Chemotherapeutic Drugs Inhibiting Topoisomerase 1 Activity Impede Cytokine-Induced and NF-κB p65-Regulated Gene Expression

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
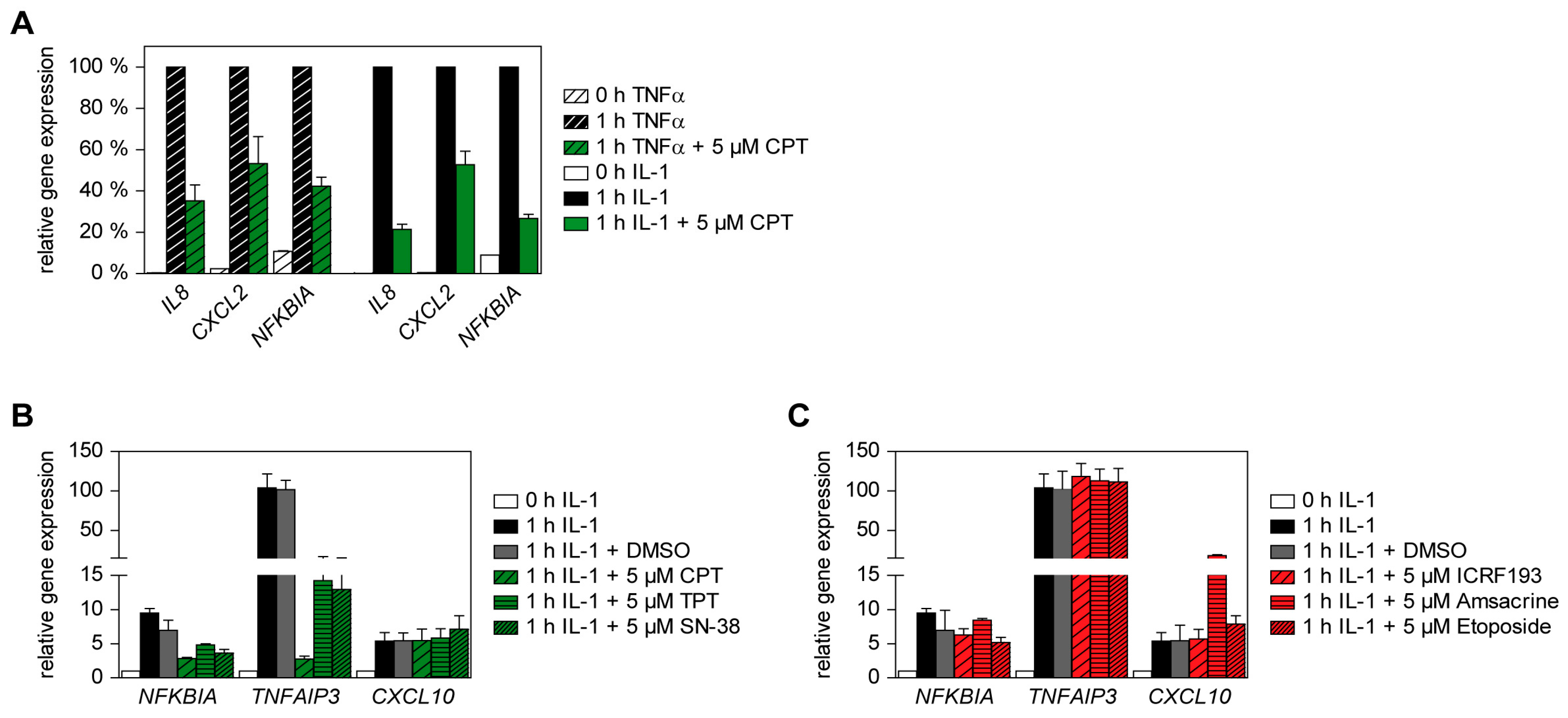
2.1. Effects of Various Clinically Used TOP1 and TOP2 Inhibitors on TNFα-Triggered Gene Expression
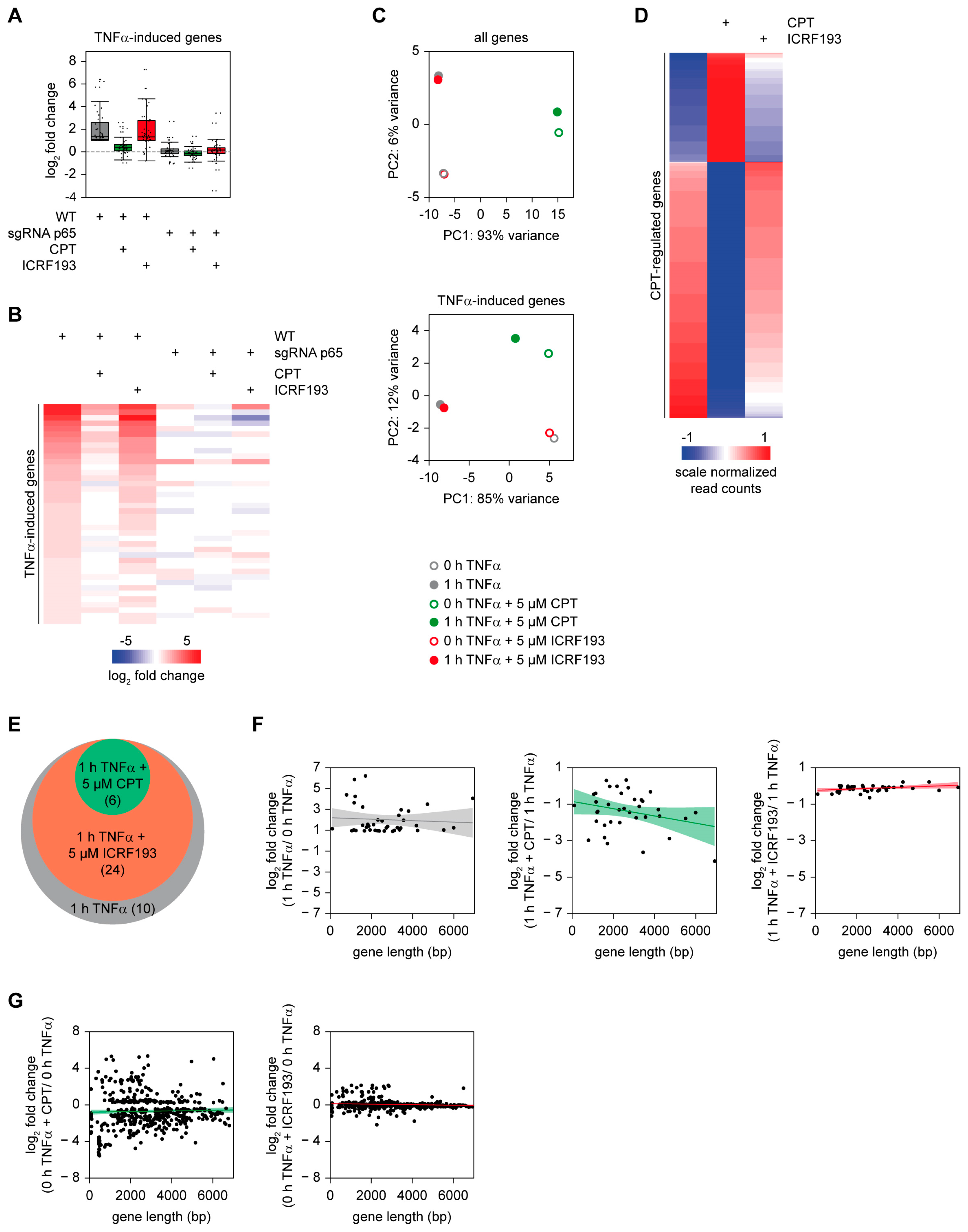
2.2. A General and Supportive Role of TOP1 for the Induction of the TNFα-Triggered Gene Response
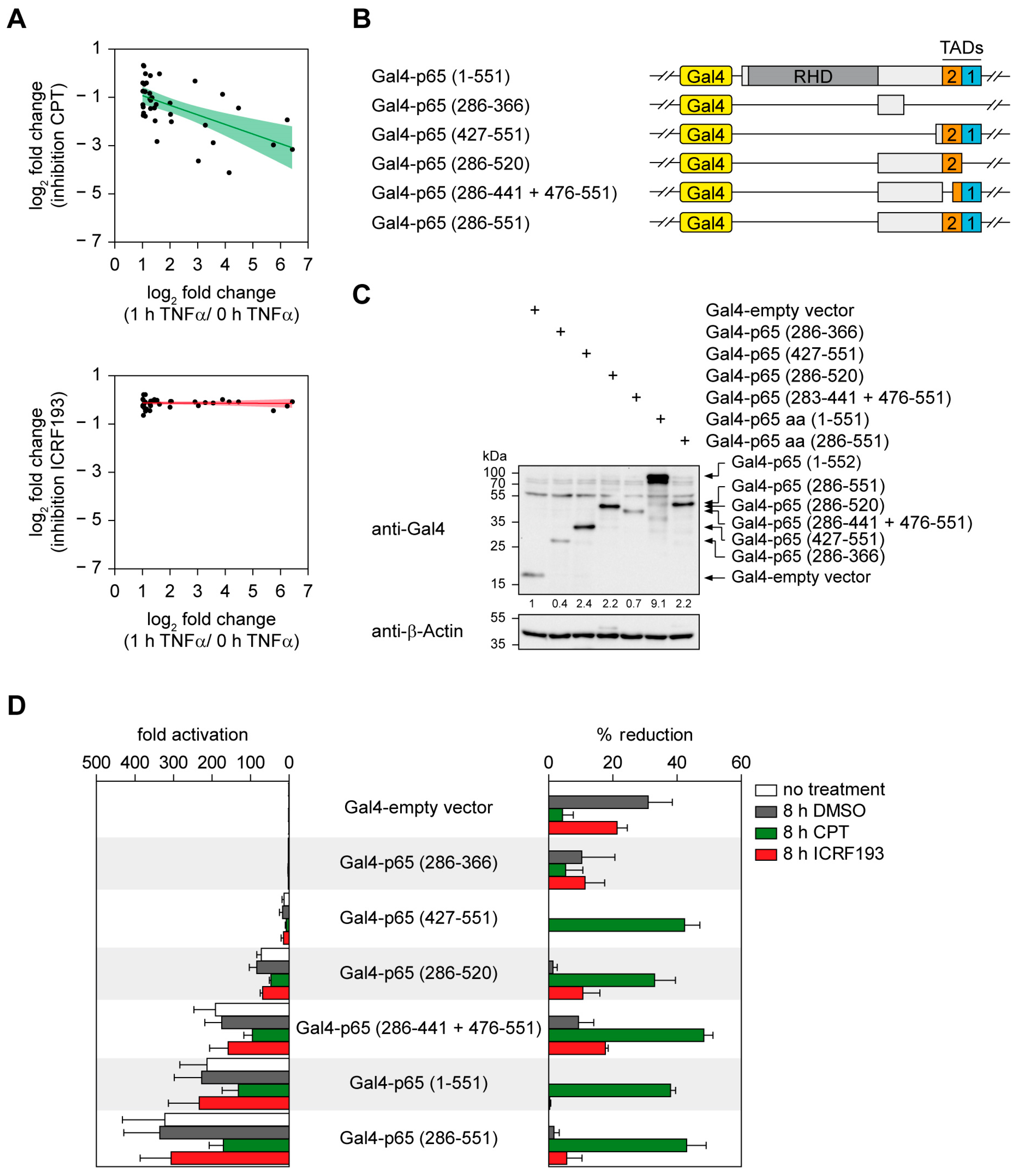
2.3. The Positive Effect of TOP1 on Gene Expression Does Not Strictly Correlate with the Magnitude of Gene Induction
2.4. IL-1-Triggered Gene Expression Is Also Supported by TOP1
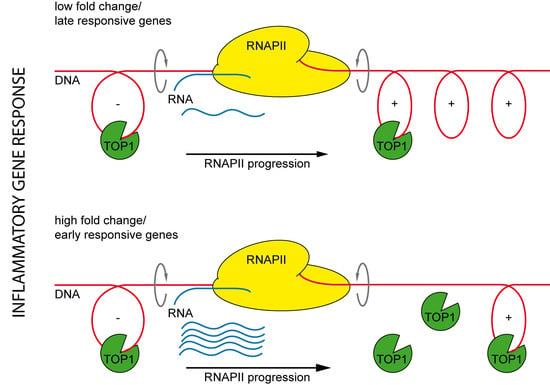
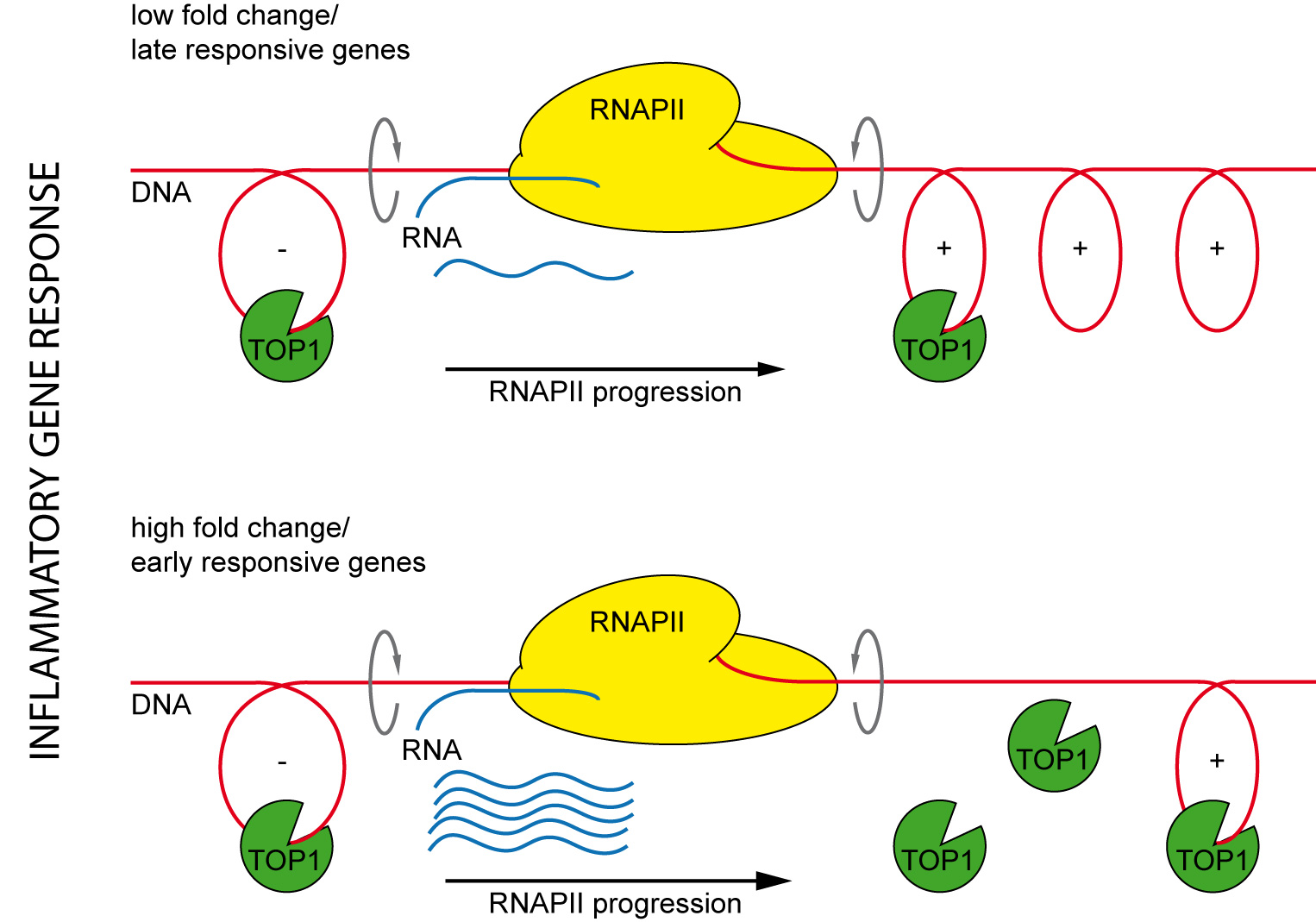
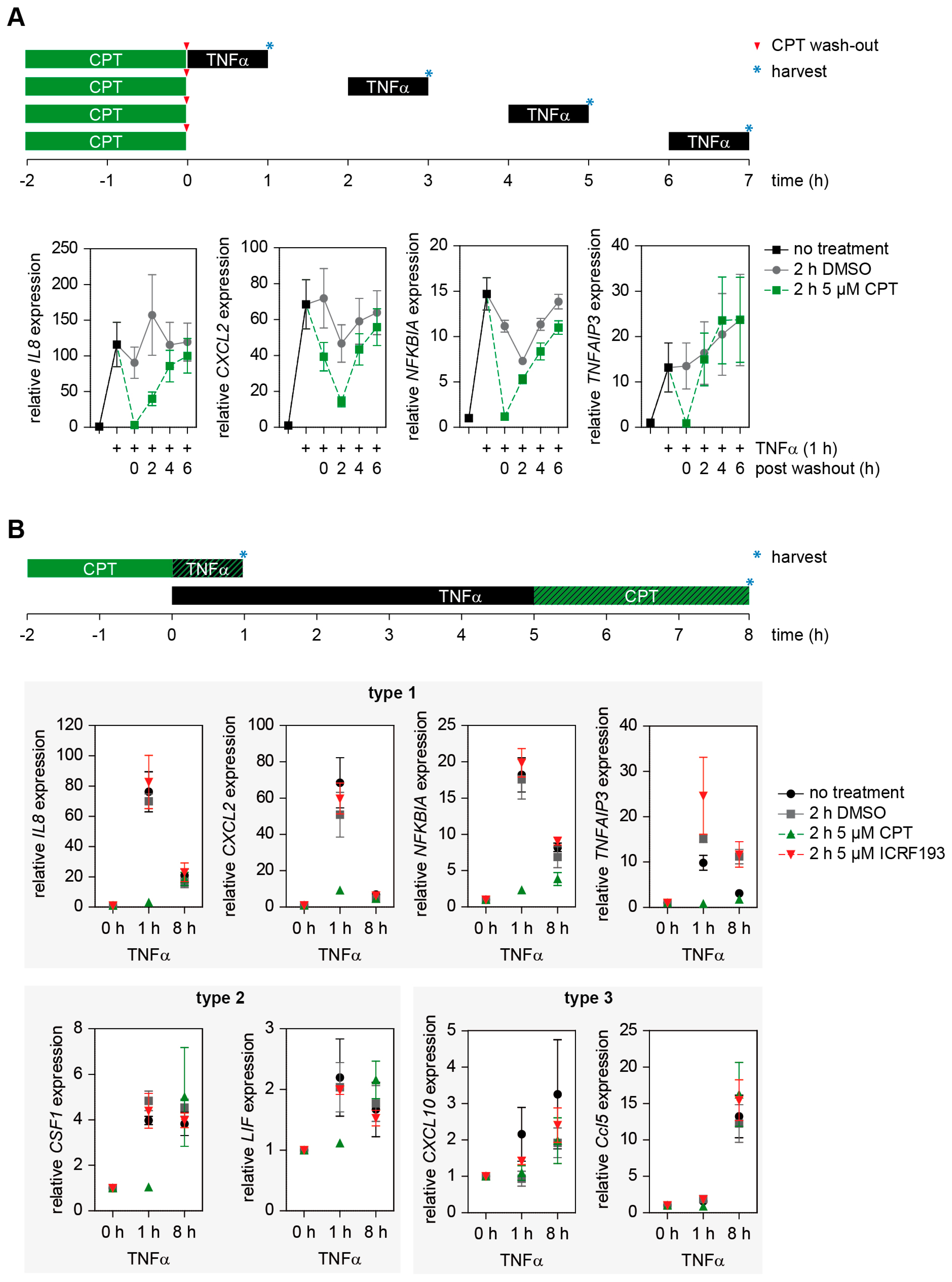
2.5. The Effect of TOP1 on Inflammatory Gene Expression Is Fully Reversible and Preferentially Affects Early Induced Genes
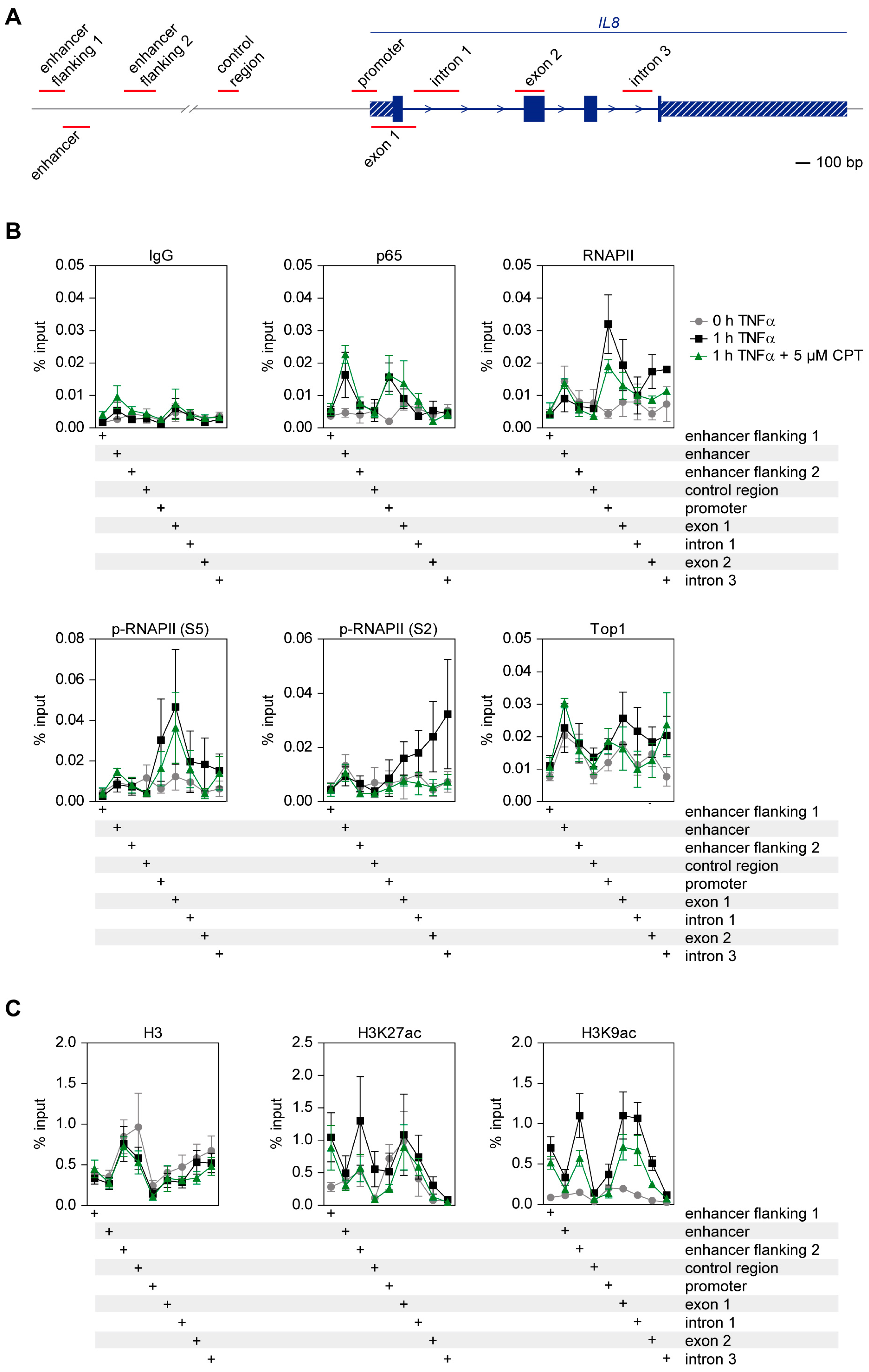
2.6. CPT Interferes with TNFα-Induced Histone H3 Acetylation and Transcription Elongation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfections
4.2. Cell Extraction and Western Blotting
4.3. Quantification of Gene Expression by RT-qPCR
4.4. Luciferase Reporter Assays
4.5. RNA-Seq and Bioinformatics Analysis
4.6. Chromatin-Immunoprecipitation (ChIP)
4.7. Antibodies, Plasmids, Oligonucleotides and Reagents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, M.; Patel, H.; Lai, C.F.; Nguyen, V.T.M.; Nevedomskaya, E.; Harrod, A.; Russell, R.; Remenyi, J.; Ochocka, A.M.; Thomas, R.S.; et al. Apobec3b-mediated cytidine deamination is required for estrogen receptor action in breast cancer. Cell Rep. 2015, 13, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.M.; Lees-Miller, S.P. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011, 32, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. Brca1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase ii-DNA complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase i. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar]
- Li, T.K.; Liu, L.F. Tumor cell death induced by topoisomerase-targeting drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 53–77. [Google Scholar] [CrossRef]
- Masuda, N.; Fukuoka, M.; Kusunoki, Y.; Matsui, K.; Takifuji, N.; Kudoh, S.; Negoro, S.; Nishioka, M.; Nakagawa, K.; Takada, M. Cpt-11: A new derivative of camptothecin for the treatment of refractory or relapsed small-cell lung cancer. J. Clin. Oncol. 1992, 10, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Baranello, L.; Wojtowicz, D.; Cui, K.; Devaiah, B.N.; Chung, H.J.; Chan-Salis, K.Y.; Guha, R.; Wilson, K.; Zhang, X.; Zhang, H.; et al. Rna polymerase ii regulates topoisomerase 1 activity to favor efficient transcription. Cell 2016, 165, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Veloso, A.; Biewen, B.; Paulsen, M.T.; Berg, N.; Carmo de Andrade Lima, L.; Prasad, J.; Bedi, K.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Genome-wide transcriptional effects of the anti-cancer agent camptothecin. PLoS ONE 2013, 8, e78190. [Google Scholar] [CrossRef]
- Bansal, K.; Yoshida, H.; Benoist, C.; Mathis, D. The transcriptional regulator aire binds to and activates super-enhancers. Nat. Immunol. 2017, 18, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Kozbial, P.; Li, W.; Tan, Y.; Liu, Z.; Suter, T.; Ohgi, K.A.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Ligand-dependent enhancer activation regulated by topoisomerase-i activity. Cell 2015, 160, 367–380. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Weber, A.; Roxlau, T.; Gaestel, M.; Kracht, M. Signal integration, crosstalk mechanisms and networks in the function of inflammatory cytokines. Biochim. Biophys. Acta 2011, 1813, 2165–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Henkel, T.; Machleidt, T.; Alkalay, I.; Kronke, M.; Ben-Neriah, Y.; Baeuerle, P.A. Rapid proteolysis of i kappa b-alpha is necessary for activation of transcription factor nf-kappa b. Nature 1993, 365, 182–185. [Google Scholar] [CrossRef]
- Renner, F.; Schmitz, M.L. Autoregulatory feedback loops terminating the nf-kappab response. Trends Biochem. Sci. 2009, 34, 128–135. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Baeuerle, P.A. The p65 subunit is responsible for the strong transcription activating potential of nf-kappa b. EMBO J. 1991, 10, 3805–3817. [Google Scholar] [CrossRef]
- Callegari, A.; Sieben, C.; Benke, A.; Suter, D.M.; Fierz, B.; Mazza, D.; Manley, S. Single-molecule dynamics and genome-wide transcriptomics reveal that nf-kb (p65)-DNA binding times can be decoupled from transcriptional activation. PLoS Genet. 2019, 15, e1007891. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, L.; Raiola, L.; Chabot, P.R.; Cyr, N.; Arseneault, G.; Legault, P.; Omichinski, J.G. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of nf-kappab and transcription regulatory factors. Nucleic Acids Res. 2017, 45, 5564–5576. [Google Scholar] [CrossRef] [PubMed]
- Paal, K.; Baeuerle, P.A.; Schmitz, M.L. Basal transcription factors tbp and tfiib and the viral coactivator e1a 13s bind with distinct affinities and kinetics to the transactivation domain of nf-kappab p65. Nucleic Acids Res. 1997, 25, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, M.; Hintermair, C.; Voss, K.; Eick, D. Dynamic phosphorylation patterns of rna polymerase ii ctd during transcription. Biochim. Biophys. Acta 2013, 1829, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Paul, W.E.; Seder, R.A. Lymphocyte responses and cytokines. Cell 1994, 76, 241–251. [Google Scholar] [CrossRef]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, M.R.; Vincent, J.L.; Deviere, J.; Alegre, M.; Kahn, R.J.; Dupont, E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 1993, 103, 565–575. [Google Scholar] [CrossRef]
- Ulloa, L.; Tracey, K.J. The “cytokine profile”: A code for sepsis. Trends Mol. Med. 2005, 11, 56–63. [Google Scholar] [CrossRef]
- Khobta, A.; Ferri, F.; Lotito, L.; Montecucco, A.; Rossi, R.; Capranico, G. Early effects of topoisomerase i inhibition on rna polymerase ii along transcribed genes in human cells. J. Mol. Biol. 2006, 357, 127–138. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Grochow, L.B.; Hendricks, C.B.; Ettinger, D.S.; Forastiere, A.A.; Hurowitz, L.A.; McGuire, W.P.; Sartorius, S.E.; Lubejko, B.G.; Kaufmann, S.H.; et al. Phase i and pharmacologic study of topotecan: A novel topoisomerase i inhibitor. J. Clin. Oncol. 1992, 10, 647–656. [Google Scholar] [CrossRef]
- Kaneda, N.; Nagata, H.; Furuta, T.; Yokokura, T. Metabolism and pharmacokinetics of the camptothecin analogue cpt-11 in the mouse. Cancer Res. 1990, 50, 1715–1720. [Google Scholar] [PubMed]
- Riedlinger, T.; Liefke, R.; Meier-Soelch, J.; Jurida, L.; Nist, A.; Stiewe, T.; Kracht, M.; Schmitz, M.L. Nf-kappab p65 dimerization and DNA-binding is important for inflammatory gene expression. FASEB J. 2019, 33, 4188–4202. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to ap-1. J. Leukoc. Biol. 1998, 63, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Scoggin, S.; Gaynor, R.B.; Williams, N.S. Identification of nf-kappa b-regulated genes induced by tnfalpha utilizing expression profiling and rna interference. Oncogene 2003, 22, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Mabb, A.M.; Simon, J.M.; King, I.F.; Lee, H.M.; An, L.K.; Philpot, B.D.; Zylka, M.J. Topoisomerase 1 regulates gene expression in neurons through cleavage complex-dependent and -independent mechanisms. PLoS ONE 2016, 11, e0156439. [Google Scholar] [CrossRef] [PubMed]
- Stielow, B.; Sapetschnig, A.; Wink, C.; Kruger, I.; Suske, G. Sumo-modified sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep. 2008, 9, 899–906. [Google Scholar] [CrossRef]
- Jurida, L.; Soelch, J.; Bartkuhn, M.; Handschick, K.; Muller, H.; Newel, D.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Bhuju, S.; et al. The activation of il-1-induced enhancers depends on tak1 kinase activity and nf-kappab p65. Cell Rep. 2015, 10, 726–739. [Google Scholar] [CrossRef]
- Herben, V.M.; ten Bokkel Huinink, W.W.; Beijnen, J.H. Clinical pharmacokinetics of topotecan. Clin. Pharmacokinet. 1996, 31, 85–102. [Google Scholar] [CrossRef]
- Hoffmann, A.; Leung, T.H.; Baltimore, D. Genetic analysis of nf-kappab/rel transcription factors defines functional specificities. EMBO J. 2003, 22, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Kawano, Y. Rhythmic binding of topoisomerase i impacts on the transcription of bmal1 and circadian period. Nucleic Acids Res. 2012, 40, 9482–9492. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Begum, N.A.; Taniguchi, T.; Taniguchi, H.; Kobayashi, M.; Honjo, T. Chromatin remodeller smarca4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat. Commun. 2016, 7, 10549. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone h3k27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone h3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Rialdi, A.; Campisi, L.; Zhao, N.; Lagda, A.C.; Pietzsch, C.; Ho, J.S.Y.; Martinez-Gil, L.; Fenouil, R.; Chen, X.; Edwards, M.; et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 2016, 352, aad7993. [Google Scholar] [CrossRef]
- Jin, J.; Xu, W.; Wan, B.; Wang, X.; Zhou, Z.; Miao, Y.; Lv, T.; Song, Y. Topotecan alleviates lipopolysaccharide-mediated acute lung injury via the nf-kappab signaling pathway. J. Surg. Res. 2019, 235, 83–92. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. MMBR 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Seufzer, B.J.; Shumway, S.D.; Kurama, T.; Boothman, D.A.; Miyamoto, S. Nf-kappab activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J. Biol. Chem. 2000, 275, 9501–9509. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. Nf-kappab inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic atm-traf6-ciap1 module links nuclear DNA damage signaling to ubiquitin-mediated nf-kappab activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.M.; Fredsoe, J.; Roedgaard, M.; Andreasen, L.; Mundbjerg, K.; Kruhoffer, M.; Brinch, M.; Schierup, M.H.; Bjergbaek, L.; Andersen, A.H. DNA topoisomerases maintain promoters in a state competent for transcriptional activation in saccharomyces cerevisiae. PLoS Genet. 2012, 8, e1003128. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase iibeta-mediated dsdna break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Flatten, K.S.; Peterson, K.L.; Beito, T.G.; Schneider, P.A.; Perkins, A.L.; Harki, D.A.; Kaufmann, S.H. Immunodetection of human topoisomerase i-DNA covalent complexes. Nucleic Acids Res. 2016, 44, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Sakasai, R.; Iwabuchi, K. The distinctive cellular responses to DNA strand breaks caused by a DNA topoisomerase i poison in conjunction with DNA replication and rna transcription. Genes Genet. Syst. 2016, 90, 187–194. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, N.; Galvin, M.; Morgan, J.G. Physical mapping of the cxc chemokine locus on human chromosome 4. Cytogenet. Cell Genet. 1999, 84, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Ribeyre, C.; Zellweger, R.; Chauvin, M.; Bec, N.; Larroque, C.; Lopes, M.; Constantinou, A. Nascent DNA proteomics reveals a chromatin remodeler required for topoisomerase i loading at replication forks. Cell Rep. 2016, 15, 300–309. [Google Scholar] [CrossRef]
- Song, L.N.; Bowen, C.; Gelmann, E.P. Structural and functional interactions of the prostate cancer suppressor protein nkx3.1 with topoisomerase i. Biochem. J. 2013, 453, 125–136. [Google Scholar] [CrossRef]
- Mialon, A.; Sankinen, M.; Soderstrom, H.; Junttila, T.T.; Holmstrom, T.; Koivusalo, R.; Papageorgiou, A.C.; Johnson, R.S.; Hietanen, S.; Elenius, K.; et al. DNA topoisomerase i is a cofactor for c-jun in the regulation of epidermal growth factor receptor expression and cancer cell proliferation. Mol. Cell. Biol. 2005, 25, 5040–5051. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Ferri, F.; Fogli, M.V.; Russo, A.; Lotito, L.; Baranello, L. The effects of camptothecin on rna polymerase ii transcription: Roles of DNA topoisomerase i. Biochimie 2007, 89, 482–489. [Google Scholar] [CrossRef]
- Marinello, J.; Chillemi, G.; Bueno, S.; Manzo, S.G.; Capranico, G. Antisense transcripts enhanced by camptothecin at divergent cpg-island promoters associated with bursts of topoisomerase i-DNA cleavage complex and r-loop formation. Nucleic Acids Res. 2013, 41, 10110–10123. [Google Scholar] [CrossRef] [PubMed]
- Durand-Dubief, M.; Persson, J.; Norman, U.; Hartsuiker, E.; Ekwall, K. Topoisomerase i regulates open chromatin and controls gene expression in vivo. EMBO J. 2010, 29, 2126–2134. [Google Scholar] [CrossRef] [PubMed]
- Munschauer, M.; Nguyen, C.T.; Sirokman, K.; Hartigan, C.R.; Hogstrom, L.; Engreitz, J.M.; Ulirsch, J.C.; Fulco, C.P.; Subramanian, V.; Chen, J.; et al. The norad lncrna assembles a topoisomerase complex critical for genome stability. Nature 2018, 561, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic effects of topoisomerase i inhibition on r-loops and short transcripts at active promoters. PLoS ONE 2016, 11, e0147053. [Google Scholar] [CrossRef]
- Longo, P.A.; Kavran, J.M.; Kim, M.S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (pei). Methods Enzymol. 2013, 529, 227–240. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. Deeptools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An r package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedlinger, T.; Bartkuhn, M.; Zimmermann, T.; Hake, S.B.; Nist, A.; Stiewe, T.; Kracht, M.; Schmitz, M.L. Chemotherapeutic Drugs Inhibiting Topoisomerase 1 Activity Impede Cytokine-Induced and NF-κB p65-Regulated Gene Expression. Cancers 2019, 11, 883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060883
Riedlinger T, Bartkuhn M, Zimmermann T, Hake SB, Nist A, Stiewe T, Kracht M, Schmitz ML. Chemotherapeutic Drugs Inhibiting Topoisomerase 1 Activity Impede Cytokine-Induced and NF-κB p65-Regulated Gene Expression. Cancers. 2019; 11(6):883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060883
Chicago/Turabian StyleRiedlinger, Tabea, Marek Bartkuhn, Tobias Zimmermann, Sandra B. Hake, Andrea Nist, Thorsten Stiewe, Michael Kracht, and M. Lienhard Schmitz. 2019. "Chemotherapeutic Drugs Inhibiting Topoisomerase 1 Activity Impede Cytokine-Induced and NF-κB p65-Regulated Gene Expression" Cancers 11, no. 6: 883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11060883