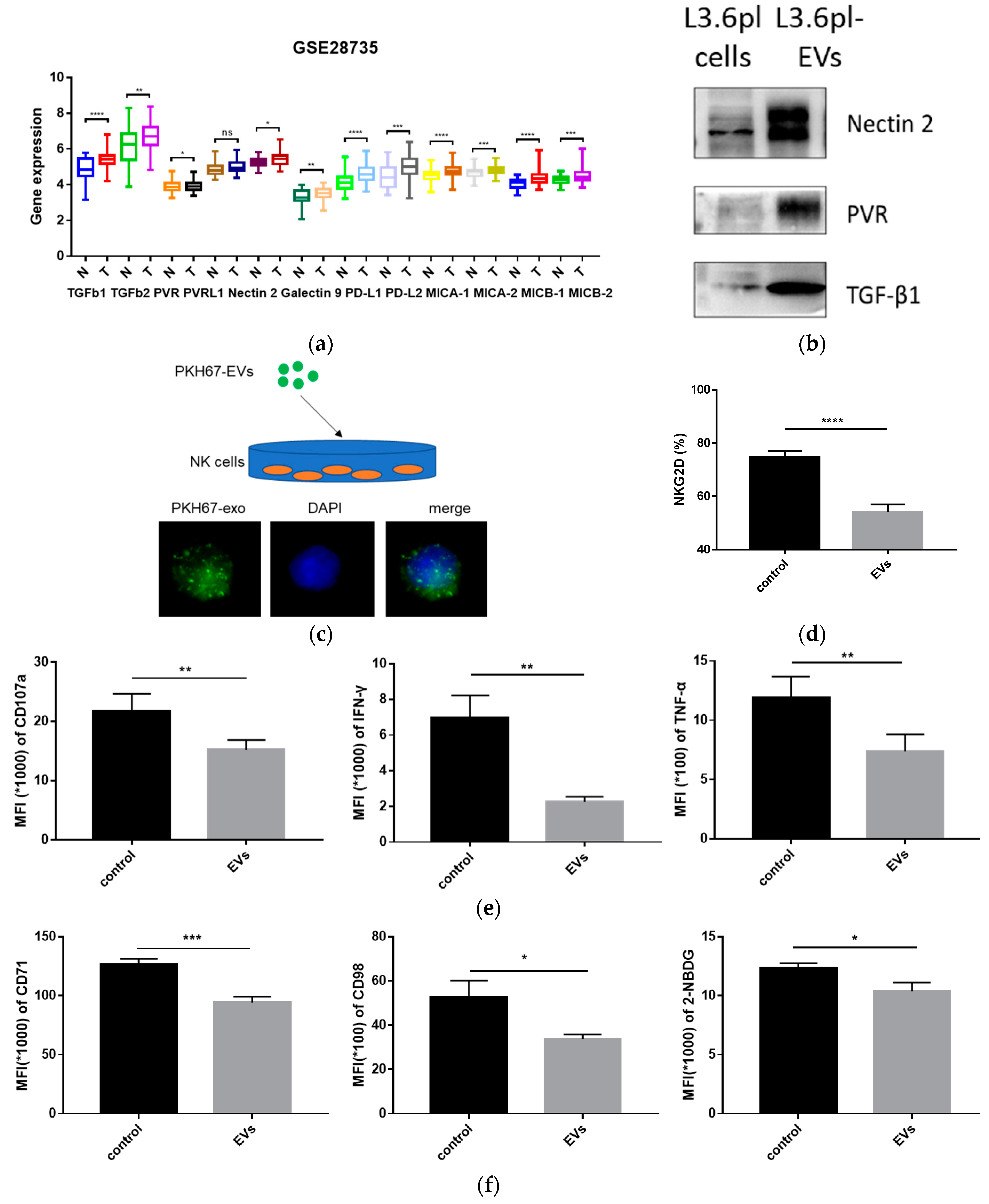
2.4. Pancreatic Cancer-Derived EVs Carry Immune Regulatory Factors
2.5. Pancreatic Cancer-Derived EVs Inhibit NK Cell Function
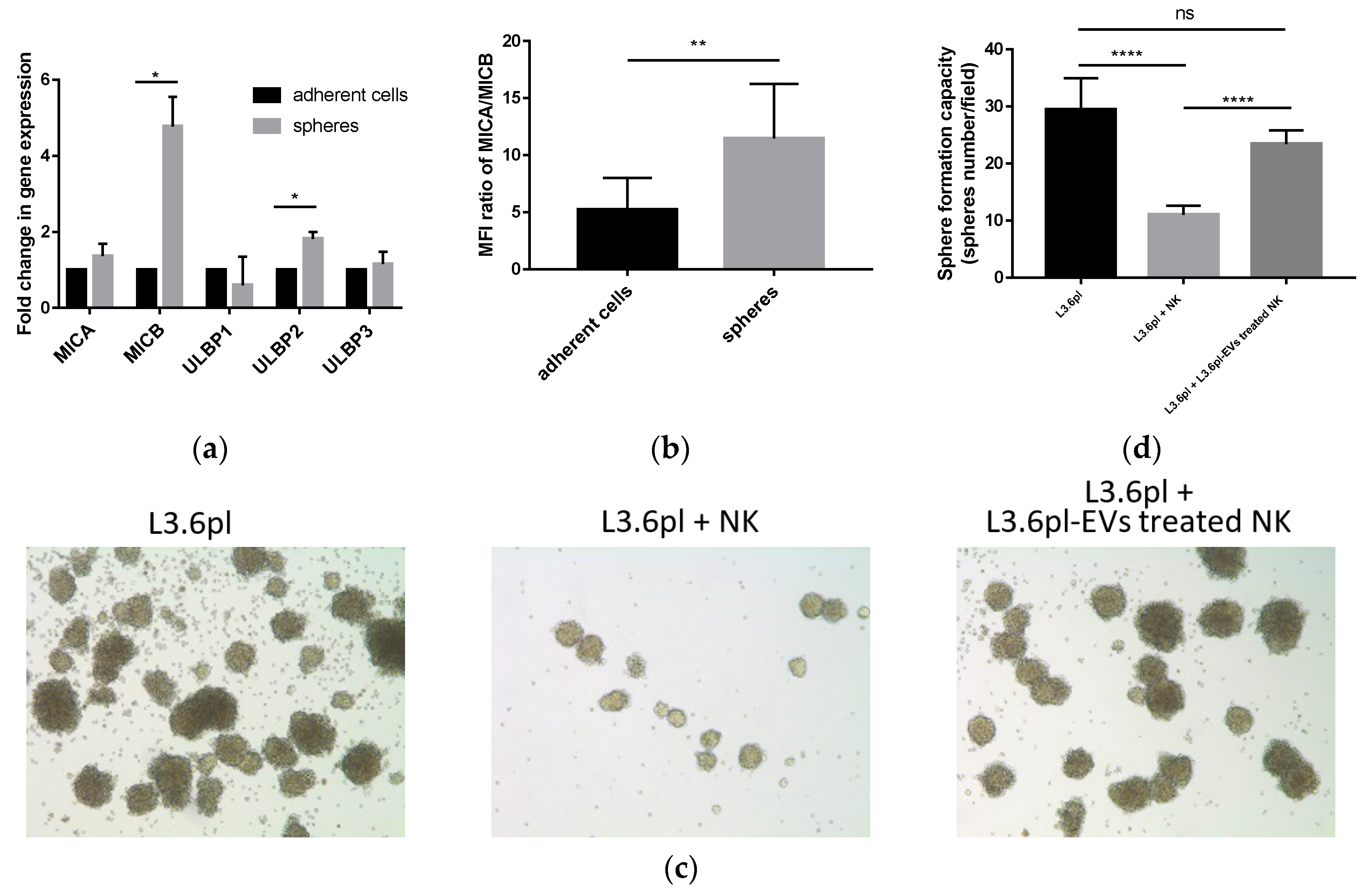
2.6. Pancreatic Cancer-Derived EVs Impair NK Cell Cytotoxicity
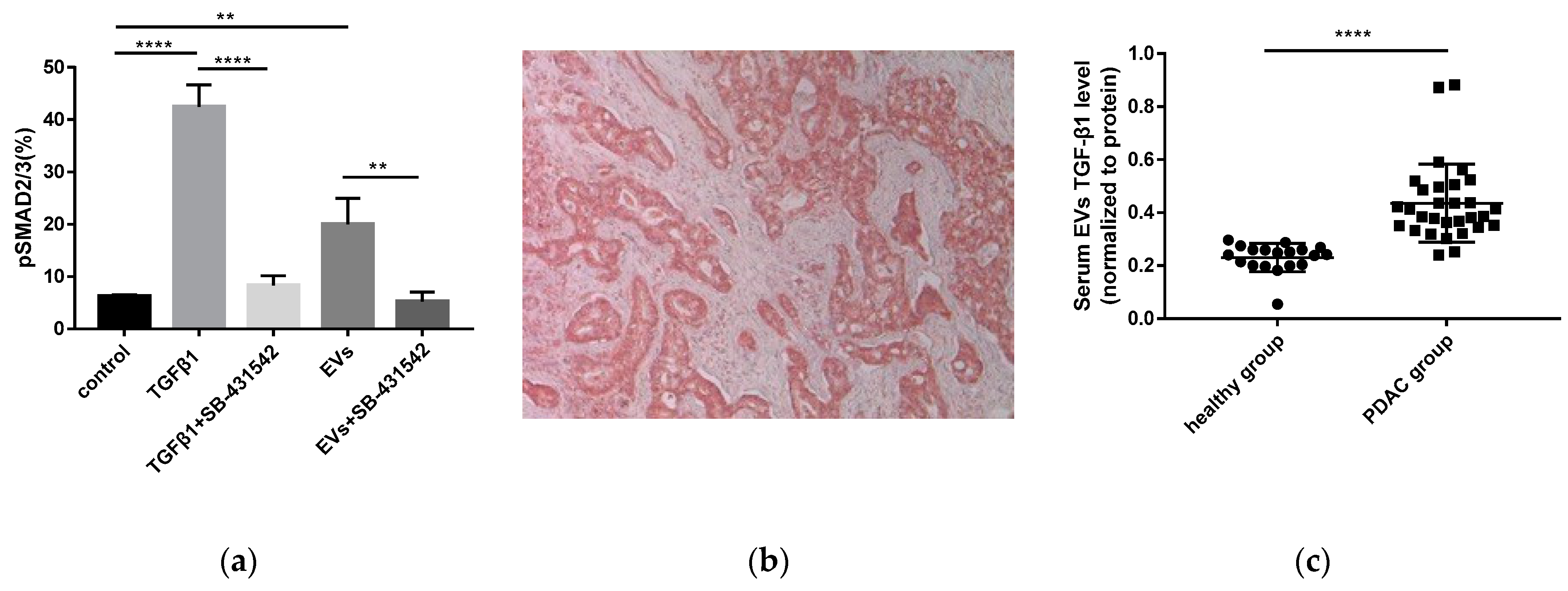
2.7. Pancreatic Cancer-Derived EVs Phosphorylate Smad2/3 in NK Cells
2.8. Increased TGF-β1 Levels in Serum EVs of Patients with PDAC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. EVs’ Isolation
4.3. Nanoparticle Tracking Analysis
4.4. Transmission Electron Microscopy
4.5. Western Blot
4.6. Flow Cytometry of EVs
4.7. Flow Cytometry of Cells
4.8. Mass Spectrometry of EVs
4.9. In Vitro EVs’ Uptake Assay
4.10. In Vivo Distribution of EVs
4.11. In Vitro NK Cell Cytotoxicity Assay
4.12. Sphere Formation Assay
4.13. RNA Isolation, cDNA Synthesis, and qRT-PCR
4.14. Human Studies
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Kommalapati, A.; Tella, S.H.; Goyal, G. Contemporary Management of Localized Resectable Pancreatic Cancer. Cancers (Basel) 2018, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gillam, A.; Lacy, J.; Hosein, P.J.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754. [Google Scholar] [CrossRef] [PubMed]
- S, E.L.A.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lotvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Saari, H.; Lazaro-Ibanez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release Off. J. Control. Release Soc. 2015, 220, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release Off. J. Control. Release Soc. 2019, 294, 165–175. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Gurlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350. [Google Scholar] [CrossRef]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J. Immunol. 2015. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; Andre, P.; Dieu-Nosjean, M.C.; Alifano, M.; Regnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Tseng, H.C. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J. Cancer 2011, 2, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Sheppard, S.; Ferry, A.; Guedes, J.; Guerra, N. The Paradoxical Role of NKG2D in Cancer Immunity. Front. Immunol. 2018, 9, 1808. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Wang, R.; Jaw, J.J.; Stutzman, N.C.; Zou, Z.; Sun, P.D. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012, 91, 299–309. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Rouce, R.H.; Shaim, H.; Sekine, T.; Weber, G.; Ballard, B.; Ku, S.; Barese, C.; Murali, V.; Wu, M.F.; Liu, H.; et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 2016, 30, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Zhao, S.; Ren, L.; Wang, L.; Chen, Z.; Hoffman, R.M.; Zhou, J. Pancreatic cancer-derived exosomes promote tumor metastasis and liver pre-metastatic niche formation. Oncotarget 2017, 8, 63461–63483. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Q.; Zhen, Q.; Zhao, Y.; Liu, N.; Li, T.; Hao, Y.; Zhang, Y.; Luo, C.; Wu, X. Negative regulation of tumor-infiltrating NK cell in clear cell renal cell carcinoma patients through the exosomal pathway. Oncotarget 2017, 8, 37783–37795. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef]
- Yadav, D.K.; Bai, X.; Yadav, R.K.; Singh, A.; Li, G.; Ma, T.; Chen, W.; Liang, T. Liquid biopsy in pancreatic cancer: The beginning of a new era. Oncotarget 2018, 9, 26900–26933. [Google Scholar] [CrossRef]
- Theodoraki, M.N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical Significance of PD-L1(+) Exosomes in Plasma of Head and Neck Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 896–905. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018, 4, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.-H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers 2019, 11, 874. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060874
Zhao J, Schlößer HA, Wang Z, Qin J, Li J, Popp F, Popp MC, Alakus H, Chon S-H, Hansen HP, et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers. 2019; 11(6):874. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060874
Chicago/Turabian StyleZhao, Jiangang, Hans A. Schlößer, Zhefang Wang, Jie Qin, Jiahui Li, Felix Popp, Marie Christine Popp, Hakan Alakus, Seung-Hun Chon, Hinrich P. Hansen, and et al. 2019. "Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer" Cancers 11, no. 6: 874. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060874